Neuronal Activity and Its Role in Controlling Antioxidant Genes

 ,
, 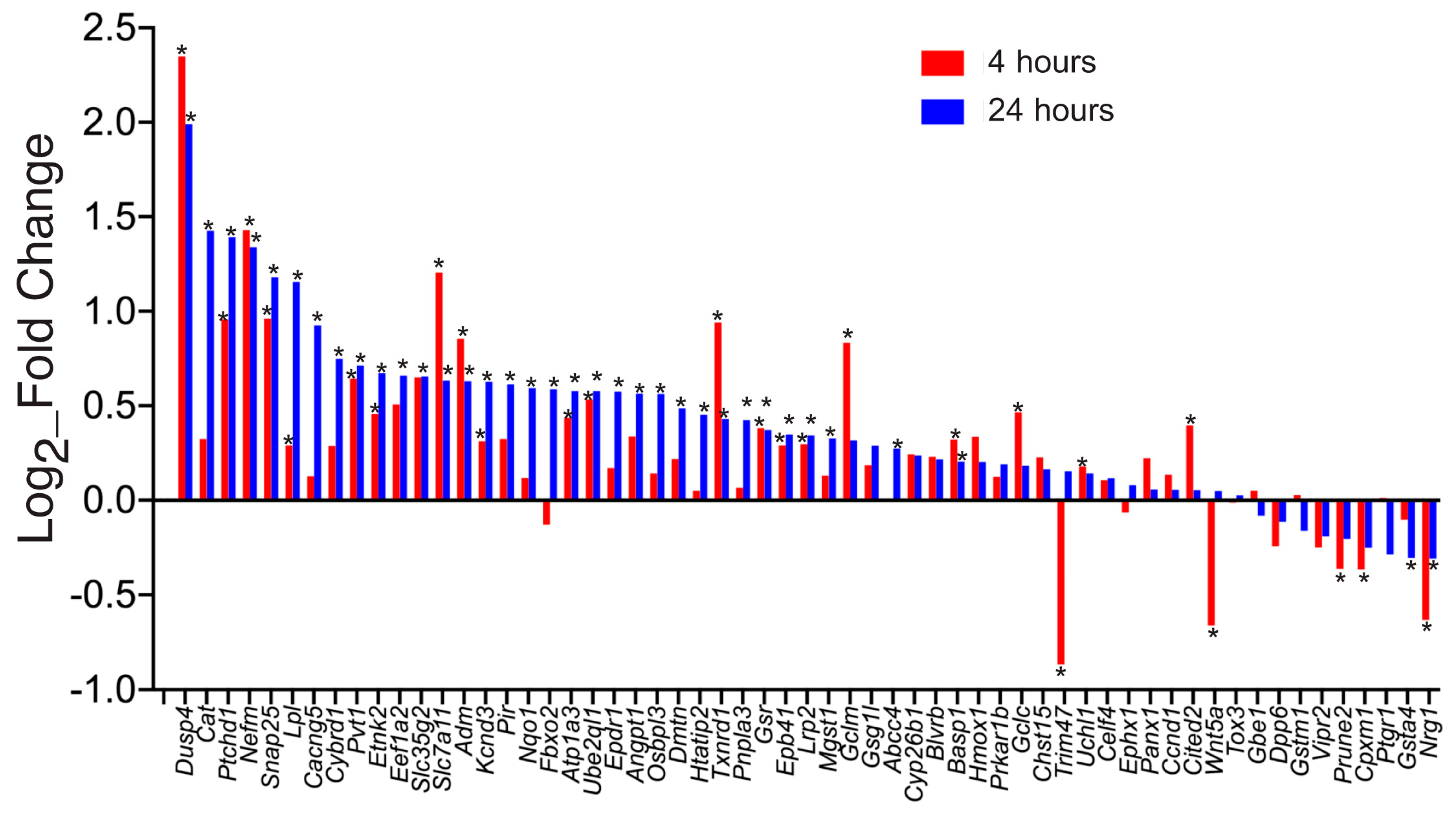{kind=link}
Abstract
:1. The Nrf2 Pathway Is Weak in Neurons
2. The Forebrain Neuronal Nrf2 Pathway Is Developmentally Shut off
3. Metabolic Adaption Induced by Neuronal Ca2+ Signals
4. Neuronal Regulation of Antioxidant Defenses
5. Neuronal Ca2+ Signals Can Induce Nrf2 Target Genes Independently of Nrf2
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Fernandez-Fernandez, S.; Almeida, A.; Bolanos, J.P. Antioxidant and bioenergetic coupling between neurons and astrocytes. Biochem. J. 2012, 443, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dringen, R.; Pawlowski, P.G.; Hirrlinger, J. Peroxide detoxification by brain cells. J. Neurosci. Res. 2005, 79, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Johnson, D.A.; Wong, G.; Kraft, A.D.; Jiang, L.; Erb, H.; Johnson, J.A.; Murphy, T.H. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J. Neurosci. 2003, 23, 3394–3406. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Hirrlinger, J. Glutathione pathways in the brain. Biol. Chem. 2003, 384, 505–516. [Google Scholar] [CrossRef]
- Vargas, M.R.; Johnson, J.A. The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev. Mol. Med. 2009, 11, e17. [Google Scholar] [CrossRef] [Green Version]
- Gupta, K.; Patani, R.; Baxter, P.; Serio, A.; Story, D.; Tsujita, T.; Hayes, J.D.; Pedersen, R.A.; Hardingham, G.E.; Chandran, S. Human embryonic stem cell derived astrocytes mediate non-cell-autonomous neuroprotection through endogenous and drug-induced mechanisms. Cell Death Differ. 2012, 19, 779–787. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Hernandez, J.I.; Almeida, A.; Delgado-Esteban, M.; Fernandez, E.; Bolanos, J.P. Knockdown of glutamate-cysteine ligase by small hairpin RNA reveals that both catalytic and modulatory subunits are essential for the survival of primary neurons. J. Biol. Chem. 2005, 280, 38992–39001. [Google Scholar] [CrossRef] [Green Version]
- Cuevas, C.; Huenchuguala, S.; Munoz, P.; Villa, M.; Paris, I.; Mannervik, B.; Segura-Aguilar, J. Glutathione transferase-M2-2 secreted from glioblastoma cell protects SH-SY5Y cells from aminochrome neurotoxicity. Neurotox. Res. 2015, 27, 217–228. [Google Scholar] [CrossRef]
- Dreger, H.; Westphal, K.; Weller, A.; Baumann, G.; Stangl, V.; Meiners, S.; Stangl, K. Nrf2-dependent upregulation of antioxidative enzymes: A novel pathway for proteasome inhibitor-mediated cardioprotection. Cardiovasc. Res. 2009, 83, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Zhu, H.; Jia, Z.; Zhang, L.; Yamamoto, M.; Misra, H.P.; Trush, M.A.; Li, Y. Antioxidants and phase 2 enzymes in macrophages: Regulation by Nrf2 signaling and protection against oxidative and electrophilic stress. Exp. Biol. Med. (Maywood) 2008, 233, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Higgins, L.G.; Kelleher, M.O.; Eggleston, I.M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Transcription factor Nrf2 mediates an adaptive response to sulforaphane that protects fibroblasts in vitro against the cytotoxic effects of electrophiles, peroxides and redox-cycling agents. Toxicol. Appl. Pharmacol. 2009, 237, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; McMahon, M.; Chowdhry, S.; Dinkova-Kostova, A.T. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid. Redox Signal. 2010, 13, 1713–1748. [Google Scholar] [CrossRef]
- McMahon, M.; Lamont, D.J.; Beattie, K.A.; Hayes, J.D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc and alkenals. Proc. Natl. Acad. Sci. USA 2010, 107, 18838–18843. [Google Scholar] [CrossRef] [Green Version]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [Green Version]
- Bell, K.F.; Al-Mubarak, B.; Fowler, J.H.; Baxter, P.S.; Gupta, K.; Tsujita, T.; Chowdhry, S.; Patani, R.; Chandran, S.; Horsburgh, K.; et al. Mild oxidative stress activates Nrf2 in astrocytes, which contributes to neuroprotective ischemic preconditioning. Proc. Natl. Acad. Sci. USA 2011, 108. [Google Scholar] [CrossRef] [Green Version]
- Bell, K.F.; Fowler, J.H.; Al-Mubarak, B.; Horsburgh, K.; Hardingham, G.E. Activation of Nrf2-regulated glutathione pathway genes by ischemic preconditioning. Oxid. Med. Cell Longev. 2011, 2011, 689524. [Google Scholar] [CrossRef] [Green Version]
- Bell, K.F.; Al-Mubarak, B.; Martel, M.A.; McKay, S.; Wheelan, N.; Hasel, P.; Markus, N.M.; Baxter, P.; Deighton, R.F.; Serio, A.; et al. Neuronal development is promoted by weakened intrinsic antioxidant defences due to epigenetic repression of Nrf2. Nat. Commun. 2015, 6, 7066. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Blasco, D.; Santofimia-Castano, P.; Gonzalez, A.; Almeida, A.; Bolanos, J.P. Astrocyte NMDA receptors’ activity sustains neuronal survival through a Cdk5-Nrf2 pathway. Cell Death Differ. 2015, 22, 1877–1889. [Google Scholar] [CrossRef] [Green Version]
- Hall, C.N.; Klein-Flugge, M.C.; Howarth, C.; Attwell, D. Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J. Neurosci. 2012, 32, 8940–8951. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [Green Version]
- Llorente-Folch, I.; Rueda, C.B.; Amigo, I.; del Arco, A.; Saheki, T.; Pardo, B.; Satrustegui, J. Calcium-regulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1-malate aspartate shuttle in intact cortical neurons. J. Neurosci. 2013, 33, 13957–13971. [Google Scholar] [CrossRef] [PubMed]
- Gellerich, F.N.; Gizatullina, Z.; Gainutdinov, T.; Muth, K.; Seppet, E.; Orynbayeva, Z.; Vielhaber, S. The control of brain mitochondrial energization by cytosolic calcium: The mitochondrial gas pedal. IUBMB Life 2013, 65, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Ca(2+)-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem. J. 1992, 283, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Glancy, B.; Balaban, R.S. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef] [Green Version]
- Hasel, P.; McKay, S.; Qiu, J.; Hardingham, G.E. Selective dendritic susceptibility to bioenergetic, excitotoxic and redox perturbations in cortical neurons. Biochim. Biophys. Acta 2015, 1853, 2066–2076. [Google Scholar] [CrossRef] [Green Version]
- Bas-Orth, C.; Schneider, J.; Lewen, A.; McQueen, J.; Hasenpusch-Theil, K.; Theil, T.; Hardingham, G.E.; Bading, H.; Kann, O. The mitochondrial calcium uniporter is crucial for the generation of fast cortical network rhythms. J. Cereb. Blood Flow. Metab. 2019. [Google Scholar] [CrossRef]
- Poyton, R.O.; Ball, K.A.; Castello, P.R. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol. Metab. 2009, 20, 332–340. [Google Scholar] [CrossRef]
- Hongpaisan, J.; Winters, C.A.; Andrews, S.B. Calcium-dependent mitochondrial superoxide modulates nuclear CREB phosphorylation in hippocampal neurons. Mol. Cell Neurosci. 2003, 24, 1103–1115. [Google Scholar] [CrossRef]
- Hongpaisan, J.; Winters, C.A.; Andrews, S.B. Strong calcium entry activates mitochondrial superoxide generation, upregulating kinase signaling in hippocampal neurons. J. Neurosci. 2004, 24, 10878–10887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, A.M.; Suh, S.W.; Won, S.J.; Narasimhan, P.; Kauppinen, T.M.; Lee, H.; Edling, Y.; Chan, P.H.; Swanson, R.A. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 2009, 12, 857–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dryanovski, D.I.; Guzman, J.N.; Xie, Z.; Galteri, D.J.; Volpicelli-Daley, L.A.; Lee, V.M.; Miller, R.J.; Schumacker, P.T.; Surmeier, D.J. Calcium entry and alpha-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J. Neurosci. 2013, 33, 10154–10164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, P.S.; Bell, K.F.; Hasel, P.; Kaindl, A.M.; Fricker, M.; Thomson, D.; Cregan, S.P.; Gillingwater, T.H.; Hardingham, G.E. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat. Commun. 2015, 6, 6761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, P.S.; Hardingham, G.E. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 2016, 100, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardingham, G.E.; Lipton, S.A. Regulation of neuronal oxidative and nitrosative stress by endogenous protective pathways and disease processes. Antioxid. Redox Signal. 2011, 14, 1421–1424. [Google Scholar] [CrossRef]
- Nguyen, T.; Yang, C.S.; Pickett, C.B. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress. Free Radic. Biol. Med. 2004, 37, 433–441. [Google Scholar] [CrossRef]
- Soriano, F.X.; Baxter, P.; Murray, L.M.; Sporn, M.B.; Gillingwater, T.H.; Hardingham, G.E. Transcriptional regulation of the AP-1 and Nrf2 target gene sulfiredoxin. Mol. Cells 2009, 27, 279–282. [Google Scholar] [CrossRef] [Green Version]
- Lewerenz, J.; Maher, P. Control of redox state and redox signaling by neural antioxidant systems. Antioxid. Redox Signal. 2011, 14, 1449–1465. [Google Scholar] [CrossRef]
- Deighton, R.F.; Markus, N.M.; Al-Mubarak, B.; Bell, K.F.; Papadia, S.; Meakin, P.J.; Chowdhry, S.; Hayes, J.D.; Hardingham, G.E. Nrf2 target genes can be controlled by neuronal activity in the absence of Nrf2 and astrocytes. Proc. Natl. Acad. Sci. USA 2014, 111, E1818–E1820. [Google Scholar] [CrossRef] [Green Version]
- Papadia, S.; Soriano, F.X.; Leveille, F.; Martel, M.A.; Dakin, K.A.; Hansen, H.H.; Kaindl, A.; Sifringer, M.; Fowler, J.; Stefovska, V.; et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat. Neurosci. 2008, 11, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Baxter, P.; Kassubek, R.; Albrecht, P.; Van Liefferinge, J.; Westhoff, M.A.; Halatsch, M.E.; Karpel-Massler, G.; Meakin, P.J.; Hayes, J.D.; et al. Phosphoinositide 3-kinases upregulate system xc(-) via eukaryotic initiation factor 2alpha and activating transcription factor 4-A pathway active in glioblastomas and epilepsy. Antioxid. Redox Signal. 2014, 20, 2907–2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; McQueen, J.; Bilican, B.; Dando, O.; Magnani, D.; Punovuori, K.; Selvaraj, B.T.; Livesey, M.; Haghi, G.; Heron, S.; et al. Evidence for evolutionary divergence of activity-dependent gene expression in developing neurons. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilican, B.; Livesey, M.R.; Haghi, G.; Qiu, J.; Burr, K.; Siller, R.; Hardingham, G.E.; Wyllie, D.J.; Chandran, S. Physiological normoxia and absence of EGF is required for the long-term propagation of anterior neural precursors from human pluripotent cells. PLoS ONE 2014, 9, e85932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livesey, M.R.; Bilican, B.; Qiu, J.; Rzechorzek, N.M.; Haghi, G.; Burr, K.; Hardingham, G.E.; Chandran, S.; Wyllie, D.J. Maturation of AMPAR composition and the GABAAR reversal potential in hPSC-derived cortical neurons. J. Neurosci. 2014, 34, 4070–4075. [Google Scholar] [CrossRef] [Green Version]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. Neurosci. 2008, 28, 13574–13581. [Google Scholar] [CrossRef]
- Soriano, F.X.; Hardingham, G.E. Compartmentalized NMDA receptor signalling to survival and death. J. Physiol. 2007, 584, 381–387. [Google Scholar] [CrossRef]
- Wahl, A.S.; Buchthal, B.; Rode, F.; Bomholt, S.F.; Freitag, H.E.; Hardingham, G.E.; Ronn, L.C.; Bading, H. Hypoxic/ischemic conditions induce expression of the putative pro-death gene Clca1 via activation of extrasynaptic N-methyl-D-aspartate receptors. Neuroscience 2009, 158, 344–352. [Google Scholar] [CrossRef]
- Gupta, K.; Hardingham, G.E.; Chandran, S. NMDA receptor-dependent glutamate excitotoxicity in human embryonic stem cell-derived neurons. Neurosci. Lett. 2013, 543, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Bell, K.F.; Hardingham, G.E. CNS peroxiredoxins and their regulation in health and disease. Antioxid. Redox Signal. 2011, 14, 1467–1477. [Google Scholar] [CrossRef]
- Turrigiano, G. Homeostatic synaptic plasticity: Local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 2012, 4, a005736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardingham, N.R.; Hardingham, G.E.; Fox, K.D.; Jack, J.J. Presynaptic efficacy directs normalization of synaptic strength in layer 2/3 rat neocortex after paired activity. J. Neurophysiol. 2007, 97, 2965–2975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardingham, N.R.; Read, J.C.; Trevelyan, A.J.; Nelson, J.C.; Jack, J.J.; Bannister, N.J. Quantal analysis reveals a functional correlation between presynaptic and postsynaptic efficacy in excitatory connections from rat neocortex. J. Neurosci. 2010, 30, 1441–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, K.F.; Hardingham, G.E. The influence of synaptic activity on neuronal health. Curr. Opin. Neurobiol. 2011, 21, 299–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mubarak, B.; Soriano, F.X.; Hardingham, G.E. Synaptic NMDAR activity suppresses FOXO1 expression via a cis-acting FOXO binding site: FOXO1 is a FOXO target gene. Channels Austin. Tex. 2009, 3, 233–238. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, G.J.; Stevenson, P.; Ward, G.; Papadia, S.; Bading, H.; Chawla, S.; Privalsky, M.; Hardingham, G.E. Nuclear Ca2+ and CaM kinase IV specify hormonal- and Notch-responsiveness. J. Neurochem. 2005, 93, 171–185. [Google Scholar] [CrossRef]
- Baxter, P.S.; Martel, M.A.; McMahon, A.; Kind, P.C.; Hardingham, G.E. Pituitary adenylate cyclase-activating peptide induces long-lasting neuroprotection through the induction of activity-dependent signaling via the cyclic AMP response element-binding protein-regulated transcription co-activator 1. J. Neurochem. 2011, 118, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.J.; Zou, M.; Lu, L.; Lau, D.; Ditzel, D.A.; Delucinge-Vivier, C.; Aso, Y.; Descombes, P.; Bading, H. Nuclear calcium signaling controls expression of a large gene pool: Identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet. 2009, 5, e1000604. [Google Scholar] [CrossRef]
- Soriano, F.X.; Leveille, F.; Papadia, S.; Bell, K.F.; Puddifoot, C.; Hardingham, G.E. Neuronal activity controls the antagonistic balance between PGC-1α and SMRT in regulating antioxidant defences. Antioxid. Redox Signal. 2011, 14, 1425–1436. [Google Scholar] [CrossRef] [Green Version]
- Gomes, J.R.; Costa, J.T.; Melo, C.V.; Felizzi, F.; Monteiro, P.; Pinto, M.J.; Inacio, A.R.; Wieloch, T.; Almeida, R.D.; Graos, M.; et al. Excitotoxicity downregulates TrkB.FL signaling and upregulates the neuroprotective truncated TrkB receptors in cultured hippocampal and striatal neurons. J. Neurosci. 2012, 32, 4610–4622. [Google Scholar] [CrossRef] [Green Version]
- Puddifoot, C.; Martel, M.A.; Soriano, F.X.; Camacho, A.; Vidal-Puig, A.; Wyllie, D.J.; Hardingham, G.E. PGC-1alpha negatively regulates extrasynaptic NMDAR activity and excitotoxicity. J. Neurosci. 2012, 32, 6995–7000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.J.; Steijaert, M.N.; Lau, D.; Schutz, G.; Delucinge-Vivier, C.; Descombes, P.; Bading, H. Decoding NMDA Receptor Signaling: Identification of Genomic Programs Specifying Neuronal Survival and Death. Neuron 2007, 53, 549–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, J.; Dando, O.; Febery, J.A.; Fowler, J.H.; Chandran, S.; Hardingham, G.E. Neuronal Activity and Its Role in Controlling Antioxidant Genes. Int. J. Mol. Sci. 2020, 21, 1933. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061933
Qiu J, Dando O, Febery JA, Fowler JH, Chandran S, Hardingham GE. Neuronal Activity and Its Role in Controlling Antioxidant Genes. International Journal of Molecular Sciences. 2020; 21(6):1933. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061933
Chicago/Turabian StyleQiu, Jing, Owen Dando, James A. Febery, Jill H. Fowler, Siddharthan Chandran, and Giles E. Hardingham. 2020. "Neuronal Activity and Its Role in Controlling Antioxidant Genes" International Journal of Molecular Sciences 21, no. 6: 1933. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21061933