Mechanistic Chromatographic Column Characterization for the Analysis of Flavonoids Using Quantitative Structure-Retention Relationships Based on Density Functional Theory

 ,
,  , , ,
, , ,
Abstract
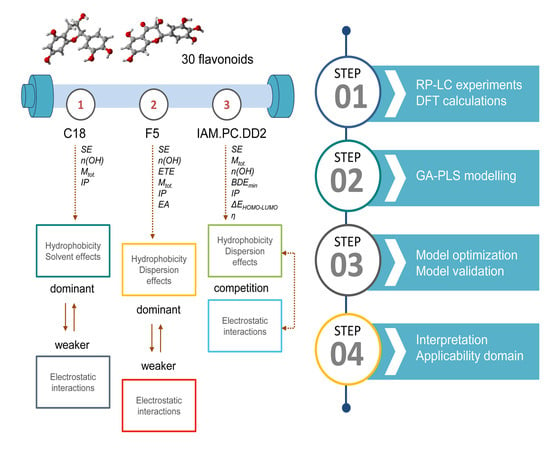
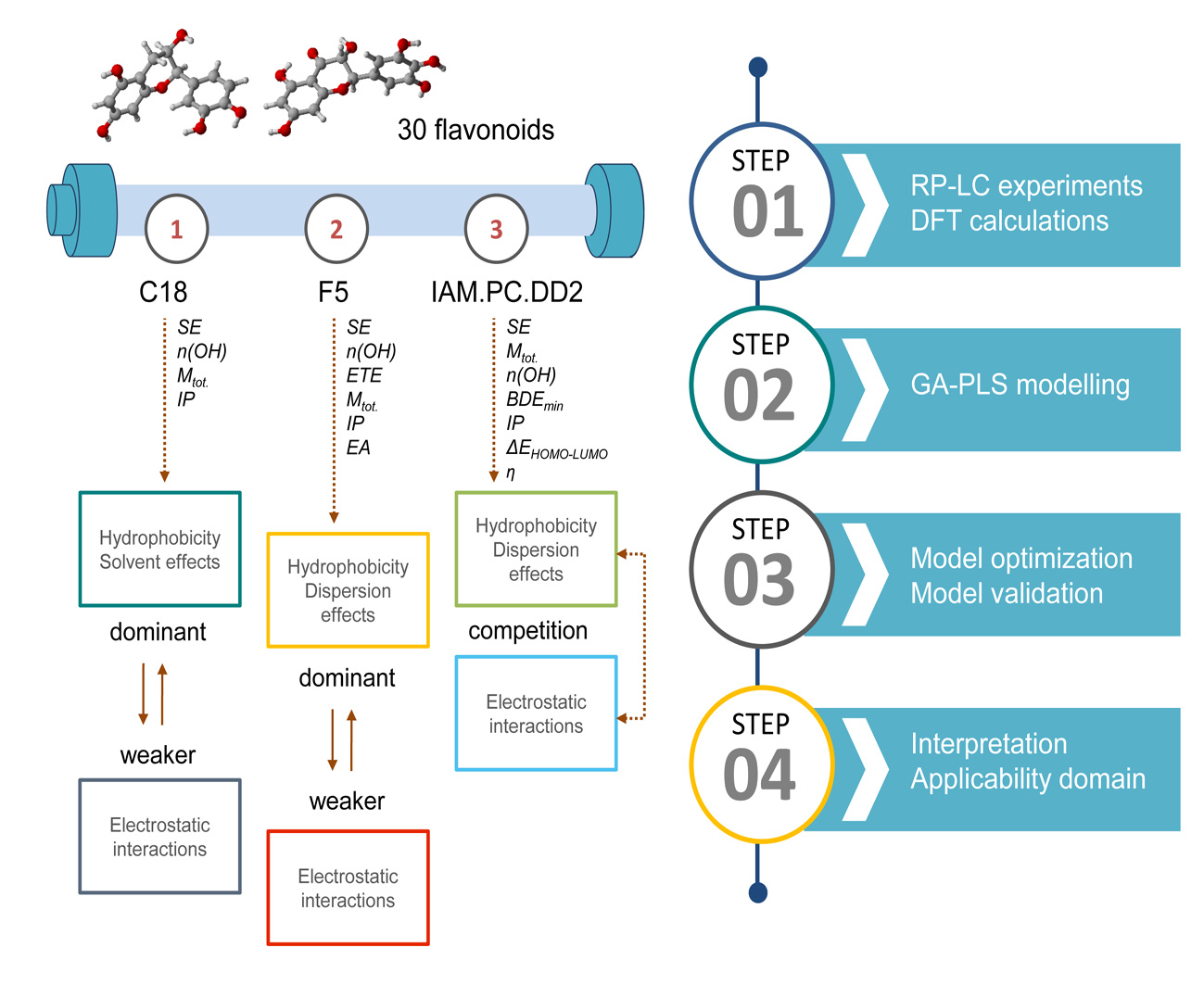
:
1. Introduction
2. Results and Discussion
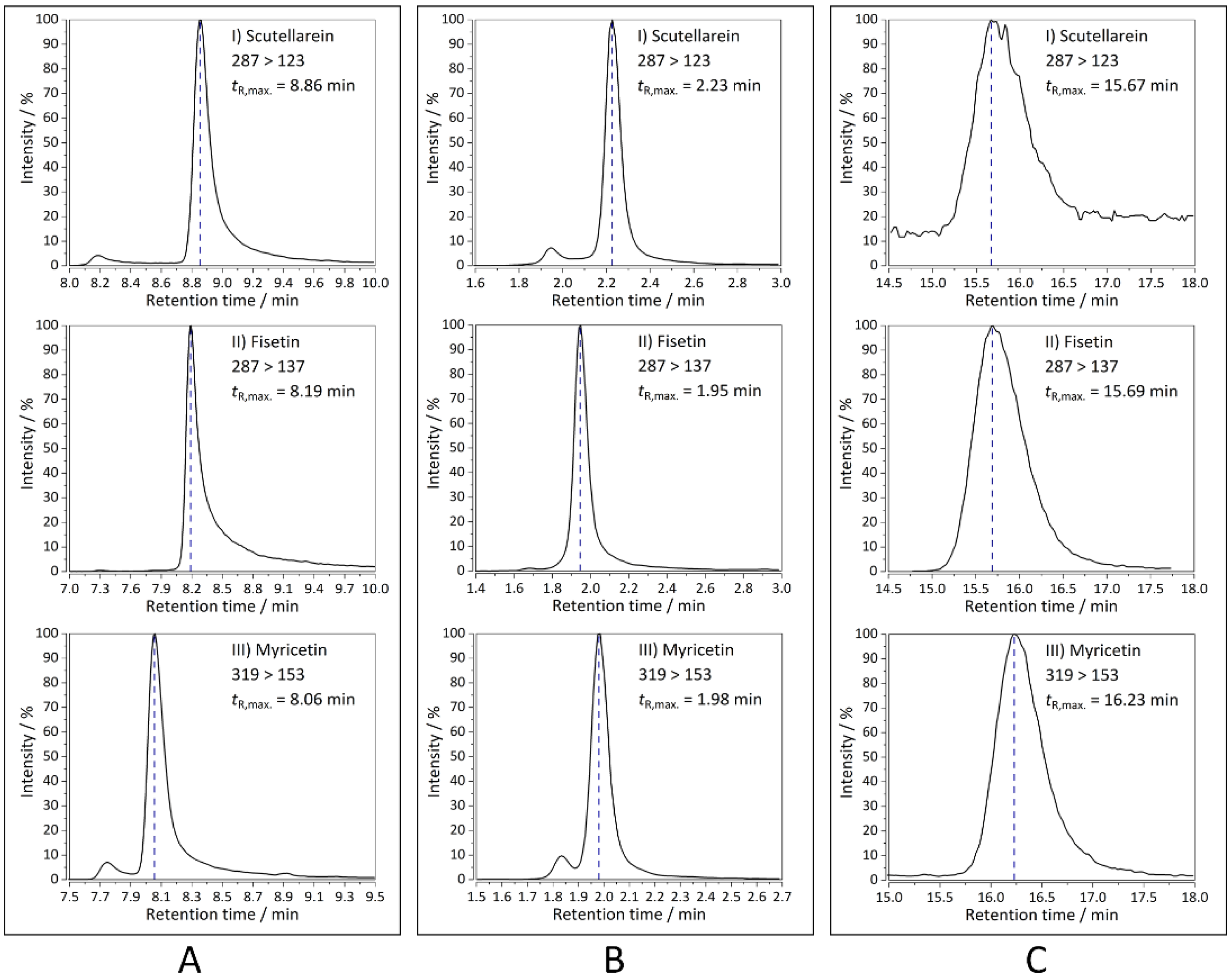
2.1. HPLC-MS/MS Analyses
2.2. Consensus Genetic Algorithm-Partial Least Squares (GA-PLS) QSRR Model for the K-C18 Column
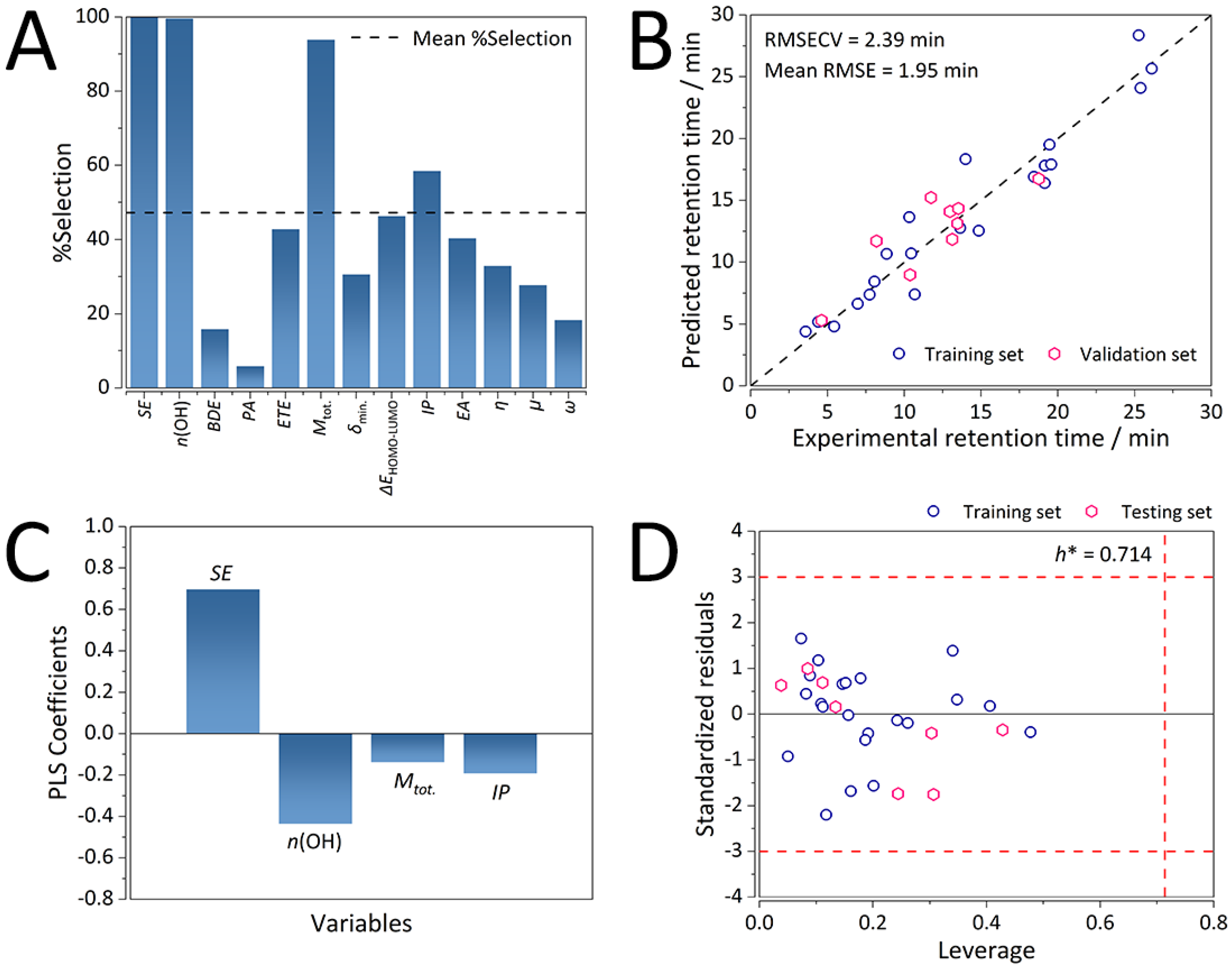
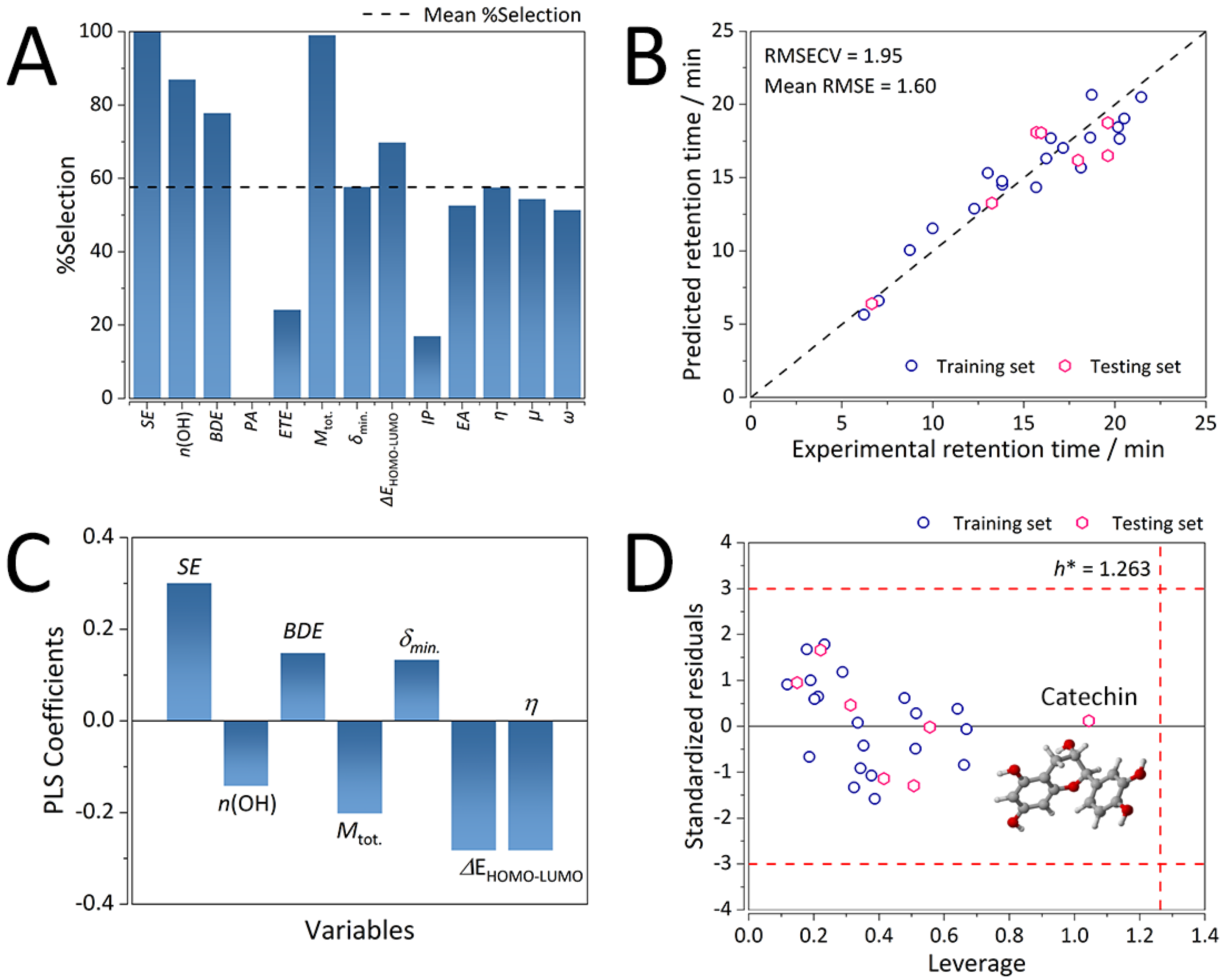
2.2.1. Selected Molecular Descriptors and Predictive Ability for the Consensus K-C18 GA-PLS QSRR Model
2.2.2. Mechanistic Interpretations of the K-C18 Consensus GA-PLS QSRR Model
2.2.3. Chemical Domain of Applicability of the K-C18 Consensus GA-PLS QSRR Model
2.3. Selected Molecular Descriptors and Predictive Ability for the Consensus K-F5 GA-PLS QSRR Model
2.3.1. Mechanistic Interpretations of the K-F5 Consensus GA-PLS QSRR Model
2.3.2. Chemical Domain of Applicability of the K-F5 Consensus GA-PLS QSRR Model
2.4. Selected Molecular Descriptors and Predictive Ability for the Consensus IAM.PC.DD2 GA-PLS QSRR Model
2.4.1. Mechanistic Interpretations of the IAM.PC.DD2 Consensus GA-PLS QSRR Model
2.4.2. Chemical Domain of Applicability of the IAM.PC.DD2 Consensus GA-PLS QSRR Model
3. Materials and Methods
3.1. Reagents and Chemicals
3.2. HPLC-MS/MS Conditions
3.3. Chromatographic Columns
3.4. Mechanistic QSRR Model Development
3.5. Theoretical Methods
3.5.1. Partial Least Squares (PLS)
3.5.2. Genetic Algorithms (GAs)
3.5.3. Optimization of GA-PLS Parameters
3.5.4. Consensus Modeling
3.5.5. Molecular Descriptors for Mechanistic QSRR Modeling
3.5.6. Hydrophobicity and the Solvent Effect
3.5.7. Antioxidant Activity
3.5.8. Electrostatic Effects
3.5.9. Mechanistic QSRR Model Validation
3.5.10. Leave-One-Out Cross-Validation (LOO-CV)
3.5.11. External Validation
3.5.12. Chemical Domain of Applicability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singh, B.; Kumar, A.; Malik, A.K. Flavonoids biosynthesis in plants and its further analysis by capillary electrophoresis. Electrophoresis 2017, 38, 820–832. [Google Scholar] [CrossRef]
- Williams, C.A.; Grayer, R.J. Anthocyanins and other flavonoids. Nat. Prod. Rep. 2004, 21, 539–573. [Google Scholar] [CrossRef]
- De Villiers, A.; Venter, P.; Pasch, H. Recent advances and trends in the liquid-chromatography-mass spectrometry analysis of flavonoids. J. Chromatogr. A 2015, 1430, 16–78. [Google Scholar] [CrossRef]
- Kaliszan, R. QSRR: Quantitative Structure-(Chromatographic) Retention Relationships. Chem. Rev. 2007, 107, 3212–3246. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Jing, L.; Qiu, F.; Zhang, H.; Huhman, D.; Zhou, Z.; Sumner, L.W. Construction of an Ultrahigh Pressure Liquid Chromatography-Tandem Mass Spectral Library of Plant Natural Products and Comparative Spectral Analyses. Anal. Chem. 2015, 87, 7373–7381. [Google Scholar] [CrossRef] [PubMed]
- Zapadka, M.; Kaczmarek, M.; Kupcewicz, B.; Dekowski, P.; Walkowiak, A.; Kokotkiewicz, A.; Łuczkiewicz, M.; Buciński, A. An application of QSRR approach and multiple linear regression method for lipophilicity assessment of flavonoids. J. Pharm. Biomed. Anal. 2019, 164, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Welerowicz, T.; Buszewski, B. The effect of stationary phase on lipophilicity determination of β-blockers using reverse-phase chromatographic systems. Biomed. Chromatogr. 2005, 19, 725–736. [Google Scholar] [CrossRef]
- Giaginis, C.; Tsantili-Kakoulidou, A. Quantitative structure-retention relationships as useful tool to characterize chromatographic systems and their potential to simulate biological processes. Chromatographia 2013, 76, 211–226. [Google Scholar] [CrossRef]
- Santoro, A.L.; Carrilho, E.; Lancas, F.M.; Montanari, C.A. Quantitative structure – retention relationships of flavonoids unraveled by immobilized artificial membrane chromatography. Eur. J. Pharm. Sci. 2016, 88, 147–157. [Google Scholar] [CrossRef]
- Wen, Y.; Amos, R.I.J.; Talebi, M.; Szucs, R.; Dolan, J.W.; Pohl, C.A.; Haddad, P.R. Retention Index Prediction Using Quantitative Structure–Retention Relationships for Improving Structure Identification in Nontargeted Metabolomics. Anal. Chem. 2018, 90, 9434–9440. [Google Scholar] [CrossRef]
- Buszewski, B.; Gadzała-Kopciuch, R.M.; Markuszewski, M.; Kaliszan, R. Chemically Bonded Silica Stationary Phases: Synthesis, Physicochemical Characterization, and Molecular Mechanism of Reversed-Phase HPLC Retention. Anal. Chem. 1997, 69, 3277–3284. [Google Scholar] [CrossRef]
- Žuvela, P.; Skoczylas, M.; Jay Liu, J.; Ba̧czek, T.; Kaliszan, R.; Wong, M.W.; Buszewski, B. Column Characterization and Selection Systems in Reversed-Phase High-Performance Liquid Chromatography. Chem. Rev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Buszewski, B.; Walczak, J.; Skoczylas, M.; Haddad, P.R. High performance liquid chromatography as a molecular probe in quantitative structure-retention relationships studies of selected lipid classes on polar-embedded stationary phases. J. Chromatogr. A 2019, 1585, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Sagandykova, G.N.; Pomastowski, P.P.; Kaliszan, R.; Buszewski, B. Modern analytical methods for consideration of natural biological activity. TrAC-Trends Anal. Chem. 2018, 198–213. [Google Scholar] [CrossRef]
- Talebi, M.; Schuster, G.; Shellie, R.A.; Szucs, R.; Haddad, P.R. Performance comparison of partial least squares-related variable selection methods for quantitative structure retention relationships modelling of retention times in reversed-phase liquid chromatography. J. Chromatogr. A 2015, 1424, 69–76. [Google Scholar] [CrossRef]
- Žuvela, P.; Liu, J.J.; Macur, K.; Bączek, T. Molecular Descriptor Subset Selection in Theoretical Peptide Quantitative Structure–Retention Relationship Model Development Using Nature-Inspired Optimization Algorithms. Anal. Chem. 2015, 87, 9876–9883. [Google Scholar] [CrossRef]
- Kaliszan, R. Quantitative structure-retention relationships applied to reversed-phase high-performance liquid chromatography. J. Chromatogr. A 1993, 656, 417–435. [Google Scholar] [CrossRef]
- Kaliszan, R. Quantitative structure-retention relationships. Anal. Chem. 1992, 64, 619–631. [Google Scholar] [CrossRef]
- Akbar, J.; Iqbal, S.; Batool, F.; Karim, A.; Chan, K.W. Predicting Retention Times of Naturally Occurring Phenolic Compounds in Reversed-Phase Liquid Chromatography: A Quantitative Structure Retention Relationship (QSRR) Approach. Int. J. Mol. Sci. 2012, 13, 15387–15400. [Google Scholar] [CrossRef]
- Wiczling, P.; Markuszewski, M.J.; Kaliszan, R. Determination of pKa by pH gradient reversed-phase HPLC. Anal. Chem. 2004, 76. [Google Scholar] [CrossRef]
- Tache, F.; Naşcu-Briciu, R.D.; Sârbu, C.; Micǎle, F.; Medvedovici, A. Estimation of the lipophilic character of flavonoids from the retention behavior in reversed phase liquid chromatography on different stationary phases: A comparative study. J. Pharm. Biomed. Anal. 2012, 57, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Taraji, M.; Haddad, P.R.; Amos, R.I.J.; Talebi, M.; Szucs, R.; Dolan, J.W.; Pohl, C.A. Rapid Method Development in Hydrophilic Interaction Liquid Chromatography for Pharmaceutical Analysis Using a Combination of Quantitative Structure–Retention Relationships and Design of Experiments. Anal. Chem. 2017, 89, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Ba̧czek, T.; Kaliszan, R. Predictive approaches to gradient retention based on analyte structural descriptors from calculation chemistry. J. Chromatogr. A 2003, 987, 29–37. [Google Scholar] [CrossRef]
- Levy, R.M.; Zhang, L.Y.; Gallicchio, E.; Felts, A.K. On the Nonpolar Hydration Free Energy of Proteins: Surface Area and Continuum Solvent Models for the Solute−Solvent Interaction Energy. J. Am. Chem. Soc. 2003, 125, 9523–9530. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Bell, D.S.; Jones, A.D. Solute attributes and molecular interactions contributing to “U-shape” retention on a fluorinated high-performance liquid chromatography stationary phase. J. Chromatogr. A 2005, 1073, 99–109. [Google Scholar] [CrossRef]
- Croes, K.; Steffens, A.; Marchand, D.H.; Snyder, L.R. Relevance of π-π and dipole-dipole interactions for retention on cyano and phenyl columns in reversed-phase liquid chromatography. J. Chromatogr. A 2005, 1098, 123–130. [Google Scholar] [CrossRef]
- Janas, P.; Bocian, S.; Jandera, P.; Kowalkowski, T.; Buszewski, B. Separation of flavonoids on different phenyl-bonded stationary phases-the influence of polar groups in stationary phase structure. J. Chromatogr. A 2016, 1429, 198–206. [Google Scholar] [CrossRef]
- Yang, M.; Fazio, S.; Munch, D.; Drumm, P. Impact of methanol and acetonitrile on separations based on π-π interactions with a reversed-phase phenyl column. J. Chromatogr. A 2005, 1097, 124–129. [Google Scholar] [CrossRef]
- Emenike, B.U.; Spinelle, R.A.; Rosario, A.; Shinn, D.W.; Yoo, B. Solvent Modulation of Aromatic Substituent Effects in Molecular Balances Controlled by CH-π Interactions. J. Phys. Chem. A 2018, 122, 909–915. [Google Scholar] [CrossRef]
- Cockroft, S.L.; Hunter, C.A. Desolvation and substituent effects in edge-to-face aromatic interactions. Chem. Commun. 2009, 3961–3963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsopelas, F.; Tsagkrasouli, M.; Poursanidis, P.; Pitsaki, M.; Vasios, G.; Danias, P.; Panderi, I.; Tsantili-Kakoulidou, A.; Giaginis, C. Retention behavior of flavonoids on immobilized artificial membrane chromatography and correlation with cell-based permeability. Biomed. Chromatogr. 2018, 32, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, G.E.; Galan, L. de Preparation and Chromatographic Properties of Some Chemically Bonded Phases For Reversed-Phase Liquid Chromatography. J. Liq. Chromatogr. 1978, 1, 561–586. [Google Scholar] [CrossRef]
- Žuvela, P.; David, J.; Yang, X.; Huang, D.; Wong, M.W. Non-Linear Quantitative Structure–Activity Relationships Modelling, Mechanistic Study and In-Silico Design of Flavonoids as Potent Antioxidants. Int. J. Mol. Sci. 2019, 20, 2328. [Google Scholar] [CrossRef] [Green Version]
- Zuvela, P.; David, J.; Wong, M.W. Interpretation of ANN-Based QSAR models for prediction of antioxidant activity of flavonoids. J. Comput. Chem. 2018. [Google Scholar]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Musialik, M.; Kuzmicz, R.; Pawłowski, T.S.; Litwinienko, G. Acidity of Hydroxyl Groups: An Overlooked Influence on Antiradical Properties of Flavonoids. J. Org. Chem. 2009, 74, 2699–2709. [Google Scholar] [CrossRef]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. Development and use of quantum mechanical molecular models. 76. AM1: a new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G*basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, 1133–1138. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Ou, B.; Prior, R.L. The Chemistry behind Antioxidant Capacity Assays. J. Agric. Food Chem. 2005, 53, 1841–1856. [Google Scholar] [CrossRef] [PubMed]
- Al-Haj, M.A.; Kaliszan, R.; Nasal, A. Test Analytes for Studies of the Molecular Mechanism of Chromatographic Separations by Quantitative Structure−Retention Relationships. Anal. Chem. 1999, 71, 2976–2985. [Google Scholar] [CrossRef] [PubMed]
- Kaliszan, R.; Marszałł, M.P.; Jan Markuszewski, M.; Bączek, T.; Pernak, J. Suppression of deleterious effects of free silanols in liquid chromatography by imidazolium tetrafluoroborate ionic liquids. J. Chromatogr. A 2004, 1030, 263–271. [Google Scholar] [CrossRef]
- Kennard, R.W.; Stone, L.A. Computer Aided Design of Experiments. Technometrics 1969, 11, 137–148. [Google Scholar] [CrossRef]
- Leardi, R. Application of genetic algorithm-PLS for feature selection in spectral data sets. J. Chemom. 2000, 14, 643–655. [Google Scholar] [CrossRef]
- Žuvela, P.; Lin, K.; Shu, C.; Zheng, W.; Lim, C.M.; Huang, Z. Fiber-Optic Raman Spectroscopy with Nature-Inspired Genetic Algorithms Enhances Real-Time in Vivo Detection and Diagnosis of Nasopharyngeal Carcinoma. Anal. Chem. 2019, 91, 8101–8108. [Google Scholar] [CrossRef]
- Eriksson, L.; Trygg, J.; Wold, S. CV-ANOVA for significance testing of PLS and OPLS® models. J. Chemom. 2008, 22, 594–600. [Google Scholar] [CrossRef]
- Wold, H. Estimation of principal components and related models by iterative least squares. In Multivariate Analysis; Krishnaiah, P.K., Ed.; Academic Press: New York, NY, USA, 1966; pp. 391–420. [Google Scholar]
- Wold, S.; Sjöström, M.; Eriksson, L. PLS-regression: A basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- de Jong, S. SIMPLS: An alternative approach to partial least squares regression. Chemom. Intell. Lab. Syst. 1993, 18, 251–263. [Google Scholar] [CrossRef]
- Holland, J.H. Genetic Algorithms. Sci. Am. 1992, 267, 66–72. [Google Scholar] [CrossRef]
- Forrest, S. Genetic algorithms: Principles of natural selection applied to computation. Science 1993, 261, 872–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietta, P.-G. Flavonoids as Antioxidants. J. Nat. Prod. 2000, 63, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Litwinienko, G.; Ingold, K.U. Abnormal Solvent Effects on Hydrogen Atom Abstractions. 1. The Reactions of Phenols with 2,2-Diphenyl-1-picrylhydrazyl (dpph • ) in Alcohols. J. Org. Chem. 2003, 68, 3433–3438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litwinienko, G.; Ingold, K.U. Abnormal Solvent Effects on Hydrogen Atom Abstraction. 2. Resolution of the Curcumin Antioxidant Controversy. The Role of Sequential Proton Loss Electron Transfer. J. Org. Chem. 2004, 69, 5888–5896. [Google Scholar] [CrossRef]
- Litwinienko, G.; Ingold, K.U. Solvent Effects on the Rates and Mechanisms of Reaction of Phenols with Free Radicals. Acc. Chem. Res. 2007, 40, 222–230. [Google Scholar] [CrossRef]
- Bartmess, J.E. Thermodynamics of the Electron and the Proton. J. Phys. Chem. 1994, 98, 6420–6424. [Google Scholar] [CrossRef]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Pearson, R.G. Chemical hardness and density functional theory. J. Chem. Sci. 2005, 117, 369–377. [Google Scholar] [CrossRef]
- Koopmans, T. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den Einzelnen Elektronen Eines Atoms. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Anderson, L.N.; Oviedo, M.B.; Wong, B.M. Accurate Electron Affinities and Orbital Energies of Anions from a Nonempirically Tuned Range-Separated Density Functional Theory Approach. J. Chem. Theory Comput. 2017, 13, 1656–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baerends, E.J.; Gritsenko, O.V.; Van Meer, R. The Kohn-Sham gap, the fundamental gap and the optical gap: The physical meaning of occupied and virtual Kohn-Sham orbital energies. Phys. Chem. Chem. Phys. 2013, 15, 16408–16425. [Google Scholar] [CrossRef]
- Eriksson, L.; Jaworska, J.; Worth, A.P.; Cronin, M.T.D.; McDowell, R.M.; Gramatica, P. Methods for reliability and uncertainty assessment and for applicability evaluations of classification- and regression-based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef] [Green Version]
- Nikolova-Jeliazkova, N.; Jaworska, J. An approach to determining applicability domains for QSAR group contribution models: An analysis of SRC KOWWIN. ATLA Altern. Lab. Anim. 2005, 33, 461–470. [Google Scholar] [CrossRef]
- Atkinson, A.C. Plots, Transformations and Regression. An Introduction to Graphical Methods of Diagnostic Regression Analysis; Clarendon Press: Oxford, UK, 1985. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Retention | Coefficients | tR (K-C18) | tR (K-F5) | tR (IAM.PC.DD2) |
|---|---|---|---|---|
| tR (K-C18) | R | 1 | ||
| p | n.a. | |||
| tR (K-F5) | R | 0.93 | 1 | |
| p | 1.42 × 10−13 | n.a. | ||
| tR (IAM.PC.DD2) | R | 0.81 | 0.79 | 1 |
| p | 2.81 × 10−7 | 1.22 × 10−6 | n.a. |
| Source | SS | df | MS | F | Prob. > F |
|---|---|---|---|---|---|
| Total | 19.33 | 20 | 0.966 | 36.27 | 1.27 × 10−4 |
| Fit | 18.15 | 6 | 3.024 | ||
| Residual | 1.18 | 14 | 0.083 |
| Source | SS | df | MS | F | Prob. > F |
|---|---|---|---|---|---|
| Total | 19.36 | 20 | 0.97 | 49.84 | 3.6 × 10−6 |
| Fit | 18.81 | 8 | 2.35 | ||
| Residual | 0.55 | 12 | 0.05 |
| Source | SS | df | MS | F | Prob. > F |
|---|---|---|---|---|---|
| Total | 17.46 | 18 | 0.97 | 39.39 | 1.05 × 10−4 |
| Fit | 16.75 | 6 | 2.79 | ||
| Residual | 0.71 | 12 | 0.07 |
| # | Column Name | Length / mm | Internal Diameter (ID) / mm | Particle Size /μm | Carbon Load / % | Pore Size / Å | Surface Area / m−2 g | Ligand Type * | Surface coverage density (αRP) / µmol/m2 ** |
|---|---|---|---|---|---|---|---|---|---|
| 1 | K-C18 | 150 | 4.6 | 5 | 12 | 100 | 200 | C18 | 3.23 |
| 2 | K-F5 | 100 | 2.1 | 2.6 | 9 | 100 | 200 | C-F5 | 5.11 |
| 3 | IAM.PC.DD2 | 150 | 4.6 | 10 | 7 | 300 | 110 | diacylated PC | 1.53 |
| Name | Description |
|---|---|
| Solvation energy (SE) | defined in Equation (6) |
| Number of hydroxyl groups (n(OH)) | number of OH-groups in flavonoid structure |
| Minimum bond dissociation enthalpy (BDEmin) | parameter of the first oxidation step of SPLET mechanism, defined in Equation (7) |
| Proton affinity (PA) | PA is the negative quantity of proton-gain enthalpy, which is a standard enthalpy of the reaction: A− (g) +H+(g) → HA(g) |
| Electron transfer enthalpy (ETE) | parameter of the first oxidation step of SPLET mechanism, defined in Equation (8) |
| Excess charge of the most negatively charged atom (δmin) | shows the ability of analytes to participate in polar interactions with the phases of the charge transfer and hydrogen bonding |
| Total dipole moment Mtot. | accounts for the dipole-dipole and dipole-induced dipole attractive interactions of the analyte with mobile and stationary phases |
| HOMO-LUMO energy gap (ΔEHOMO-LUMO) | the difference between the HOMO and LUMO energies GAP = ɛ LUMO − ɛ HOMO, where ɛ LUMO and ɛ HOMO are the energies of the lowest unoccupied molecular orbital and the highest occupied molecular orbital, respectively |
| Ionization potential (IP) | ionization potential (or ionization energy) is defined as the energy needed to extract one electron from a chemical system, i.e., IP = E(Nel) − E (Nel − 1), where Nel is the number of electrons of the system |
| Electronic chemical potential (µ) | negative of electronegativity |
| Electrophilicity (ω) | electrophilicity can be defined as ω = (EHOMO+ELUMO)2/2(ELUMO − EHOMO) |
| Global hardness (η) | can be defined as resistance to charge transfer, Equation (10) |
| Electron affinity (EA) | EA is the energy released when an electron attaches to a gas-phase atom: E(g) + e−(g) → E−(g) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buszewski, B.; Žuvela, P.; Sagandykova, G.; Walczak-Skierska, J.; Pomastowski, P.; David, J.; Wong, M.W. Mechanistic Chromatographic Column Characterization for the Analysis of Flavonoids Using Quantitative Structure-Retention Relationships Based on Density Functional Theory. Int. J. Mol. Sci. 2020, 21, 2053. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062053
Buszewski B, Žuvela P, Sagandykova G, Walczak-Skierska J, Pomastowski P, David J, Wong MW. Mechanistic Chromatographic Column Characterization for the Analysis of Flavonoids Using Quantitative Structure-Retention Relationships Based on Density Functional Theory. International Journal of Molecular Sciences. 2020; 21(6):2053. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062053
Chicago/Turabian StyleBuszewski, Bogusław, Petar Žuvela, Gulyaim Sagandykova, Justyna Walczak-Skierska, Paweł Pomastowski, Jonathan David, and Ming Wah Wong. 2020. "Mechanistic Chromatographic Column Characterization for the Analysis of Flavonoids Using Quantitative Structure-Retention Relationships Based on Density Functional Theory" International Journal of Molecular Sciences 21, no. 6: 2053. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21062053