Kv1.1 Channelopathies: Pathophysiological Mechanisms and Therapeutic Approaches

 ,
,  and
and
Abstract
:1. Kv1.1 Channel Expression and Physiological Roles
2. Diseases Associated with Kv1.1 Channel Dysfunction
2.1. Clinical Aspects and Therapeutic Management of Episodic Ataxia Type 1
2.2. Functional Consequences of Episodic Ataxia Type 1 Mutations and Genotype-Phenotype Correlation
2.3. Kv1.1 Channel Involvement in Epilepsy
3. Kv1.1-Targeted Pharmacological Approaches
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of potassium channels. Cell. Mol. Life Sci. 2015, 72, 3677–3693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005, 309, 897–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Wang, Q.; Ni, F.; Ma, J. Structure of the full-length Shaker potassium channel Kv1.2 by normal-mode-based X-ray crystallographic refinement. Proc. Natl. Acad. Sci. USA 2010, 107, 11352–11357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, S.B.; Tao, X.; Campbell, E.B.; MacKinnon, R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 2007, 450, 376–382. [Google Scholar] [CrossRef]
- Herson, P.S.; Virk, M.; Rustay, N.R.; Bond, C.T.; Crabbe, J.C.; Adelman, J.P.; Maylie, J. A mouse model of episodic ataxia type-1. Nat. Neurosci. 2003, 6, 378–383. [Google Scholar] [CrossRef]
- Brunetti, O.; Imbrici, P.; Botti, F.M.; Pettorossi, V.E.; D’Adamo, M.C.; Valentino, M.; Zammit, C.; Mora, M.; Gibertini, S.; Di Giovanni, G.; et al. Kv1.1 knock-in ataxic mice exhibit spontaneous myokymic activity exacerbated by fatigue, ischemia and low temperature. Neurobiol. Dis. 2012, 47, 310–321. [Google Scholar] [CrossRef]
- Lorincz, A.; Nusser, Z. Cell-type-dependent molecular composition of the axon initial segment. J. Neurosci. 2008, 28, 14329–14340. [Google Scholar] [CrossRef]
- Vacher, H.; Mohapatra, D.P.; Trimmer, J.S. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol. Rev. 2008, 88, 1407–1447. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Kunkel, D.D.; Martin, T.M.; Schwartzkroin, P.A.; Tempel, B.L. Heteromultimeric K1 channels in terminal and juxtaparanodal regions of neurons. Nature 1993, 365, 75–79. [Google Scholar] [CrossRef]
- Wang, H.; Kunkel, D.D.; Schwartzkroin, P.A.; Tempel, B.L. Localization of Kv1.1 and Kv1.2, two K1 channel proteins, to synaptic terminals, somata, and dendrites in the mouse brain. J. Neurosci. 1994, 14, 4588–4599. [Google Scholar] [CrossRef]
- Smart, S.L.; Lopantsev, V.; Zhang, C.L.; Robbins, C.A.; Wang, H.; Chiu, S.Y.; Schwartzkroin, P.A.; Messing, A.; Tempel, B.L. Deletion of the Kv1.1 potassium channel causes epilepsy in mice. Neuron 1998, 20, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Trimmer, J.S. Subcellular localization of K+ channels in mammalian brain neurons: Remarkable precision in the midst of extraordinary complexity. Neuron 2015, 85, 238–256. [Google Scholar] [CrossRef] [Green Version]
- Dodson, P.D.; Forsythe, I.D. Presynaptic K+ channels: Electrifying regulators of synaptic terminal excitability. Trends Neurosci. 2004, 27, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Rettig, J.; Heinemann, S.H.; Wunder, F.; Lorra, C.; Parcej, D.N.; Dolly, J.O.; Pongs, O. Inactivation properties of voltage-gated K+ channels altered by presence of beta-subunit. Nature 1994, 369, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, S.H.; Rettig, J.; Graack, H.R.; Pongs, O. Functional characterization of Kv channel beta-subunits from rat brain. J. Physiol. 1996, 493 Pt 3, 625–633. [Google Scholar] [CrossRef]
- Pongs, O.; Schwarz, J.R. Ancillary subunits associated with voltage-dependent K+ channels. Physiol. Rev. 2010, 90, 755–796. [Google Scholar] [CrossRef] [Green Version]
- Southan, A.P.; Robertson, B. Patch-clamp recordings from cerebellar basket cell bodies and their presynaptic terminals reveal an asymmetric distribution of voltage-gated potassium channels. J. Neurosci. 1998, 18, 948–955. [Google Scholar] [CrossRef]
- Begum, R.; Bakiri, Y.; Volynski, K.E.; Kullmann, D.M. Action potential broadening in a presynaptic channelopathy. Nat. Commun. 2016, 7, 12102. [Google Scholar] [CrossRef] [Green Version]
- D’Adamo, M.C.; Imbrici, P.; Sponcichetti, F.; Pessia, M. Mutations in the KCNA1 gene associated with episodic ataxia type-1 syndrome impair heteromeric voltage-gated K(+) channel function. FASEB J. 1999, 13, 1335–1345. [Google Scholar] [CrossRef]
- Geiger, J.R.; Jonas, P. Dynamic control of presynaptic Ca(2+) inflow by fast-inactivating K(+) channels in hippocampal mossy fiber boutons. Neuron 2000, 28, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Simeone, T.A.; Simeone, K.A.; Samson, K.K.; Kim, D.Y.; Rho, J.M. Loss of the Kv1.1 potassium channel promotes pathologic sharp waves and high frequency oscillations in vitro hippocampal slices. Neurobiol. Dis. 2013, 54, 68–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Messing, A.; Chiu, S.Y. Determinants of excitability at transition zones in Kv1.1-deficient myelinated nerves. J. Neurosci. 1999, 19, 5768–5781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, J.; Padilla, F.; Dandonneau, M.; Lavebratt, C.; Lesage, F.; Noël, J.; Delmas, P. Kv1.1 channels act as mechanical brake in the senses of touch and pain. Neuron 2013, 77, 899–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ison, J.R.; Allen, P.D. Deficits in responding to brief noise offsets in Kcna1−/−mice reveal a contribution of this gene to precise temporal processing seen previously only for stimulus onsets. J. Assoc. Res. Otolaryngol. 2012, 13, 351–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, U.; Thumfart, J.O.; Klöcker, N.; Sailer, C.A.; Bildl, W.; Biniossek, M.; Dehn, D.; Deller, T.; Eble, S.; Abbass, K.; et al. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron 2006, 49, 697–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seagar, M.; Russier, M.; Caillard, O.; Maulet, Y.; Fronzaroli-Molinieres, L.; De San Feliciano, M.; Boumedine-Guignon, N.; Rodriguez, L.; Zbili, M.; Usseglio, F.; et al. LGI1 tunes intrinsic excitability by regulating the density of axonal Kv1 channels. Proc. Natl. Acad. Sci. USA 2017, 114, 7719–7724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petit-Pedrol, M.; Sell, J.; Planaguma, J.; Mannara, F.; Radosevic, M.; Haselmann, H.; Ceanga, M.; Sabater, L.; Spatola, M.; Soto, D.; et al. LGI1 antibodies alter Kv1.1 and AMPA receptors changing synaptic excitability, plasticity and memory. Brain 2018, 141, 3144–3159. [Google Scholar] [CrossRef]
- Lancaster, E.; Burnor, E.; Zhang, J.; Lancaster, E. ADAM23 is a negative regulator of Kv1.1/Kv1.4 potassium currents. Neurosci. Lett. 2019, 704, 159–163. [Google Scholar] [CrossRef]
- Kim, E.; Niethammer, M.; Rothschild, A.; Jan, Y.N.; Sheng, M. Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature 1995, 378, 85–88. [Google Scholar] [CrossRef]
- Raab-Graham, K.F.; Haddick, P.C.; Jan, Y.N.; Jan, L.Y. Activity- and mTOR-dependent suppression of Kv1.1 channel mRNA translation in dendrites. Science 2006, 314, 144–148. [Google Scholar] [CrossRef]
- Nguyen, L.H.; Anderson, A.E. mTOR-dependent alterations of Kv1.1 subunit expression in the neuronal subset-specific Pten knockout mouse model of cortical dysplasia with epilepsy. Sci. Rep. 2018, 8, 3568. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Tiwari, D. Regulation of Ion Channels by MicroRNAs and the Implication for Epilepsy. Curr. Neurol. Neurosci. Rep. 2018, 18, 60. [Google Scholar] [CrossRef]
- Cusimano, A.; D’Adamo, M.C.; Pessia, M. An episodic ataxia type-1 mutation in the S1 segment sensitises the hKv1.1 potassium channel to extracellular Zn2+. FEBS Lett. 2004, 576, 237–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poliak, S.; Gollan, L.; Martinez, R.; Custer, A.; Einheber, S.; Salzer, J.L.; Trimmer, J.S.; Shrager, P.; Peles, E. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 1999, 24, 1037–1047. [Google Scholar] [CrossRef] [Green Version]
- Imbrici, P.; Tucker, S.J.; D’Adamo, M.C.; Pessia, M. Role of RPTPα and tyrosine phosphorylation in the serotonergic inhibition of voltage-dependent potassium channels. Pflügers Archiv. Eur. J. Physiol. 2000, 441, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Lavebratt, C.; Almgren, M.; Portwood, N.; Forsberg, L.E.; Bränström, R.; Berglund, E.; Falkmer, S.; Sundler, F.; Wierup, N.; et al. Evidence for presence and functional effects of Kv1.1 channels in β-cells: General survey and results from mceph/mceph mice. PLoS ONE 2011, 6, e18213. [Google Scholar] [CrossRef] [Green Version]
- Dukes, I.D.; Philipson, L.H. K+ channels: Generating excitement in pancreatic beta-cells. Diabetes 1996, 45, 845–853. [Google Scholar] [CrossRef]
- Choi, B.H.; Hahn, S.J. Kv1.3: A potential pharmacological target for diabetes. Acta Pharmacol. Sin. 2010, 31, 1031–1035. [Google Scholar] [CrossRef] [Green Version]
- Tricarico, D.; Rolland, J.F.; Cannone, G.; Mele, A.; Cippone, V.; Laghezza, A.; Carbonara, G.; Fracchiolla, G.; Tortorella, P.; Loiodice, F.; et al. Structural nucleotide analogs are potent activators/inhibitors of pancreatic β cell KATP channels: An emerging mechanism supporting their use as antidiabetic drugs. J. Pharmacol. Exp. Ther. 2012, 340, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Firth, A.L.; Remillard, C.V.; Platoshyn, O.; Fantozzi, I.; Ko, E.A.; Yuan, J.X. Functional ion channels in human pulmonary artery smooth muscle cells: Voltage-dependent cation channels. Pulm. Circ. 2011, 1, 48–71. [Google Scholar] [CrossRef] [Green Version]
- Glaudemans, B.; van der Wijst, J.; Scola, R.H.; Lorenzoni, P.J.; Heister, A.; van der Kemp, A.W.; Knoers, N.V.; Hoenderop, J.G.; Bindels, R.J. A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J. Clin. Investig. 2009, 119, 936–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Wijst, J.; Konrad, M.; Verkaart, S.A.J.; Tkaczyk, M.; Latta, F.; Altmüller, J.; Thiele, H.; Beck, B.; Schlingmann, K.P.; de Baaij, J.H.F. A de novo KCNA1 Mutation in a Patient with Tetany and Hypomagnesemia. Nephron 2018, 139, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Glasscock, E. Kv1.1 channel subunits in the control of neurocardiac function. Channels 2019, 13, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Glasscock, E.; Yoo, J.W.; Chen, T.T.; Klassen, T.L.; Noebels, J.L. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J. Neurosci. 2010, 30, 5167–5175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasscock, E.; Voigt, N.; McCauley, M.D.; Sun, Q.; Li, N.; Chiang, D.Y.; Zhou, X.B.; Molina, C.E.; Thomas, D.; Schmidt, C.; et al. Expression and function of Kv1.1 potassium channels in human atria from patients with atrial fibrillation. Basic Res. Cardiol. 2015, 110, 505. [Google Scholar] [CrossRef] [Green Version]
- Si, M.; Trosclair, K.; Hamilton, K.A.; Glasscock, E. Genetic ablation or pharmacological inhibition of Kv1.1 potassium channel subunits impairs atrial repolarization in mice. Am. J. Physiol. Cell Physiol. 2018, 316, C154–C161. [Google Scholar] [CrossRef]
- Van Dyke, D.H.; Griggs, R.C.; Murphy, M.J.; Goldstein, M.N. Hereditary myokymia and periodic ataxia. J. Neurol. Sci. 1975, 25, 109–118. [Google Scholar] [CrossRef]
- Litt, M.; Kramer, P.; Browne, D.; Gancher, S.; Brunt, E.R.; Root, D.; Phromchotikul, T.; Dubay, C.J.; Nutt, J. A gene for episodic ataxia/myokymia maps to chromosome 12p13. Am. J. Hum. Genet. 1994, 55, 702–709. [Google Scholar] [PubMed]
- Browne, D.L.; Gancher, S.T.; Nutt, J.G.; Brunt, E.R.P.; Smith, E.A.; Kramer, P.; Litt, M. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat. Genet. 1994, 8, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Browne, D.L.; Brunt, E.R.P.; Griggs, R.C.; Nutt, J.G.; Gancher, S.T.; Smith, E.A.; Litt, M. Identification of two new KCNA1 mutations in episodic ataxia/myokymia families. Hum. Mol. Genet. 1995, 4, 1671–1672. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, M.C.; Hasan, S.; Guglielmi, L.; Servettini, I.; Cenciarini, M.; Catacuzzeno, L.; Franciolini, F. New insights into the pathogenesis and therapeutics of episodic ataxia type 1. Front. Cell. Neurosci. 2015, 9, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graves, T.D.; Cha, Y.H.; Hahn, A.F.; Barohn, R.; Salajegheh, M.K.; Griggs, R.C.; Bundy, B.N.; Jen, J.C.; Baloh, R.W.; Hanna, M.G.; et al. Episodic ataxia type 1: Clinical characterization, quality of life and genotype-phenotype correlation. Brain 2014, 137 Pt 4, 1009–1018. [Google Scholar] [CrossRef] [Green Version]
- Jen, J.C.; Graves, T.D.; Hess, E.J.; Hanna, M.G.; Griggs, R.C.; Baloh, R.W.; CINCH investigators. Primary episodic ataxias: Diagnosis, pathogenesis and treatment. Brain 2007, 130 Pt 10, 2484–2493. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, S.E.; Tan, S.V.; Kullmann, D.M.; Griggs, R.C.; Burke, D.; Hanna, M.G.; Bostock, H. Nerve excitability studies characterize Kv1.1 fast potassium channel dysfunction in patients with episodic ataxia type 1. Brain 2010, 133 Pt 12, 3530–3540. [Google Scholar] [CrossRef] [Green Version]
- Graves, T.D.; Rajakulendran, S.; Zuberi, S.M.; Morris, H.R.; Schorge, S.; Hanna, M.G.; Kullmann, D.M. Nongenetic factors influence severity of episodic ataxia type 1 in monozygotic twins. Neurology 2010, 75, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Demos, M.K.; Macri, V.; Farrell, K.; Nelson, T.N.; Chapman, K.; Accili, E.; Armstrong, L. A novel KCNA1 mutation associated with global delay and persistent cerebellar dysfunction. Mov. Disord. 2009, 24, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Eunson, L.H.; Rea, R.; Zuberi, S.M.; Youroukos, S.; Panayiotopoulos, C.P.; Liguori, R.; Avoni, P.; McWilliam, R.C.; Stephenson, J.B.P.; Hanna, M.G.; et al. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann. Neurol. 2000, 48, 647–656. [Google Scholar] [CrossRef]
- Kinali, M.; Jungbluth, H.; Eunson, L.H.; Sewry, C.A.; Manzur, A.Y.; Mercuri, E.; Hanna, M.G.; Muntoni, F. Expanding the phenotype of potassium channelopathy: Severe neuromyotonia and skeletal deformities without prominent Episodic Ataxia. Neuromuscul. Disord. 2004, 14, 689–693. [Google Scholar] [CrossRef]
- Klein, A.; Boltshauser, E.; Jen, J.; Baloh, R.W. Episodic ataxia type 1 with distal weakness: A novel manifestation of a potassium channelopathy. Neuropediatrics 2004, 35, 147–149. [Google Scholar] [CrossRef]
- Chen, S.H.; Fu, S.J.; Huang, J.J.; Tang, C.Y. The episodic ataxia type 1 mutation I262T alters voltage-dependent gating and disrupts protein biosynthesis of human Kv1.1 potassium channels. Sci. Rep. 2016, 6, 19378. [Google Scholar] [CrossRef] [Green Version]
- Brownstein, C.A.; Beggs, A.H.; Rodan, L.; Shi, J.; Towne, M.C.; Pelletier, R.; Cao, S.; Rosenberg, P.A.; Urion, D.K.; Picker, J.; et al. Clinical heterogeneity associated with KCNA1 mutations include cataplexy and nonataxic presentations. Neurogenetics 2016, 17, 11–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Adamo, M.C.; Gallenmuller, C.; Servettini, I.; Hartl, E.; Tucker, S.J.; Arning, L.; Biskup, S.; Grottesi, A.; Guglielmi, L.; Imbrici, P.; et al. Novel phenotype associated with a mutation in the KCNA1(Kv1.1) gene. Front. Physiol. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestre, T.A.; Manole, A.; MacDonald, H.; Riazi, S.; Kraeva, N.; Hanna, M.G.; Lang, A.E.; Männikkö, R.; Yoon, G. A novel KCNA1 mutation in a family with episodic ataxia and malignant hyperthermia. Neurogenetics 2016, 17, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Imbrici, P.; Altamura, C.; Gualandi, F.; Mangiatordi, G.F.; Neri, M.; De Maria, G.; Ferlini, A.; Padovani, A.; D’Adamo, M.C.; Nicolotti, O.; et al. A novel KCNA1 mutation in a patient with paroxysmal ataxia, myokymia, painful contractures and metabolic dysfunctions. Mol. Cell. Neurosci. 2017, 83, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Shook, S.J.; Mamsa, H.; Jen, J.C.; Baloh, R.W.; Zhou, L. Novel Mutation in KCNA1 Causes Episodic Ataxia With Paroxysmal Dyspnea. Muscle Nerve 2008, 37, 399–402. [Google Scholar] [CrossRef]
- Zuberi, S.M.; Eunson, L.H.; Spauschus, A.; De Silva, R.; Tolmie, J.; Wood, N.W.; McWilliam, R.C.; Stephenson, J.B.; Stephenson, J.P.; Kullmann, D.M.; et al. A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain 1999, 122, 817–825. [Google Scholar] [CrossRef] [Green Version]
- Karalok, Z.S.; Megaro, A.; Cenciarini, M.; Guven, A.; Hasan, S.M.; Taskin, B.D.; Imbrici, P.; Ceylaner, S.; Pessia, M.; D’Adamo, M.C. Identification of a new de novo mutation underlying regressive episodic ataxia type I. Front. Neurol. 2018, 9, 587. [Google Scholar] [CrossRef]
- Yin, X.M.; Lin, J.H.; Cao, L.; Zhang, T.M.; Zeng, S.; Zhang, K.L.; Tian, W.T.; Hu, Z.M.; Li, N.; Wang, J.L.; et al. Familial paroxysmal kinesigenic dyskinesia is associated with mutations in the KCNA1 gene. Hum. Mol. Genet. 2018, 27, 625–637. [Google Scholar] [CrossRef]
- Lassche, S.; Lainez, S.; Bloem, B.R.; van de Warrenburg, B.P.; Hofmeijer, J.; Lemmink, H.H.; Hoenderop, J.G.; Bindels, R.J.; Drost, G. A novel KCNA1 mutation causing episodic ataxia type I. Muscle Nerve 2014, 50, 289–291. [Google Scholar] [CrossRef] [Green Version]
- Jen, J.C.; Wan, J. Episodic ataxias. Handb. Clin. Neurol. 2018, 155, 205–215. [Google Scholar] [CrossRef]
- Imbrici, P.; Liantonio, A.; Camerino, G.M.; De Bellis, M.; Camerino, C.; Mele, A.; Giustino, A.; Pierno, S.; De Luca, A.; Tricarico, D.; et al. Therapeutic Approaches to Genetic Ion Channelopathies and Perspectives in Drug Discovery. Front. Pharmacol. 2016, 7, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbrici, P.; Gualandi, F.; D’Adamo, M.C.; Masieri, M.T.; Cudia, P.; De Grandis, D.; Mannucci, R.; Nicoletti, I.; Tucker, S.J.; Ferlini, A.; et al. A novel KCNA1 mutation identified in an Italian family affected by episodic ataxia type 1. Neuroscience 2008, 157, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Rea, R.; Spauschus, A.; Eunson, L.H.; Hanna, M.G.; Kullmann, D.M. Variable K(+) channel subunit dysfunction in inherited mutations of KCNA1. J. Physiol. 2002, 538 Pt 1, 5–23. [Google Scholar] [CrossRef]
- Tristán-Clavijo, E.; Scholl, F.G.; Macaya, A.; Iglesias, G.; Rojas, A.M.; Lucas, M.; Castellano, A.; Martinez-Mir, A. Dominant-negative mutation p.Arg324Thr in KCNA1 impairs Kv1.1 channel function in episodic ataxia. Mov. Disord. 2016, 31, 1743–1748. [Google Scholar] [CrossRef] [PubMed]
- Imbrici, P.; Cusimano, A.; D’Adamo, M.C.; De Curtis, A.; Pessia, M. Functional characterization of an episodic ataxia type-1 mutation occurring in the S1 segment of hKv1.1 channels. Pflugers Arch. 2003, 446, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Adelman, J.P.; Bond, C.T.; Pessia, M.; Maylie, J. Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron 1995, 15, 1449–1454. [Google Scholar] [CrossRef] [Green Version]
- D’Adamo, M.C.; Liu, Z.; Adelman, J.P.; Maylie, J.; Pessia, M. Episodic ataxia type-1 mutations in the hKv1.1 cytoplasmic pore region alter the gating properties of the channel. EMBO J. 1998, 17, 1200–1207. [Google Scholar] [CrossRef]
- Hasan, S.; Bove, C.; Silvestri, G.; Mantuano, E.; Modoni, A.; Veneziano, L.; Macchioni, L.; Hunter, T.; Hunter, G.; Pessia, M.; et al. A channelopathy mutation in the voltage-sensor discloses contributions of a conserved phenylalanine to gating properties of Kv1.1 channels and ataxia. Sci. Rep. 2017, 7, 4583. [Google Scholar] [CrossRef] [Green Version]
- Petitjean, D.; Kalstrup, T.; Zhao, J.; Blunck, R. A Disease Mutation Causing Episodic Ataxia Type I in the S1 Links Directly to the Voltage Sensor and the Selectivity Filter in Kv Channels. J. Neurosci. 2015, 35, 12198–12206. [Google Scholar] [CrossRef] [Green Version]
- Imbrici, P.; Grottesi, A.; D’Adamo, M.C.; Mannucci, R.; Tucker, S.J.; Pessia, M. Contribution of the central hydrophobic residue in the PXP motif of voltage-dependent K+ channels to S6 flexibility and gating properties. Channels 2009, 3, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Imbrici, P.; D’Adamo, M.C.; Kullmann, D.M.; Pessia, M. Episodic ataxia type 1 mutations in the KCNA1 gene impair the fast inactivation properties of the human potassium channels Kv1.4-1.1/Kvbeta1.1 and Kv1.4-1.1/Kvbeta1.2. Eur. J. Neurosci. 2006, 24, 3073–3083. [Google Scholar] [CrossRef] [PubMed]
- Imbrici, P.; D’Adamo, M.C.; Grottesi, A.; Biscarini, A.; Pessia, M. Episodic ataxia type 1 mutations affect fast inactivation of K+ channels by a reduction in either subunit surface expression or affinity for inactivation domain. Am. J. Physiol. Cell Physiol. 2011, 300, C1314–C1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbrici, P.; D’Adamo, M.C.; Cusimano, A.; Pessia, M. Episodic ataxia type 1 mutation F184C alters Zn2+-induced modulation of the human K+ channel Kv1.4-Kv1.1/Kvbeta1.1. Am. J. Physiol. Cell. Physiol. 2007, 292, C778–C787. [Google Scholar] [CrossRef] [PubMed]
- Set, K.K.; Ghosh, D.; Huq, A.H.M.; Luat, A.F. Episodic Ataxia Type 1 (K-channelopathy) Manifesting as Paroxysmal Nonkinesogenic Dyskinesia: Expanding the Phenotype. Mov. Disord. Clin. Pract. 2017, 4, 784–786. [Google Scholar] [CrossRef]
- Tomlinson, S.E.; Rajakulendran, S.; Tan, S.V.; Graves, T.D.; Bamiou, D.E.; Labrum, R.W.; Burke, D.; Sue, C.M.; Giunti, P.; Schorge, S.; et al. Clinical, genetic, neurophysiological and functional study of new mutations in episodic ataxia type 1. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1107–1112. [Google Scholar] [CrossRef] [Green Version]
- Coutelier, M.; Coarelli, G.; Monin, M.L.; Konop, J.; Davoine, C.S.; Tesson, C.; Valter, R.; Anheim, M.; Behin, A.; Castelnovo, G.; et al. A panel study on patients with dominant cerebellar ataxia highlights the frequency of channelopathies. Brain 2017, 140, 1579–1594. [Google Scholar] [CrossRef]
- Zerr, P.; Adelman, J.P.; Maylie, J. Episodic ataxia mutations in Kv1.1 alter potassium channel function by dominant negative effects or haploinsufficiency. J. Neurosci. 1998, 18, 2842–2848. [Google Scholar] [CrossRef] [Green Version]
- Zerr, P.; Adelman, J.P.; Maylie, J. Characterization of three episodic ataxia mutations in the human Kv1.1 potassium channel. FEBS Lett. 1998, 431, 461–464. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; von Hehn, C.; Kaczmarek, L.K.; Ment, L.R.; Pober, B.R.; Hisama, F.M. Functional analysis of a novel potassium channel (KCNA1) mutation in hereditary myokymia. Neurogenetics 2007, 8, 131–135. [Google Scholar] [CrossRef] [Green Version]
- Rajakulendran, S.; Tan, S.V.; Matthews, E.; Tomlinson, S.E.; Labrum, R.; Sud, R.; Kullmann, D.M.; Schorge, S.; Hanna, M.G. A patient with episodic ataxia and paramyotonia congenita due to mutations in KCNA1 and SCN4A. Neurology 2009, 73, 993–995. [Google Scholar] [CrossRef] [Green Version]
- Poujois, A.; Antoine, J.C.; Combes, A.; Touraine, R.L. Chronic neuromyotonia as a phenotypic variation associated with a new mutation in the KCNA1 gene. J. Neurol. 2006, 253, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Knight, M.A.; Storey, E.; McKinlay Gardner, R.J.; Hand, P.; Forrest, S.M. Identification of a novel missense mutation L329I in the episodic ataxia type 1 gene KCNA1--a challenging problem. Hum. Mutat. 2000, 16, 374. [Google Scholar] [CrossRef]
- Lee, H.; Wang, H.; Jen, J.C.; Sabatti, C.; Baloh, R.W.; Nelson, S.F. A novel mutation in KCNA1 causes episodic ataxia without myokymia. Hum. Mutat. 2004, 24, 536. [Google Scholar] [CrossRef] [PubMed]
- Verdura, E.; Fons, C.; Schlüter, A.; Ruiz, M.; Fourcade, S.; Casasnovas, C.; Castellano, A.; Pujol, A. Complete loss of KCNA1 activity causes neonatal epileptic encephalopathy and dyskinesia. J. Med. Genet. 2020, 57, 132–137. [Google Scholar] [CrossRef]
- Rogers, A.; Golumbek, P.; Cellini, E.; Doccini, V.; Guerrini, R.; Wallgren-Pettersson, C.; Thuresson, A.C.; Gurnett, C.A. De novo KCNA1 variants in the PVP motif cause infantile epileptic encephalopathy and cognitive impairment similar to recurrent KCNA2 variants. Am. J. Med. Genet. A 2018, 176, 1748–1752. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, C.L.; Messing, A.; Chiu, S.Y. Temperature-sensitive neuromuscular transmission in Kv1.1 null mice: Role of potassium channels under the myelin sheath in young nerves. J. Neurosci. 1998, 18, 7200–7215. [Google Scholar] [CrossRef] [Green Version]
- Klassen, T.L.; Bomben, V.C.; Patel, A.; Drabek, J.; Chen, T.T.; Gu, W.; Zhang, F.; Chapman, K.; Lupski, J.R.; Noebels, J.L.; et al. High-resolution molecular genomic autopsy reveals complex sudden unexpected death in epilepsy risk profile. Epilepsia 2014, 55, e6–e12. [Google Scholar] [CrossRef] [Green Version]
- Syrbe, S.; Hedrich, U.B.S.; Riesch, E.; Djémié, T.; Müller, S.; Møller, R.S.; Maher, B.; Hernandez-Hernandez, L.; Synofzik, M.; Caglayan, H.S.; et al. De novo loss- or gain-of-function mutations in KCNA2 cause epileptic encephalopathy. Nat. Genet. 2015, 47, 393–399. [Google Scholar] [CrossRef]
- Masnada, S.; Hedrich, U.B.S.; Gardella, E.; Schubert, J.; Kaiwar, C.; Klee, E.W.; Lanpher, B.C.; Gavrilova, R.H.; Synofzik, M.; Bast, T.; et al. Clinical spectrum and genotype–phenotype associations of KCNA2-related encephalopathies. Brain 2017, 140, 2337–2354. [Google Scholar] [CrossRef] [Green Version]
- Dhaibar, H.; Gautier, N.M.; Chernyshev, O.Y.; Dominic, P.; Glasscock, E. Cardiorespiratory profiling reveals primary breathing dysfunction in Kcna1-null mice: Implications for sudden unexpected death in epilepsy. Neurobiol. Dis. 2019, 127, 502–511. [Google Scholar] [CrossRef]
- Moore, B.M.; Jerry Jou, C.; Tatalovic, M.; Kaufman, E.S.; Kline, D.D.; Kunze, D.L. The Kv1.1 null mouse, a model of sudden unexpected death in epilepsy (SUDEP). Epilepsia 2014, 55, 1808–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rho, J.M.; Szot, P.; Tempel, B.L.; Schwartzkroin, P.A. Developmental seizure susceptibility of kv1.1 potassium channel knockout mice. Dev. Neurosci. 1999, 21, 320–327. [Google Scholar] [CrossRef]
- Heeroma, J.H.; Henneberger, C.; Rajakulendran, S.; Hanna, M.G.; Schorge, S.; Kullmann, D.M. Episodic ataxia type 1 mutations differentially affect neuronal excitability and transmitter release. Dis. Model. Mech. 2009, 2, 612–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trosclair, K.; Dhaibar, H.A.; Gautier, N.M.; Mishra, V.; Glasscock, E. Neuron-specific Kv1.1 deficiency is sufficient to cause epilepsy, premature death, and cardiorespiratory dysregulation. Neurobiol. Dis. 2020, 137, 104759. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; Sakamoto, Y.; Nishio, T.; Baulac, S.; Kuwamura, M.; Ohno, Y.; Takizawa, A.; Kaneko, S.; Serikawa, T.; Mashimo, T. Kcna1-mutant rats dominantly display myokymia, neuromyotonia and spontaneous epileptic seizures. Brain Res. 2012, 1435, 154–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersson, S.; Persson, A.S.; Johansen, J.E.; Ingvar, M.; Nilsson, J.; Klement, G.; Arhem, P.; Schalling, M.; Lavebratt, C. Truncation of the Shaker-like voltage-gated potassium channel, Kv1.1, causes megencephaly. Eur. J. Neurosci. 2003, 18, 3231–3240. [Google Scholar] [CrossRef] [PubMed]
- Sosanya, N.M.; Brager, D.H.; Wolfe, S.; Niere, F.; Raab-Graham, K.F. Rapamycin reveals an mTOR-independent repression of Kv1.1 expression during epileptogenesis. Neurobiol. Dis. 2015, 73, 96–105. [Google Scholar] [CrossRef]
- Zhou, Y.D.; Lee, S.; Jin, Z.; Wright, M.; Smith, S.E.; Anderson, M.P. Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat. Med. 2009, 15, 1208–1214. [Google Scholar] [CrossRef]
- Binks, S.N.M.; Klein, C.J.; Waters, P.; Pittock, S.J.; Irani, S.R. LGI1, CASPR2 and related antibodies: A molecular evolution of the phenotypes. J. Neurol. Neurosurg. Psychiatry 2018, 89, 526–534. [Google Scholar] [CrossRef] [Green Version]
- Streit, A.K.; Derst, C.; Wegner, S.; Heinemann, U.; Zahn, R.K.; Decher, N. RNA editing of Kv1.1 channels may account for reduced ictogenic potential of 4-aminopyridine in chronic epileptic rats. Epilepsia 2011, 52, 645–648. [Google Scholar] [CrossRef]
- Krestel, H.; Raffel, S.; von Lehe, M.; Jagella, C.; Moskau-Hartmann, S.; Becker, A.; Elger, C.E.; Seeburg, P.H.; Nirkko, A. Differences between RNA and DNA due to RNA editing in temporal lobe epilepsy. Neurobiol. Dis. 2013, 56, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Chahal, C.A.A.; Salloum, M.N.; Alahdab, F.; Gottwald, J.A.; Tester, D.J.; Anwer, L.A.; So, E.L.; Murad, M.H.; St Louis, E.K.; Ackerman, M.J.; et al. Systematic Review of the Genetics of Sudden Unexpected Death in Epilepsy: Potential Overlap With Sudden Cardiac Death and Arrhythmia-Related Genes. J. Am. Heart Assoc. 2020, 9, e012264. [Google Scholar] [CrossRef] [PubMed]
- Wacker, S.J.; Jurkowski, W.; Simmons, K.J.; Fishwick, C.W.; Johnson, A.P.; Madge, D.; Lindahl, E.; Rolland, J.F.; de Groot, B.L. Identification of selective inhibitors of the potassium channel Kv1.1-1.2((3)) by high-throughput virtual screening and automated patch clamp. ChemMedChem 2012, 7, 1775–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finol-Urdaneta, R.K.; Belovanovic, A.; Micic-Vicovac, M.; Kinsella, G.K.; McArthur, J.R.; Al-Sabi, A. Marine Toxins Targeting Kv1 Channels: Pharmacological Tools and Therapeutic Scaffolds. Mar. Drugs 2020, 18, 173. [Google Scholar] [CrossRef] [Green Version]
- Niespodziany, I.; Mullier, B.; André, V.M.; Ghisdal, P.; Jnoff, E.; Moreno-Delgado, D.; Swinnen, D.; Sands, Z.; Wood, M.; Wolff, C. Discovery of a small molecule modulator of the Kv1.1/Kvβ1 channel complex that reduces neuronal excitability and in vitro epileptiform activity. CNS Neurosci. Ther. 2019, 25, 442–451. [Google Scholar] [CrossRef]
- Lu, Q.; Peevey, J.; Jow, F.; Monaghan, M.M.; Mendoza, G.; Zhang, H.; Wu, J.; Kim, C.Y.; Bicksler, J.; Greenblatt, L.; et al. Disruption of Kv1.1 N-type inactivation by novel small molecule inhibitors (disinactivators). Bioorg. Med. Chem. 2008, 16, 3067–3075. [Google Scholar] [CrossRef]
- Ottosson, N.E.; Wu, X.; Nolting, A.; Karlsson, U.; Lund, P.E.; Ruda, K.; Svensson, S.; Konradsson, P.; Elinder, F. Resin-acid derivatives as potent electrostatic openers of voltage-gated K channels and suppressors of neuronal excitability. Sci. Rep. 2015, 5, 13278. [Google Scholar] [CrossRef] [Green Version]
- Ottosson, N.E.; Silverå Ejneby, M.; Wu, X.; Yazdi, S.; Konradsson, P.; Lindahl, E.; Elinder, F. A drug pocket at the lipid bilayer-potassium channel interface. Sci. Adv. 2017, 3, e1701099. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Suzuki, Y.; Yamamura, H.; Ohya, S.; Muraki, K.; Imaizumi, Y. Molecular mechanisms underlying pimaric acid-induced modulation of voltage-gated K+ channels. J. Pharmacol. Sci. 2017, 133, 223–231. [Google Scholar] [CrossRef]
- Silverå Ejneby, M.; Wu, X.; Ottosson, N.E.; Münger, E.P.; Lundström, I.; Konradsson, P.; Elinder, F. Atom-by-atom tuning of the electrostatic potassium-channel modulator dehydroabietic acid. J. Gen. Physiol. 2018, 150, 731–750. [Google Scholar] [CrossRef] [Green Version]
- Snowball, A.; Chabrol, E.; Wykes, R.C.; Shekh-Ahmad, T.; Cornford, J.H.; Lieb, A.; Hughes, M.P.; Massaro, G.; Rahim, A.A.; Hashemi, K.S.; et al. Epilepsy Gene Therapy Using an Engineered Potassium Channel. J. Neurosci. 2019, 39, 3159–3169. [Google Scholar] [CrossRef] [Green Version]
- Colasante, G.; Qiu, Y.; Massimino, L.; Di Berardino, C.; Cornford, J.H.; Snowball, A.; Weston, M.; Jones, S.P.; Giannelli, S.; Lieb, A.; et al. In vivo CRISPRa decreases seizures and rescues cognitive deficits in a rodent model of epilepsy. Brain 2020, 143, 891–905. [Google Scholar] [CrossRef] [Green Version]
- Boison, D. New insights into the mechanisms of the ketogenic diet. Curr. Opin. Neurol. 2017, 30, 187–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-McGill, K.J.; Jackson, C.F.; Bresnahan, R.; Levy, R.G.; Cooper, P.N. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst. Rev. 2018, 11, CD001903. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio-Simeone, K.A.; Wilke, J.C.; Milligan, H.L.; Allen, C.N.; Rho, J.M.; Maganti, R.K. Ketogenic diet treatment abolishes seizure periodicity and improves diurnal rhythmicity in epileptic Kcna1-null mice. Epilepsia 2009, 50, 2027–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeone, K.A.; Matthews, S.A.; Rho, J.M.; Simeone, T.A. Ketogenic diet treatment increases longevity in Kcna1-null mice, a model of sudden unexpected death in epilepsy. Epilepsia 2016, 57, e178–e182. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.Y.; Simeone, K.A.; Simeone, T.A.; Pandya, J.D.; Wilke, J.C.; Ahn, Y.; Geddes, J.W.; Sullivan, P.G.; Rho, J.M. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann. Neurol. 2015, 78, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Simeone, T.A.; Matthews, S.A.; Samson, K.K.; Simeone, K.A. Regulation of brain PPARgamma2 contributes to ketogenic diet anti-seizure efficacy. Exp. Neurol. 2017, 287 Pt 1, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Vegh, V.; Reutens, D.C. Persistent sodium current blockers can suppress seizures caused by loss of low-threshold D-type potassium currents: Predictions from an in silico study of Kv1 channel disorders. Epilepsia Open 2020, 5, 86–96. [Google Scholar] [CrossRef]
- Xu, L.; Enyeart, J.A.; Enyeart, J.J. Neuroprotective agent riluzole dramatically slows inactivation of Kv1.4 potassium channels by a voltage-dependent oxidative mechanism. J. Pharmacol. Exp. Ther. 2001, 299, 227–237. [Google Scholar]
- Al-Sabi, A.; Daly, D.; Hoefer, P.; Kinsella, G.K.; Metais, C.; Pickering, M.; Herron, C.; Kaza, S.K.; Nolan, K.; Dolly, J.O. A Rational Design of a Selective Inhibitor for Kv1.1 Channels Prevalent in Demyelinated Nerves That Improves Their Impaired Axonal Conduction. J. Med. Chem. 2017, 60, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Arnold, R.; Huynh, W.; Kiernan, M.C.; Krishnan, A.V. Ion Channel Modulation as a Therapeutic Approach in Multiple Sclerosis. Curr. Med. Chem. 2015, 22, 4366–4378. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
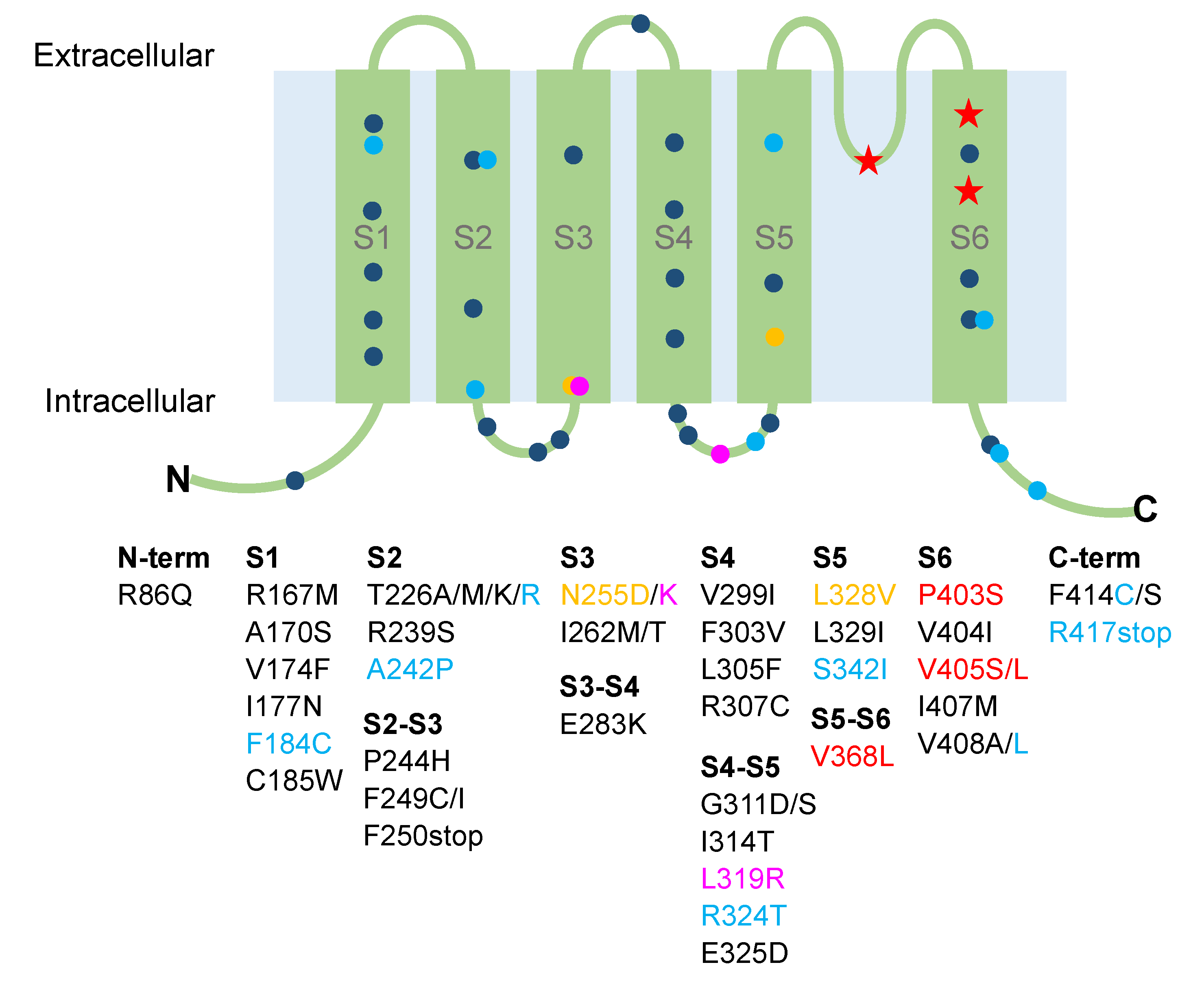
| Mutation | Position | Clinical Symptoms | Functional Defects | Treatment | References |
|---|---|---|---|---|---|
| R86Q | N-term | Severe stiffness, muscle cramps, pain | NA | Clonazepam ineffective | [84] |
| R167M | S1 | Ataxia, dysarthria, neuromyotonia | Non-functional channels and dominant-negative effect | NA | [85] |
| A170S | S1 | Cerebellar ataxia | NA | NA | [86] |
| V174F | S1 | Ataxia, myokymia, paroxsymal choreoathetosis | Non-functional channels | ACTZ and CBZ ineffective, phenytoin effective | [49,76,87] |
| I177N | S1 | Ataxia, myokymia | Reduced current density and dominant-negative effect, positive shift of voltage-dependent activation, slower activation, faster deactivation | NA | [75] |
| F184C | S1 | Severe ataxia, myokymia, tremors, weakness, stiffness, visual disturbances, epilepsy | Reduced current density and positive shift of voltage-dependent activation | Phenytoin partially effective | [47,49,52,76,87] |
| C185W | S1 | Ataxia, myokymia, stiffness, migraine, hyperthermia, short-sleep duration | Non-functional channels and dominant-negative effect | NA | [54,62,85] |
| T226A/M | S2 | Ataxia, myokymia | Reduced surface expression, positive shift of voltage dependence of activation, slower deactivation, slower activation | NA | [88] |
| T226K | S2 | Myokymia, leg hypertrophy, stiffness | Non-functional channels and dominant-negative effect | CBZ effective | [89] |
| T226R | S2 | Ataxia, myokymia, and epilepsy | Reduced current density | CBZ and ACTZ effective; phenobarbital, phenytoin, and valproate ineffective | [57,66] |
| T226R | S2 | Hypercontracted posture, skeletal deformities, stiffness | NA | NA | [58] |
| T226R | S2 | Cataplexy without ataxia, sleep disturbances | NA | ACTZ discontinued | [61] |
| R239S | S2 | Ataxia, myokymia | Non-functional channels | NA | [49,76,87] |
| A242P | S2 | Ataxia, myokymia, epilepsy | Reduced current density | ACTZ ineffective, lamotrigine effective | [57,85] |
| P244H | S2–S3 | Neuromyotonia without ataxia | Similar to WT | NA | [57] |
| F249C | S2–S3 | Ataxia, malignant hyperthermia | Nonfunctional channels | NA | [63] |
| F249I | S2–S3 | Ataxia and myokymia | Nonfunctional | NA | [49,76,87] |
| F250stop | S2–S3 | Ataxia, myokymia, paroxysmal shortness of breath | NA | NA | [65] |
| N255D | S3 | Hypomagnesemia | Reduced current density and dominant-negative effect | NA | [41] |
| N255K | S3 | Paroxysmal kinesigenic dyskinesia | Reduced current density, dominant-negative effect, positive shift of voltage-dependent activation | NA | [68] |
| I262M de novo | S3 | Ataxia, myokymia, stiffness, tremor, lower limb spasticity | Reduced current density and dominant-negative effect | Sodium valproate, diaminopyridine and phenytoin ineffective; ACTZ and CBZ worsened tremor; gabapentin and clonazepam effective for muscle stiffness | [69] |
| I262T | S3 | Ataxia, distal weakness, paresis of foot extensors, stiffness | Reduced current density and dominant-negative effect | NA | [59,60] |
| E283K | S3–S4 | Ataxia, myokymia, metabolic alterations, altered mechanosensation | Reduced current density, positive shift of voltage-dependent activation, slower activation | CBZ effective | [64] |
| V299I | S4 | Generalized myokymia and paramyotonya (due to SCN4A mutation) | Reduced current density and dominant-negative effect, positive shift of voltage-dependent activation | NA | [90] |
| F303V | S4 | Ataxia, myokymia, dizziness, slurred speech | Reduced current density, positive shift of voltage-dependent activation, slower activation, faster deactivation, increased C-type inactivation | NA | [78] |
| L305F | S4 | Remittent ataxia, neuromyotonya, cramps, stiffness, hypertrophy | NA | Clonazepam, CBZ, and amitriptyline ineffective | [91] |
| R307C | S4 | Ataxia, myokymia, headache, visual disturbance, nausea, weakness, slurred speech | Non-functional channels and dominant-negative effect | NA | [55] |
| G311D de novo | S4–S5 | Remittent ataxia, myokymia, diplopia | Reduced current density | ACTZ, oxcarbazepine, and valproate discontinued | [67] |
| G311S | S4–S5 | Ataxia | Reduced current density, positive shift of voltage-dependent activation | NA | [88] |
| I314T | S4–S5 | Cataplexy without ataxia, sleep disturbances | NA | ACTZ discontinued | [61] |
| L319R | S4–S5 | Paroxysmal kinesigenic dyskinesia without ataxia, dysarthria, seizure | Reduced current density and dominant-negative effect, positive shift of voltage-dependent activation | CBZ and oxcarbazepine effective | [68] |
| R324T | S4–S5 | Ataxia, epilepsy, and signs of paroxysmal kinesigenic dyskinesia | Reduced current density | ACTZ ineffective, CBZ effective | [74] |
| E325D | S4–S5 | Ataxia, myokymia | Reduced current, positive shift of voltage-dependent activation | ACTZ discontinued | [50,76,77,87] |
| L328V | S5 | Hypomagnesemia, muscle cramps, tetany | Reduced current and dominant-negative effect | Mg2+ and Ca2+ supplements | [42] |
| L329I | S5 | Ataxia | NA | NA | [92] |
| S342I | S5 | Ataxia, dizziness, slurred speech, seizure | NA | Phenytoin effective | [93] |
| V368L | S5–S6 | Severe infantile-onset dyskinesia, motor, and intellectual disability and epileptic encephalopathy | Non-functional channels | Oxcarbazepine effective | [94] |
| P403S | S6 | Infantile-onset seizures and cognitive impairment in twins | NA | Lamotrigine effective in one boy; drug-resistant seizures in the other boy | [95] |
| V404I | S6 | Ataxia, myokymia | Positive shift of voltage-dependent activation, slower kinetic of activation | CBZ effective | [57] |
| P405L | S6 | Infantile-onset seizures and cognitive impairment | NA | ACTZ, lamotrigine and valproate effective | [95] |
| P405S | S6 | Infantile-onset seizures and cognitive impairment | NA | Drug-resistant seizures | [95] |
| I407M | S6 | Ataxia, dysarthria, blurred vision, hearing impairment, neuromyotonya | Non-functional channels and dominant-negative effect | NA | [85] |
| V408A | S6 | Ataxia, myokymia | Faster activation and deactivation, increased C-type inactivation | NA | [49,76,87] |
| V408L | S6 | Progressive cerebellar ataxia, cognitive delay, seizures, stiffness, postural abnormalities | Faster C-type inactivation | Phenytoin effective | [56] |
| F414C | C-term | Ataxia, isolated photosensitive generalized tonic–clonic seizure | Nonfunctional channels and dominant-negative effect | ACTZ, oxcarbazepine, clozapam ineffective | [72] |
| F414S | C-term | Ataxia, myokymia, tremors, weakness, headache, visual disturbance, nausea, slurred speech | Nonfunctional channels | Variable response to CBZ in family | [55] |
| R417stop | C-term | Severe ataxia, epilepsy, periocular myokymia | Nonfunctional channels and dominant-negative effect | CBZ and ACTZ partially effective; lamotrigine, vigabatrin, clonazepam ineffective | [57,73] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Adamo, M.C.; Liantonio, A.; Rolland, J.-F.; Pessia, M.; Imbrici, P. Kv1.1 Channelopathies: Pathophysiological Mechanisms and Therapeutic Approaches. Int. J. Mol. Sci. 2020, 21, 2935. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082935
D’Adamo MC, Liantonio A, Rolland J-F, Pessia M, Imbrici P. Kv1.1 Channelopathies: Pathophysiological Mechanisms and Therapeutic Approaches. International Journal of Molecular Sciences. 2020; 21(8):2935. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082935
Chicago/Turabian StyleD’Adamo, Maria Cristina, Antonella Liantonio, Jean-Francois Rolland, Mauro Pessia, and Paola Imbrici. 2020. "Kv1.1 Channelopathies: Pathophysiological Mechanisms and Therapeutic Approaches" International Journal of Molecular Sciences 21, no. 8: 2935. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21082935