Synthesis and Cytotoxic Activity Evaluation of New Cu(I) Complexes of Bis(pyrazol-1-yl) Acetate Ligands Functionalized with an NMDA Receptor Antagonist

 , , , , and
, , , , and
Abstract
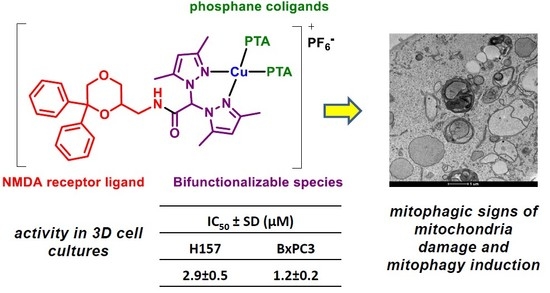
:
1. Introduction
2. Results and Discussion
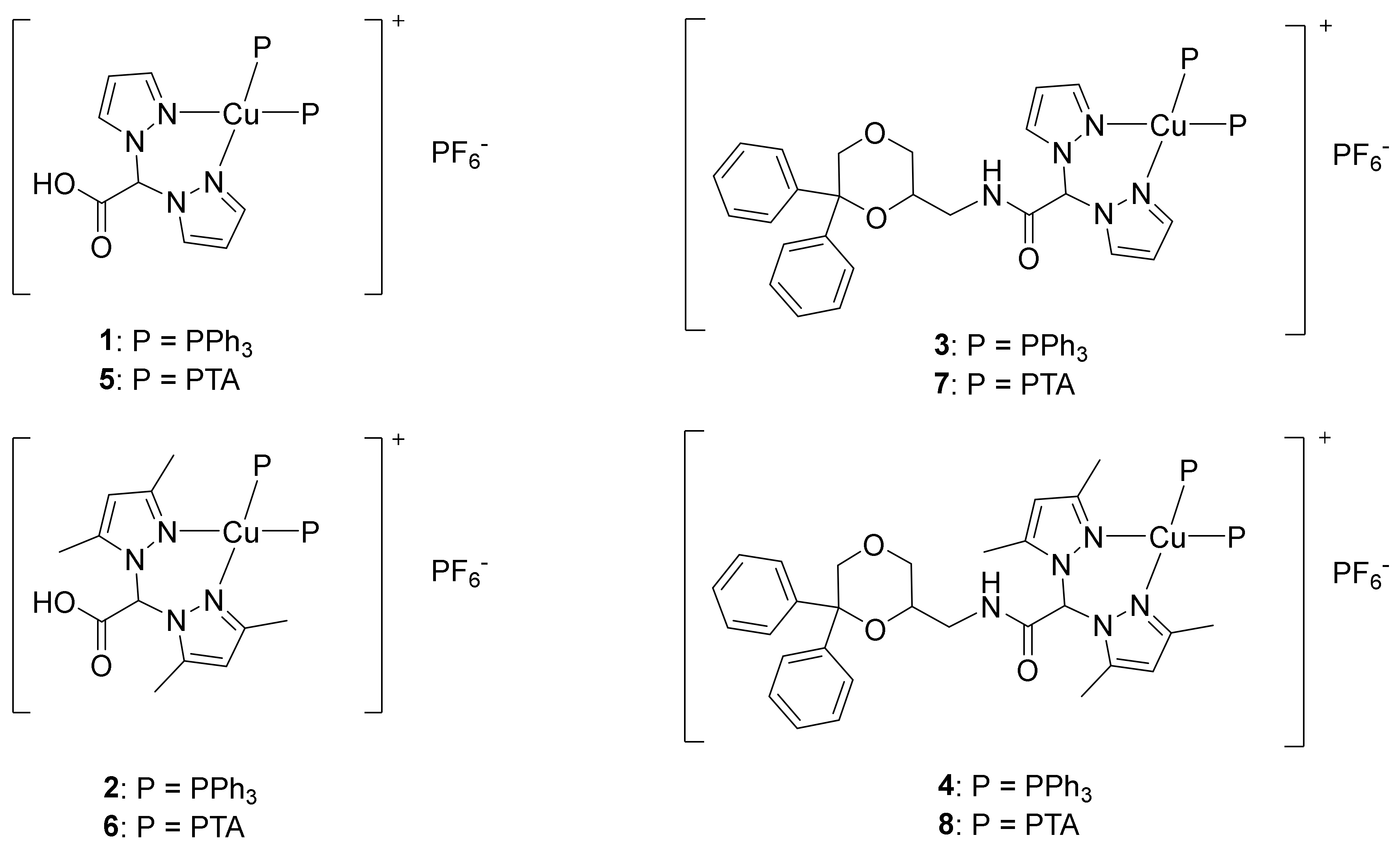
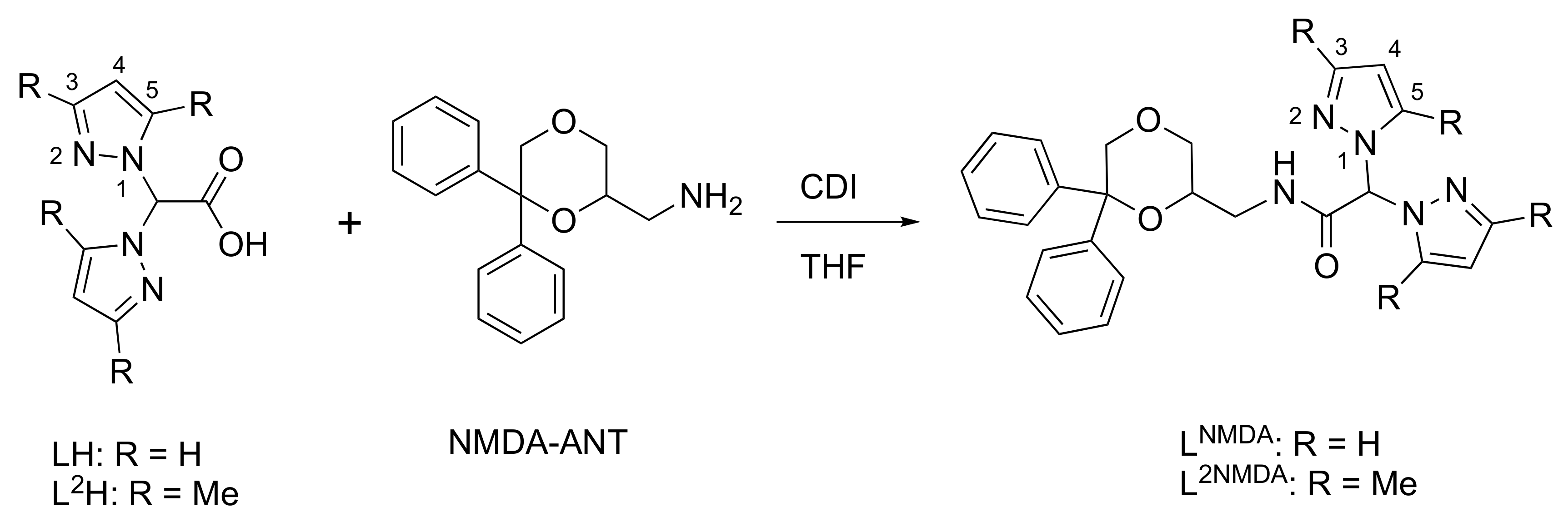
2.1. Synthesis and Characterization
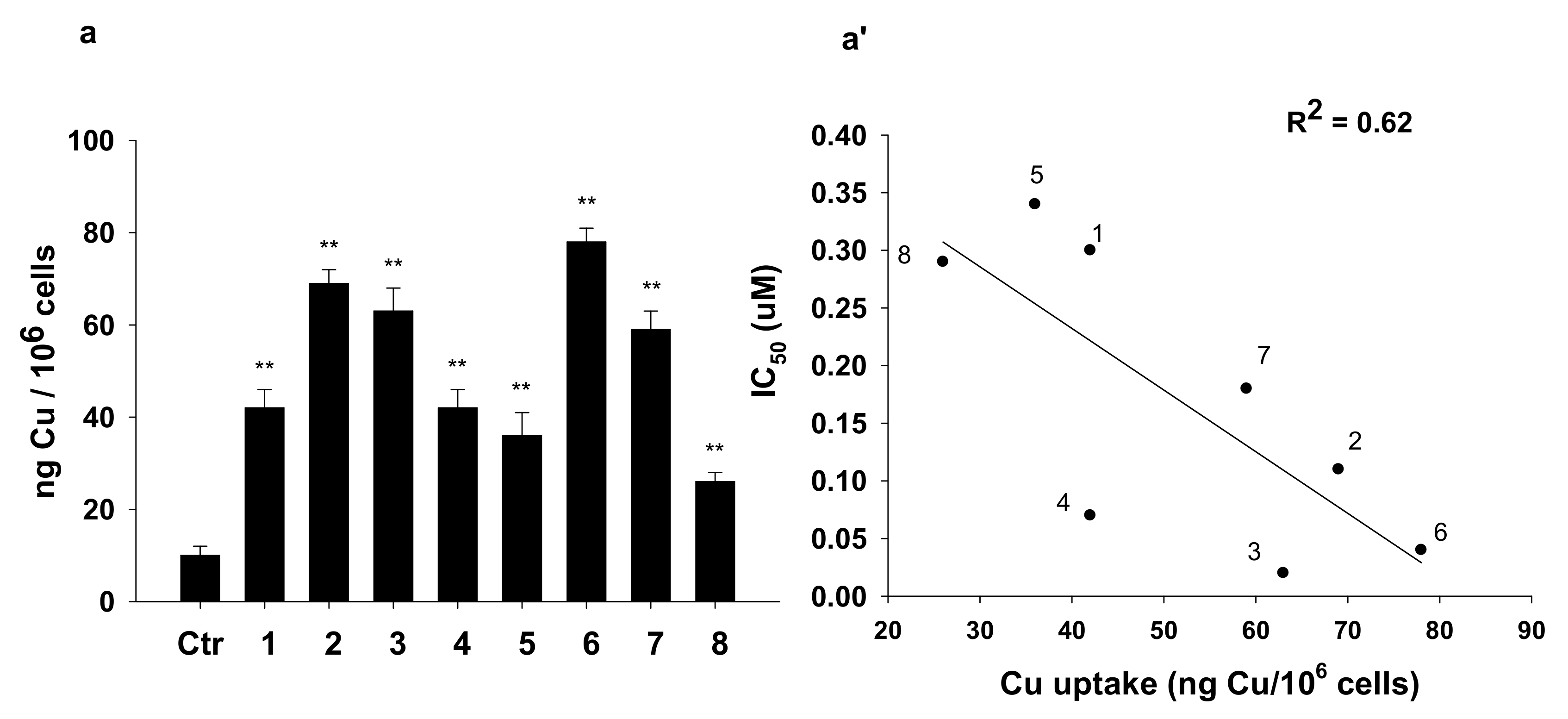
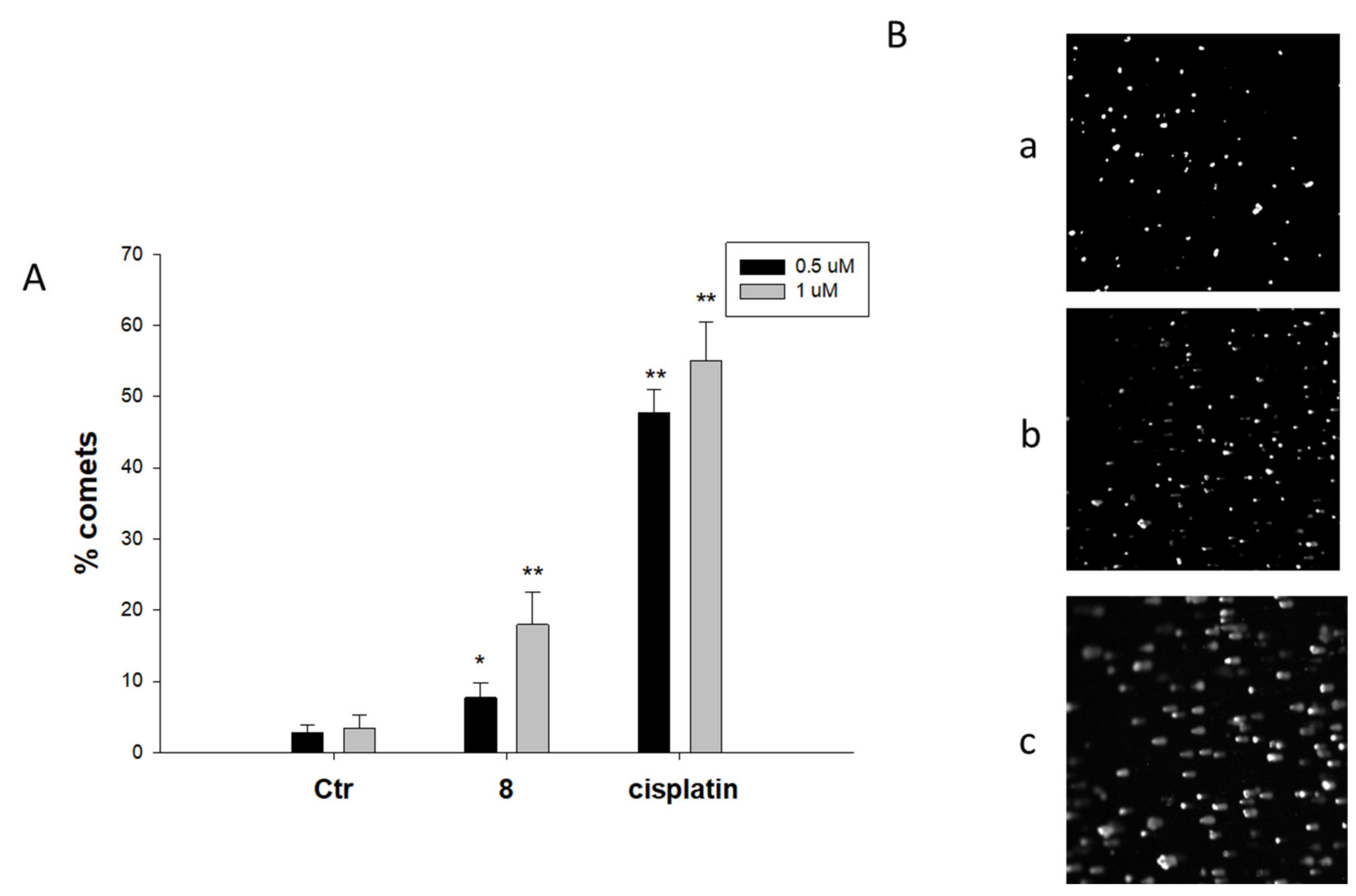
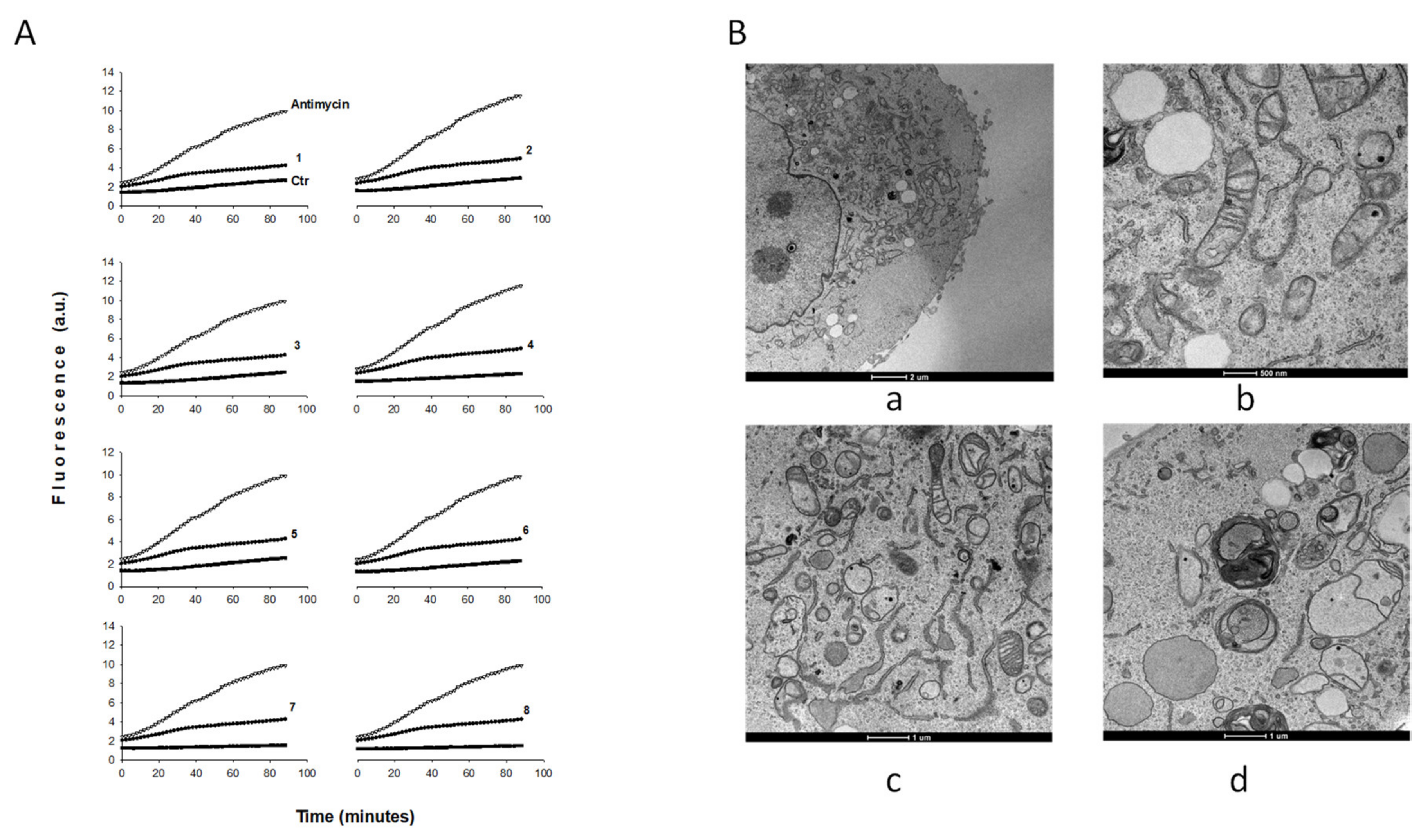
2.2. Biological Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. Materials and General Methods
3.1.2. Synthesis of [(LH)Cu(PPh3)2]PF6 (1)
3.1.3. Synthesis of [(L2H)Cu(PPh3)2]PF6 (2)
3.1.4. Synthesis of [(LNMDA)Cu(PPh3)2]PF6 (3)
3.1.5. Synthesis of [(L2NMDA)Cu(PPh3)2]PF6 (4)
3.1.6. Synthesis of [(LH)Cu(PTA)2]PF6 (5)
3.1.7. Synthesis of [(L2H)Cu(PTA)2]PF6 (6)
3.1.8. Synthesis of [(LNMDA)Cu(PTA)2]PF6 (7)
3.1.9. Synthesis of [(L2NMDA)Cu(PTA)2]PF6 (8)
3.2. Experiments with Cultured Human Cells
3.2.1. Cell Cultures
3.2.2. MTT Assay
3.2.3. Spheroid Cultures
3.2.4. APH Assay
3.2.5. Comet Assay
3.2.6. ROS Production
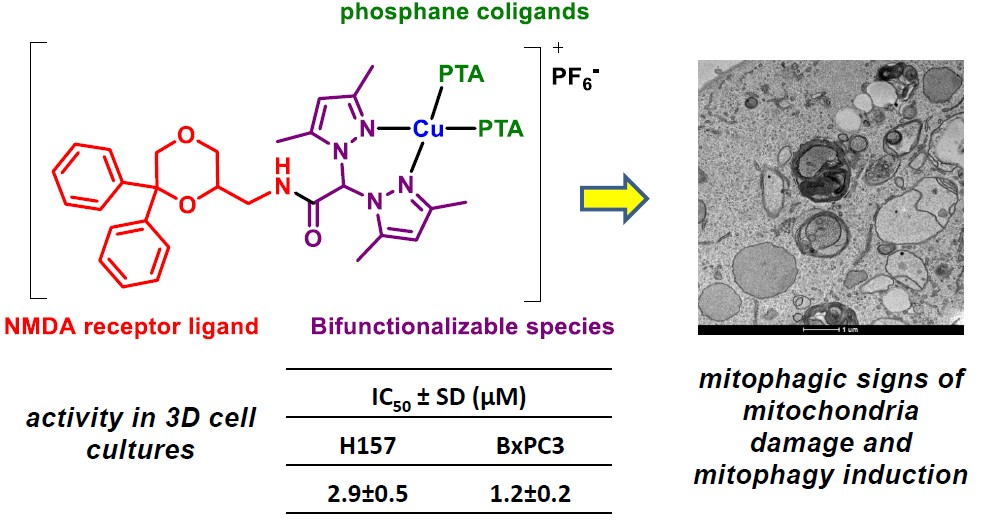
3.2.7. TEM Analyses
3.2.8. Cellular Uptake
3.2.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PPh3 | triphenylphosphine |
| PTA | 1,3,5-triaza-7-phosphaadamantane |
| MDR | multi-drug-resistant |
| TEM | transmission electron microscopy |
| ROS | reactive oxygen species |
| CDI | carbonyldiimidazole |
| APH | acid phosphatase |
| ER | endoplasmic reticulum |
References
- Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.; Marzano, C. Advances in Copper Complexes as Anticancer Agents. Chem Rev. 2014, 114, 815–862. [Google Scholar] [CrossRef] [PubMed]
- Tisato, F.; Marzano, C.; Porchia, M.; Pellei, M.; Santini, C. Copper in Diseases and Treatments, and Copper-Based Anticancer Strategies. Med. Res. Rev. 2010, 30, 708–749. [Google Scholar] [CrossRef] [PubMed]
- Wehbe, M.; Leung, A.W.Y.; Abrams, M.J.; Orvig, C.; Bally, M.B. A Perspective—Can copper complexes be developed as a novel class of therapeutics? Dalton Trans. 2017, 46, 10758–10773. [Google Scholar] [CrossRef]
- Kellett, A.; Molphy, Z.; McKee, V.; Slator, C. Recent Advances in Anticancer Copper Compounds. RSC Metallobiol. 2019, 14, 91–119. [Google Scholar]
- Öhrvik, H.; Aaseth, J.; Horn, N. Orchestration of dynamic copper navigation – new and missing pieces. Metallomics 2017, 9, 1204–1229. [Google Scholar] [CrossRef] [PubMed]
- Allardyce, C.S.; Dyson, P.J. Metal-based drugs that break the rules. Dalton Trans. 2016, 45, 3201–3209. [Google Scholar] [CrossRef] [Green Version]
- Spreckelmeyer, S.; Orvig, C.; Casini, A. Cellular Transport Mechanisms of Cytotoxic Metallodrugs: An Overview beyond Cisplatin. Molecules 2014, 19, 15584–15610. [Google Scholar] [CrossRef] [Green Version]
- Barilli, A.; Atzeri, C.; Bassanetti, I.; Ingoglia, F.; Dall’Asta, V.; Bussolati, O.; Maffini, M.; Mucchino, C.; Marchiò, L. Oxidative Stress Induced by Copper and Iron Complexes with 8-Hydroxyquinoline Derivatives Causes Paraptotic Death of HeLa Cancer Cells. Mol. Pharm. 2014, 11, 1151–1163. [Google Scholar] [CrossRef]
- Zaki, M.; Arjmand, F.; Tabassum, S. Current and future potential of metallo drugs: Revisiting DNA-binding of metal containing molecules and their diverse mechanism of action. Inorg. Chim. Acta 2016, 444, 1–22. [Google Scholar] [CrossRef]
- Medici, S.; Peana, M.; Nurchi, V.M.; Lachowicz, J.I.; Crisponi, G.; Zoroddu, M.A. Noble metals in medicine: Latest advances. Coord. Chem. Rev. 2015, 284, 329–350. [Google Scholar] [CrossRef]
- Laws, K.; Bineva-Todd, G.; Eskandari, A.; Lu, C.; O’Reilly, N.; Suntharalingam, K. A Copper(II) Phenanthroline Metallopeptide That Targets and Disrupts Mitochondrial Function in Breast Cancer Stem Cells. Angew. Chem. Int. Ed. 2018, 57, 287–291. [Google Scholar] [CrossRef]
- Gandin, V.; Ceresa, C.; Esposito, G.; Indraccolo, S.; Porchia, M.; Tisato, F.; Santini, C.; Pellei, M.; Marzano, C. Therapeutic potential of the phosphino Cu(I) complex (HydroCuP) in the treatment of solid tumors. Sci. Rep. 2017, 7, 13936. [Google Scholar] [CrossRef] [PubMed]
- Montagner, D.; Fresch, B.; Browne, K.; Gandin, V.; Erxleben, A. A Cu(ii) complex targeting the translocator protein: In vitro and in vivo antitumor potential and mechanistic insights. Chem. Commun. 2017, 53, 134–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahendiran, D.; Kumar, R.S.; Viswanathan, V.; Velmurugan, D.; Rahiman, A.K. In vitro and in vivo anti-proliferative evaluation of bis(4′-(4-tolyl)-2,2′:6′,2″-terpyridine)copper(II) complex against Ehrlich ascites carcinoma tumors. JBIC J. Biol. Inorg. Chem. 2017, 22, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.-P.; Liu, Y.-C.; Wang, H.-L.; Qin, J.-L.; Cheng, F.-J.; Tang, S.-F.; Liang, H. Synthesis and antitumor mechanisms of a copper(ii) complex of anthracene-9-imidazoline hydrazone (9-AIH). Metallomics 2015, 7, 1124–1136. [Google Scholar] [CrossRef]
- Becco, L.; García-Ramos, J.C.; Azuara, L.R.; Gambino, D.; Garat, B. Analysis of the DNA Interaction of Copper Compounds Belonging to the Casiopeínas® Antitumoral Series. Biol. Trace Elem. Res. 2014, 161, 210–215. [Google Scholar] [CrossRef]
- Gandin, V.; Tisato, F.; Dolmella, A.; Pellei, M.; Santini, C.; Giorgetti, M.; Marzano, C.; Porchia, M. In Vitro and in Vivo Anticancer Activity of Copper(I) Complexes with Homoscorpionate Tridentate Tris(pyrazolyl)borate and Auxiliary Monodentate Phosphine Ligands. J. Med. Chem. 2014, 57, 4745–4760. [Google Scholar] [CrossRef]
- Palanimuthu, D.; Shinde, S.V.; Somasundaram, K.; Samuelson, A.G. In Vitro and in Vivo Anticancer Activity of Copper Bis(thiosemicarbazone) Complexes. J. Med. Chem. 2013, 56, 722–734. [Google Scholar] [CrossRef]
- Raman, N.; Jeyamurugan, R.; Senthilkumar, R.; Rajkapoor, B.; Franzblau, S.G. In vivo and in vitro evaluation of highly specific thiolate carrier group copper(II) and zinc(II) complexes on Ehrlich ascites carcinoma tumor model. Eur. J. Med. Chem. 2010, 45, 5438–5451. [Google Scholar] [CrossRef]
- Gandin, V.; Pellei, M.; Tisato, F.; Porchia, M.; Santini, C.; Marzano, C. A novel copper complex induces paraptosis in colon cancer cellsviathe activation of ER stress signalling. J. Cell. Mol. Med. 2012, 16, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Weekley, C.M.; He, C. Developing drugs targeting transition metal homeostasis. Curr. Opin. Chem. Biol. 2017, 37, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Silva-Platas, C.; Guerrero-Beltrán, C.E.; Carrancá, M.; Castillo, E.C.; Bernal-Ramírez, J.; Oropeza-Almazán, Y.; González, L.N.; Rojo, R.; Martínez, L.E.; Valiente-Banuet, J.; et al. Antineoplastic copper coordinated complexes (Casiopeinas) uncouple oxidative phosphorylation and induce mitochondrial permeability transition in cardiac mitochondria and cardiomyocytes. J. Bioenerg. Biomembr. 2016, 48, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Denoyer, D.; Masaldan, S.; La Fontaine, S.; Cater, M.A. Targeting copper in cancer therapy: ‘Copper That Cancer’. Metallomics 2015, 7, 1459–1476. [Google Scholar] [CrossRef] [PubMed]
- Otero, A.; Fernandez-Baeza, J.; Tejeda, J.; Antinolo, A.; Carrillo-Hermosilla, F.; Diez-Barra, E.; Lara-Sanchez, A.; Fernandez-Lopez, M.; Lanfranchi, M.; Pellinghelli, M.A. Syntheses and crystal structures of lithium and niobium complexes containing a new type of monoanionic “scorpionate” ligand. J. Chem. Soc. Dalton Trans. Inorg. Chem. 1999, 20, 3537–3539. [Google Scholar] [CrossRef]
- Beck, A.; Weibert, B.; Burzlaff, N. Monoanionic N,N,O-scorpionate ligands and their iron(II) and zinc(II) complexes: Models for mononuclear active sites of non-heme iron oxidases and zinc enzymes. Eur. J. Inorg. Chem. 2001, 2001, 521–527. [Google Scholar] [CrossRef]
- Burzlaff, N.; Hegelmann, I.; Weibert, B. Bis(pyrazol-1-yl)acetates as tripodal “scorpionate” ligands in transition metal carbonyl chemistry: Syntheses, structures and reactivity of manganese and rhenium carbonyl complexes of the type [LM(CO)3] (L = bpza, bdmpza). J. Organomet. Chem. 2001, 626, 16–23. [Google Scholar] [CrossRef]
- Marzano, C.; Pellei, M.; Colavito, D.; Alidori, S.; Lobbia, G.G.; Gandin, V.; Tisato, F.; Santini, C. Synthesis, Characterization, and in Vitro Antitumor Properties of Tris(hydroxymethyl)phosphine Copper(I) Complexes Containing the New Bis(1,2,4-triazol-1-yl)acetate Ligand. J. Med. Chem. 2006, 49, 7317–7324. [Google Scholar] [CrossRef]
- Pellei, M.; Lobbia, G.G.; Santini, C.; Spagna, R.; Camalli, M.; Fedeli, D.; Falcioni, G. Synthesis, characterization and antioxidant activity of new copper(I) complexes of scorpionate and water soluble phosphane ligands. Dalton Trans. 2004, 17, 2822–2828. [Google Scholar] [CrossRef]
- Alidori, S.; Gioia Lobbia, G.; Papini, G.; Pellei, M.; Porchia, M.; Refosco, F.; Tisato, F.; Lewis Jason, S.; Santini, C. Synthesis, in vitro and in vivo characterization of (64)Cu(I) complexes derived from hydrophilic tris(hydroxymethyl)phosphane and 1,3,5-triaza-7-phosphaadamantane ligands. J. Biol. Inorg. Chem. JBIC Publ. Soc. Biol. Inorg. Chem. 2008, 13, 307–315. [Google Scholar] [CrossRef]
- Pellei, M.; Gandin, V.; Marchiò, L.; Marzano, C.; Bagnarelli, L.; Santini, C. Syntheses and Biological Studies of Cu(II) Complexes Bearing Bis(pyrazol-1-yl)- and Bis(triazol-1-yl)-acetato Heteroscorpionate Ligands. Molecules 2019, 24, 1761. [Google Scholar] [CrossRef] [Green Version]
- Otero, A.; Lara-Sánchez, A.; Fernández-Baeza, J.; Alonso-Moreno, C.; Castro-Osma, J.A.; Márquez-Segovia, I.; Sánchez-Barba, L.F.; Rodríguez, A.M.; Garcia-Martinez, J.C. Neutral and Cationic Aluminum Complexes Supported by Acetamidate and Thioacetamidate Heteroscorpionate Ligands as Initiators for Ring-Opening Polymerization of Cyclic Esters. Organometallics 2011, 30, 1507–1522. [Google Scholar] [CrossRef]
- Otero, A.; Lara-Sánchez, A.; Fernández-Baeza, J.; Alonso-Moreno, C.; Tejeda, J.; Castro-Osma, J.A.; Márquez-Segovia, I.; Sánchez-Barba, L.F.; Rodríguez, A.M.; Gómez, M.V. Straightforward Generation of Helical Chirality Driven by a Versatile Heteroscorpionate Ligand: Self-Assembly of a Metal Helicate by Using CH π Interactions. Chem. Eur. J. 2010, 16, 8615–8619. [Google Scholar] [CrossRef] [PubMed]
- Otero, A.; Lara-Sanchez, A.; Fernandez-Baeza, J.; Martinez-Caballero, E.; Marquez-Segovia, I.; Alonso-Moreno, C.; Sanchez-Barba, L.F.; Rodriguez, A.M.; Lopez-Solera, I. New achiral and chiral NNE heteroscorpionate ligands. Synthesis of homoleptic lithium complexes as well as halide and alkyl scandium and yttrium complexes. Dalton Trans. 2010, 39, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Otero, A.; Fernandez-Baeza, J.; Lara-Sanchez, A.; Alonso-Moreno, C.; Marquez-Segovia, I.; Sanchez-Barba, L.F.; Rodriguez, A.M. Ring-opening polymerization of cyclic esters by an enantiopure heteroscorpionate rare earth initiator. Angew. Chem. Int. Ed. 2009, 48, 2176–2179. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.-Y.; Hong, J.; Du, M.; Tang, L.-F. New multidentate heteroscorpionate ligands: N-Phenyl-2,2-bis(pyrazol-1-yl)thioacetamide and ethyl 2,2-bis(pyrazol-1-yl)dithioacetate as well as their derivatives. J. Organomet. Chem. 2007, 692, 1708–1715. [Google Scholar] [CrossRef]
- Otero, A.; Fernandez-Baeza, J.; Antinolo, A.; Tejeda, J.; Lara-Sanchez, A.; Sanchez-Barba, L.; Sanchez-Molina, M.; Franco, S.; Lopez-Solera, I.; Rodriguez, A.M. Design of new heteroscorpionate ligands and their coordinative ability toward Group 4 transition metals; an efficient synthetic route to obtain enantiopure ligands. Dalton Trans. 2006, 36, 4359–4370. [Google Scholar] [CrossRef]
- Pellei, M.; Gandin, V.; Cimarelli, C.; Quaglia, W.; Mosca, N.; Bagnarelli, L.; Marzano, C.; Santini, C. Syntheses and biological studies of nitroimidazole conjugated heteroscorpionate ligands and related Cu(I) and Cu(II) complexes. J. Inorg. Biochem. 2018, 187, 33–40. [Google Scholar] [CrossRef]
- Giorgetti, M.; Tonelli, S.; Zanelli, A.; Aquilanti, G.; Pellei, M.; Santini, C. Synchrotron radiation X-ray absorption spectroscopic studies in solution and electrochemistry of a nitroimidazole conjugated heteroscorpionate copper(II) complex. Polyhedron 2012, 48, 174–180. [Google Scholar] [CrossRef]
- Pellei, M.; Papini, G.; Trasatti, A.; Giorgetti, M.; Tonelli, D.; Minicucci, M.; Marzano, C.; Gandin, V.; Aquilanti, G.; Dolmella, A.; et al. Nitroimidazole and glucosamine conjugated heteroscorpionate ligands and related copper(II) complexes. Syntheses, biological activity and XAS studies. Dalton Trans. 2011, 40, 9877–9888. [Google Scholar] [CrossRef]
- Blanke, M.L.; VanDongen, A.M.J. Activation Mechanisms of the NMDA Receptor. In Biology of the NMDA Receptor; Van Dongen, A., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009; pp. 283–331. [Google Scholar]
- Deutsch, S.I.; Tang, A.H.; Burket, J.A.; Benson, A.D. NMDA receptors on the surface of cancer cells: Target for chemotherapy? Biomed. Pharmacother. 2014, 68, 493–496. [Google Scholar] [CrossRef] [Green Version]
- Mehrotra, A.; Koiri, R.K. N-Methyl-D-Aspartate (NMDA) Receptors: Therapeutic Target against Cancer. Int. J. Immunother. Cancer Res. 2015, 1, 13–17. [Google Scholar]
- North, W.G.; Gao, G.; Memoli, V.A.; Pang, R.H.; Lynch, L. Breast cancer expresses functional NMDA receptors. Breast Cancer Res. Treat. 2010, 122, 307–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonifazi, A.; Del Bello, F.; Mammoli, V.; Piergentili, A.; Petrelli, R.; Cimarelli, C.; Pellei, M.; Schepmann, D.; Wünsch, B.; Barocelli, E.; et al. Novel potent N-methyl-d-aspartate (NMDA) receptor antagonists or σ1 receptor ligands based on properly substituted 1,4-dioxane ring. J. Med. Chem. 2015, 58, 8601–8615. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.B.; Amantini, C.; Santoni, G.; Pellei, M.; Santini, C.; Cimarelli, C.; Marcantoni, E.; Petrini, M.; Del Bello, F.; Giorgioni, G.; et al. Novel antitumor copper(ii) complexes designed to act through synergistic mechanisms of action, due to the presence of an NMDA receptor ligand and copper in the same chemical entity. New J. Chem. 2018, 42, 11878–11887. [Google Scholar] [CrossRef]
- Marzano, C.; Tisato, F.; Porchia, M.; Pellei, M.; Gandin, V. Chapter 4—Phosphine copper(I) complexes as anticancer agents: Biological characterization. Part II. In Copper(I) Chemistry of Phosphines, Functionalized Phosphines and Phosphorus Heterocycles; Balakrishna, M.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 83–107. [Google Scholar] [CrossRef]
- Tisato, F.; Porchia, M.; Santini, C.; Gandin, V.; Marzano, C. Chapter 3—Phosphine–copper(I) complexes as anticancer agents: Design, synthesis, and physicochemical characterization. Part I. In Copper(I) Chemistry of Phosphines, Functionalized Phosphines and Phosphorus Heterocycles; Balakrishna, M.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 61–82. [Google Scholar] [CrossRef]
- Kankotia, S.; Stacpoole, P.W. Dichloroacetate and cancer: New home for an orphan drug? Biochim. Biophys. Acta 2014, 1846, 617–629. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer. Res. 1988, 48, 589–601. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 ± SD (μM) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PSN-1 | BxPC3 | MCF-7 | H157 | A431 | A431-Pt | RF b | A2780 | A2780cis | RF b | A2780 ADR | RF b | |
| 1 | 3.1 ± 1.9 | 0.3 ± 0.05 | 1.0 ± 0.1 | 1.3 ± 0.3 | 0.6 ± 0.1 | 1.1 ± 0.4 | 1.7 | 0.5 ± 0.3 | 0.7 ± 0.1 | 1.6 | 0.5 ± 0.1 | 1.2 |
| 2 | 1.6 ± 0.2 | 0.1 ± 0.05 | 1.8 ± 0.01 | 1.1 ± 0.2 | 2.7 ± 0.5 | 1.3 ± 0.1 | 0.5 | 0.6 ± 0.17 | 0.7 ± 0.2 | 1.0 | 0.9 ± 0.3 | 1.5 |
| 3 | 0.4 ± 0.04 | 0.02 ± 0.01 | 0.8 ± 0.1 | 0.4 ± 0.1 | 0.2 ± 0.01 | 0.1 ± 0.01 | 0.7 | 0.1 ± 0.01 | 0.1 ± 0.01 | 0.5 | 0.2 ± 0.02 | 1.7 |
| 4 | 1.7 ± 0.7 | 0.07 ± 0.02 | 2.5 ± 0.6 | 1.1 ± 0.5 | 0.6 ± 0.1 | 1.0 ± 0.2 | 1.7 | 0.2 ± 0.1 | 0.19 ± 0.05 | 1.3 | 0.3 ± 0.1 | 1.9 |
| 5 | 1.2 ± 0.04 | 0.3 ± 0.01 | 1.0 ± 0.1 | 1.0 ± 0.1 | 2.8 ± 1.2 | 1.3 ± 0.2 | 0.5 | 0.2 ± 0.05 | 0.3 ± 0.2 | 1.5 | 0.2 ± 0.1 | 1.2 |
| 6 | 0.02 ± 0.01 | 0.04 ± 0.02 | 0.6 ± 0.03 | 2.3 ± 0.5 | 0.2 ± 0.03 | 0.3 ± 0.6 | 1.7 | 0.9 ± 0.2 | 1.1 ± 0.3 | 1.2 | 1.3 ± 0.2 | 1.5 |
| 7 | 1.0 ± 0.3 | 0.2 ± 0.02 | 0.5 ± 0.03 | 0.4 ± 0.2 | 3.3 ± 0.7 | 5.9 ± 1.5 | 1.8 | 0.4 ± 0.04 | 0.4 ± 0.1 | 1.1 | 0.4 ± 0.1 | 1.1 |
| 8 | 0.6 ± 0.3 | 0.3 ± 0.06 | 1.8 ± 0.1 | 0.8 ± 0.2 | 3.2 ± 0.3 | 1.9 ± 0.5 | 0.6 | 0.04 ± 0.01 | 0.01 ± 0.01 | 0.3 | 0.1 ± 0.02 | 1.3 |
| LH | >50 | 21.2 ± 3.2 | >50 | 19.4 ± 2.6 | >50 | - | - | >50 | - | - | - | |
| L2H | >50 | >50 | >50 | 21.4 ± 3.0 | >50 | - | - | 45.5 ± 7.3 | - | - | - | |
| NMDA-ANT | 10.4 ± 2.6 | 8.3 ± 2.7 | 12.1 ± 3.2 | 5.3 ± 0.7 | 11.2 ± 2.3 | - | - | 19.8 ± 3.3 | - | - | - | |
| LNMDA | 9.6 ± 2.3 | 13.1 ± 3.5 | 7.4 ± 2.2 | 6.6 ± 1.4 | 7.6 ± 0.9 | - | - | 15.5 ± 2.6 | - | - | - | |
| L2NMDA | >50 | 42.1 ± 3.0 | >50 | >50 | >50 | - | - | >50 | - | - | - | |
| Cisplatin | 12.1 ± 2.9 | 7.3 ± 1.2 | 8.8 ± 0.2 | 26.7 ± 3.2 | 1.4 ± 0.3 | 2.9 ± 0.6 | 2.0 | 0.45 ± 0.1 | 2.6 ± 0.2 | 5.8 | - | - |
| Doxorubicin | - | - | - | - | - | - | - | 0.004 ± 0.001 | - | 0.09 ± 0.01 | 21.5 | |
| IC50 ± SD (μM) | ||
|---|---|---|
| H157 | BxPC3 | |
| 1 | 4.7 ± 0.3 | 3.1 ± 0.5 |
| 2 | 3.3 ± 1.0 | 2.4 ± 0.2 |
| 3 | 10.9 ± 3.0 | 4.5 ± 1.3 |
| 4 | 2.7 ± 0.5 | 6.5 ± 1.1 |
| 5 | 11.9 ± 2.0 | 10.2 ± 2.3 |
| 6 | 10.0 ± 0.4 | 5.5 ± 0.9 |
| 7 | 13.0 ± 1.1 | 8.5 ± 0.9 |
| 8 | 2.9 ± 0.5 | 1.2 ± 0.2 |
| Cisplatin | 52.51 ± 1.31 | 100.5 ± 12.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellei, M.; Bagnarelli, L.; Luciani, L.; Del Bello, F.; Giorgioni, G.; Piergentili, A.; Quaglia, W.; De Franco, M.; Gandin, V.; Marzano, C.; et al. Synthesis and Cytotoxic Activity Evaluation of New Cu(I) Complexes of Bis(pyrazol-1-yl) Acetate Ligands Functionalized with an NMDA Receptor Antagonist. Int. J. Mol. Sci. 2020, 21, 2616. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072616
Pellei M, Bagnarelli L, Luciani L, Del Bello F, Giorgioni G, Piergentili A, Quaglia W, De Franco M, Gandin V, Marzano C, et al. Synthesis and Cytotoxic Activity Evaluation of New Cu(I) Complexes of Bis(pyrazol-1-yl) Acetate Ligands Functionalized with an NMDA Receptor Antagonist. International Journal of Molecular Sciences. 2020; 21(7):2616. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072616
Chicago/Turabian StylePellei, Maura, Luca Bagnarelli, Lorenzo Luciani, Fabio Del Bello, Gianfabio Giorgioni, Alessandro Piergentili, Wilma Quaglia, Michele De Franco, Valentina Gandin, Cristina Marzano, and et al. 2020. "Synthesis and Cytotoxic Activity Evaluation of New Cu(I) Complexes of Bis(pyrazol-1-yl) Acetate Ligands Functionalized with an NMDA Receptor Antagonist" International Journal of Molecular Sciences 21, no. 7: 2616. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21072616