MicroRNAs-Based Nano-Strategies as New Therapeutic Approach in Multiple Myeloma to Overcome Disease Progression and Drug Resistance

 , , , , and
, , , , and
Abstract
:1. Introduction
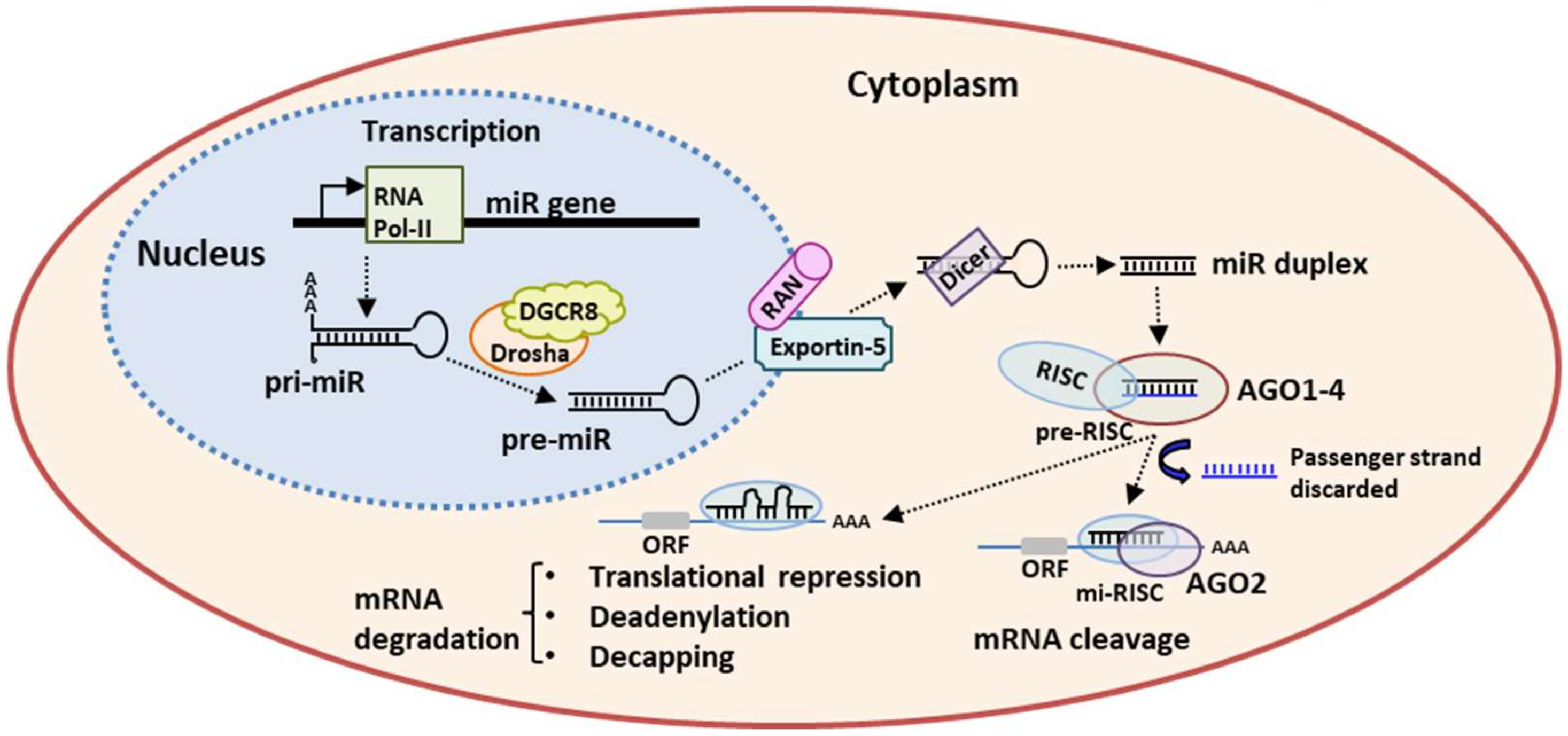
2. miRs Biogenesis and Mechanism of Action
3. miRs Deregulation in MM
4. Nanocarriers as miRs Delivery Systems
4.1. Lipid-Based Carriers
4.2. Cationic Polymer-Based Carriers
4.3. Exosomes as miRs Delivery System
4.4. miRs as Clinical-Based Therapeutic Strategies
miRs as Clinical-Based Therapeutic Strategies in MM
Author Contributions
Funding
Conflicts of Interest
References
- Anderson, K.C.; Carrasco, R.D. Pathogenesis of myeloma. Annu. Rev. Pathol. 2011, 6, 249–274. [Google Scholar] [CrossRef]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment drug resistance in multiple myeloma: Emerging new players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef] [Green Version]
- Solimando, A.G.; Da Vià, M.C.; Cicco, S.; Leone, P.; Di Lernia, G.; Giannico, D.; Desantis, V.; Frassanito, M.A.; Morizio, A.; Delgado Tascon, J.; et al. High-Risk Multiple Myeloma: Integrated Clinical and Omics Approach Dissects the Neoplastic Clone and the Tumor Microenvironment. J. Clin. Med. 2019, 8, 997. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Croce, C.M. MicroRNA dysregulation in cancer: Diagnostics, monitoring and therapeutics. EMBO Mol. Med. 2012, 4, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Chitkara, D.; Mittal, A.; Mahato, R.I. miRNAs in pancreatic cancer: Therapeutic potential, delivery challenges and strategies. Adv. Drug Deliv. Rev. 2015, 81, 34–52. [Google Scholar] [CrossRef]
- Benetatos, L.; Vartholomatos, G. Deregulated microRNAs in multiple myeloma. Cancer 2012, 118, 878–887. [Google Scholar] [CrossRef]
- Tagliaferri, P.; Rossi, M.; Di Martino, M.T.; Amodio, N.; Leone, E.; Gulla, A.; Neri, A.; Tassone, P. Promises and challenges of MicroRNA-based treatment of multiple myeloma. Curr. Cancer Drug Targets 2012, 12, 838–846. [Google Scholar] [CrossRef] [Green Version]
- Handa, H.; Murakami, Y.; Ishihara, R.; Kimura-Masuda, K.; Masuda, Y. The Role and Function of microRNA in the Pathogenesis of Multiple Myeloma. Cancers 2019, 11, 1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roccaro, A.M.; Sacco, A.; Thompson, B.; Leleu, X.; Azab, A.K.; Azab, F.; Runnels, J.; Jia, X.; Ngo, H.T.; Melhem, M.R.; et al. Micro RNAs 15a and 16 regulate tumor proliferation in multiple myeloma. Blood 2009, 113, 6669–6680. [Google Scholar] [CrossRef] [PubMed]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, Z.; Gemeinhart, R.A. Progress in microRNA delivery. J. Control. Release 2013, 172, 962–974. [Google Scholar] [CrossRef] [Green Version]
- Munker, R.; Liu, C.G.; Taccioli, C.; Alder, H.; Heerema, N. MicroRNA profiles of drug-resistant myeloma cell lines. Acta Haematol. 2010, 123, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Chitkara, D.; Singh, S.; Mittal, A. Nanocarrier-based co-delivery of small molecules and siRNA/miRNA for treatment of cancer. Ther. Deliv. 2016, 7, 245–255. [Google Scholar] [CrossRef]
- Scheideler, M.; Vidakovic, I.; Prassl, R. Lipid nanocarriers for microRNA delivery. Chem. Phys. Lipids 2020, 226, 104837. [Google Scholar] [CrossRef]
- Shi, Z.; Chen, Q.; Li, C.; Wang, L.; Qian, X.; Jiang, C.; Liu, X.; Wang, X.; Li, H.; Kang, C. MiR-124 governs glioma growth and angiogenesis and enhances chemosensitivity by targeting R-Ras and N-Ras. Neuro Oncol. 2014, 16, 1341–1353. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Ren, Y.; Shi, Z.; Long, L.; Pu, P.; Shen, J.; Yuan, X.; Kang, C. Sequence-dependent synergistic inhibition of human glioma cell lines by combined temozolomide and miR-21 inhibitor gene therapy. Mol. Pharm. 2012, 9, 2636–2645. [Google Scholar] [CrossRef]
- Gandhi, N.S.; Tekade, R.K.; Chougule, M.B. Nanocarrier mediated delivery of siRNA/miRNA in combination with chemotherapeutic agents for cancer therapy: Current progress and advances. J. Control. Release 2014, 194, 238–256. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Jeon, K.; Lee, J.T.; Kim, S.; Kim, V.N. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Singh, N.; Kumar, S.; Kumari, J.; Singh, R.; Gaba, S.; Yadav, M.C.; Grover, M.; Chaurasia, S.; Kumar, R. Identification and evolutionary analysis of polycistronic miRNA clusters in domesticated and wild wheat. Genomics 2020, 112, 2334–2348. [Google Scholar] [CrossRef] [PubMed]
- Monteys, A.M.; Spengler, R.M.; Wan, J.; Tecedor, L.; Lennox, K.A.; Xing, Y.; Davidson, B.L. Structure and activity of putative intronic miRNA promoters. RNA 2010, 16, 495–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Niaz, S. The AGO proteins: An overview. Biol. Chem. 2018, 399, 525–547. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. (Lausanne) 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Fabian, M.R.; Sonenberg, N. The mechanics of miRNA-mediated gene silencing: A look under the hood of miRISC. Nat. Struct. Mol. Biol. 2012, 19, 586–593. [Google Scholar] [CrossRef]
- Wilczynska, A.; Bushell, M. The complexity of miRNA-mediated repression. Cell Death Differ. 2015, 22, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef]
- Lionetti, M.; Agnelli, L.; Lombardi, L.; Tassone, P.; Neri, A. MicroRNAs in the pathobiology of multiple myeloma. Curr. Cancer Drug Targets 2012, 12, 823–837. [Google Scholar] [CrossRef]
- Kassambara, A.; Jourdan, M.; Bruyer, A.; Robert, N.; Pantesco, V.; Elemento, O.; Klein, B.; Moreaux, J. Global miRNA expression analysis identifies novel key regulators of plasma cell differentiation and malignant plasma cell. Nucleic Acids Res. 2017, 45, 5639–5652. [Google Scholar] [CrossRef] [PubMed]
- Pichiorri, F.; Suh, S.S.; Ladetto, M.; Kuehl, M.; Palumbo, T.; Drandi, D.; Taccioli, C.; Zanesi, N.; Alder, H.; Hagan, J.P.; et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 35, 12885–12990. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.Y.; She, X.M.; Qin, Y.; Chu, Z.B.; Chen, L.; Ai, L.S.; Zhang, L.; Hu, Y. miR-15a and miR-16 affect the angiogenesis of multiple myeloma by targeting VEGF. Carcinogenesis 2013, 34, 426–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermuller, J.; Kretzschmar, A.K.; Burger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007, 110, 1330–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, E.; Morelli, E.; Di Martino, M.T.; Amodio, N.; Foresta, U.; Gullà, A.; Rossi, M.; Neri, A.; Giordano, A.; Munshi, N.C.; et al. Targeting miR-21 inhibits in vitro and in vivo multiple myeloma cell growth. Clin. Cancer Res. 2013, 19, 2096–2106. [Google Scholar] [CrossRef] [Green Version]
- Petrocca, F.; Visone, R.; Onelli, M.R.; Shah, M.H.; Nicoloso, M.S.; de Martino, I.; Iliopoulos, D.; Pilozzi, E.; Liu, C.G.; Negrini, M.; et al. E2F1-regulated microRNAs impair TGFbeta-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell 2008, 13, 272–286. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Li, C.; Zhang, R.; Gao, X.; Qu, X.; Zhao, M.; Qiao, C.; Xu, J.; Li, J. miR-17-92 cluster microRNAs confers tumorigenicity in multiple myeloma. Cancer Lett. 2011, 309, 62–70. [Google Scholar] [CrossRef]
- Chim, C.S.; Wong, K.Y.; Qi, Y.; Loong, F.; Lam, W.L.; Wong, L.G.; Jin, D.Y.; Costello, J.F.; Liang, R. Epigenetic inactivation of the miR-34a in hematological malignancies. Carcinogenesis 2010, 31, 745–750. [Google Scholar] [CrossRef] [Green Version]
- Misso, G.; Di Martino, M.T.; De Rosa, G.; Farooqi, A.A.; Lombardi, A.; Campani, V.; Zarone, M.R.; Gullà, A.; Tagliaferri, P.; Tassone, P.; et al. Mir-34: A new weapon against cancer? Mol. Ther. Nucleic Acids 2014, 3, e194. [Google Scholar] [CrossRef]
- Di Martino, M.T.; Campani, V.; Misso, G.; Gallo Cantafio, M.E.; Gullà, A.; Foresta, U.; Guzzi, P.H.; Castellano, M.; Grimaldi, A.; Gigantino, V.; et al. In vivo activity of miR-34a mimics delivered by stable nucleic acid lipid particles (SNALPs) against multiple myeloma. PLoS ONE 2014, 9, e90005. [Google Scholar] [CrossRef]
- Amodio, N.; Stamato, M.A.; Gullà, A.M.; Morelli, E.; Romeo, E.; Raimondi, L.; Pitari, M.R.; Ferrandino, I.; Misso, G.; Caraglia, M.; et al. Therapeutic Targeting of miR-29b/HDAC4 Epigenetic Loop in Multiple Myeloma. Mol. Cancer Ther. 2016, 15, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Leotta, M.; Bellizzi, D.; Di Martino, M.T.; D’Aquila, P.; Lionetti, M.; Fabiani, F.; Leone, E.; Gullà, A.M.; Passarino, G.; et al. DNA-demethylating and anti-tumor activity of synthetic miR-29b mimics in multiple myeloma. Oncotarget 2012, 3, 1246–1258. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.K.; Wang, H.; Leng, Y.; Li, Z.L.; Yang, Y.F.; Xiao, F.J.; Li, Q.F.; Chen, X.Q.; Wang, L.S. Overexpression of microRNA-29b induces apoptosis of multiple myeloma cells through down regulating Mcl-1. Biochem. Biophys. Res. Commun. 2011, 414, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Di Martino, M.T.; Foresta, U.; Leone, E.; Lionetti, M.; Leotta, M.; Gullà, A.M.; Pitari, M.R.; Conforti, F.; Rossi, M.; et al. miR-29b sensitizes multiple myeloma cells to bortezomib-induced apoptosis through the activation of a feedback loop with the transcription factor Sp1. Cell Death Dis. 2012, 3, e436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lionetti, M.; Biasiolo, M.; Agnelli, L.; Todoerti, K.; Mosca, L.; Fabris, S.; Sales, G.; Deliliers, G.L.; Bicciato, S.; Lombardi, L.; et al. Identification of microRNA expression patterns and definition of a microRNA/mRNA regulatory network in distinct molecular groups of multiple myeloma. Blood 2009, 114, e20–e26. [Google Scholar] [CrossRef] [Green Version]
- Leotta, M.; Biamonte, L.; Raimondi, L.; Ronchetti, D.; Di Martino, M.T.; Botta, C.; Leone, E.; Pitari, M.R.; Neri, A.; Giordano, A.; et al. A p53-dependent tumor suppressor network is induced by selective miR-125a-5p inhibition in multiple myeloma cells. J. Cell Physiol. 2014, 229, 2106–2116. [Google Scholar] [CrossRef]
- Morelli, E.; Leone, E.; Cantafio, M.E.; Di Martino, M.T.; Amodio, N.; Biamonte, L.; Gullà, A.; Foresta, U.; Pitari, M.R.; Botta, C.; et al. Selective targeting of IRF4 by synthetic microRNA-125b-5p mimics induces anti-multiple myeloma activity in vitro and in vivo. Leukemia 2015, 29, 2173–2183. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Luan, Y.; Chang, H.; Chen, G. The diagnostic and prognostic value of plasma microRNA-125b-5p in patients with multiple myeloma. Oncol. Lett. 2018, 16, 4001–4007. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Li, T.; Yin, Z.; Chen, S.; Fei, J.; Shen, J.; Zhang, Y. Integrative analysis of signaling pathways and diseases associated with the miR-106b/25 cluster and their function study in berberine-induced multiple myeloma cells. Funct. Integr. Genom. 2017, 17, 253–262. [Google Scholar] [CrossRef]
- Yuan, R.; Liu, N.; Yang, J.; Peng, J.; Liu, L.; Guo, X. The expression and role of miR-181a in multiple myeloma. Medicine (Baltimore) 2018, 97, e12081. [Google Scholar] [CrossRef]
- Frassanito, M.A.; Desantis, V.; Di Marzo, L.; Craparotta, I.; Beltrame, L.; Marchini, S.; Annese, T.; Visino, F.; Arciuli, M.; Saltarella, I.; et al. Bone marrow fibroblasts overexpress miR-27b and miR-214 in step with multiple myeloma progression, dependent on tumour cell-derived exosomes. J. Pathol. 2019, 247, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Triboulet, R.; Mohseni, M.; Schlegelmilch, K.; Shrestha, K.; Camargo, F.D.; Gregory, R.I. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell 2014, 156, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.; Pitari, M.R.; Amodio, N.; Di Martino, M.T.; Conforti, F.; Leone, E.; Botta, C.; Paolino, F.M.; Del Giudice, T.; Iuliano, E.; et al. miR-29b negatively regulates human osteoclastic cell differentiation and function: Implications for the treatment of multiple myeloma-related bone disease. J. Cell Physiol. 2013, 228, 1506–1515. [Google Scholar] [CrossRef] [PubMed]
- Botta, C.; Cucè, M.; Pitari, M.R.; Caracciolo, D.; Gullà, A.; Morelli, E.; Riillo, C.; Biamonte, L.; Gallo Cantafio, M.E.; Prabhala, R.; et al. MiR-29b antagonizes the pro-inflammatory tumor-promoting activity of multiple myeloma-educated dendritic cells. Leukemia 2018, 32, 1003–1015. [Google Scholar] [CrossRef] [Green Version]
- Umezu, T.; Imanishi, S.; Yoshizawa, S.; Kawana, C.; Ohyashiki, J.H.; Ohyashiki, K. Induction of multiple myeloma bone marrow stromal cell apoptosis by inhibiting extracellular vesicle miR-10a secretion. Blood Adv. 2019, 3, 3228–3240. [Google Scholar] [CrossRef]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J. Clin. Investig. 2013, 123, 1542–1555. [Google Scholar] [CrossRef]
- De Veirman, K.; Wang, J.; Xu, S.; Leleu, X.; Himpe, E.; Maes, K.; De Bruyne, E.; Van Valckenborgh, E.; Vanderkerken, K.; Menu, E.; et al. Induction of miR-146a by multiple myeloma cells in mesenchymal stromal cells stimulates their pro-tumoral activity. Cancer Lett. 2016, 377, 17–24. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, D.Y.; Huang, L. In vivo delivery of miRNAs for cancer therapy: Challenges and strategies. Adv. Drug Deliv. Rev. 2015, 81, 128–141. [Google Scholar] [CrossRef] [Green Version]
- Kang, L.; Gao, Z.; Huang, W.; Jin, M.; Wang, Q. Nanocarrier-mediated co-delivery of chemotherapeutic drugs and gene agents for cancer treatment. Acta Pharm. Sin. B 2015, 5, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z. The guideline of the design and validation of MiRNA mimics. Methods Mol. Biol. 2011, 676, 211–223. [Google Scholar]
- Hosseinahli, N.; Aghapour, M.; Duijf, P.H.G.; Baradaran, B. Treating cancer with microRNA replacement therapy: A literature review. J. Cell Physiol. 2018, 233, 5574–5588. [Google Scholar] [CrossRef] [PubMed]
- Lima, J.F.; Cerqueira, L.; Figueiredo, C.; Oliveira, C.; Azevedo, N.F. Anti-miRNA oligonucleotides: A comprehensive guide for design. RNA Biol. 2018, 15, 338–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.G.; Brown, D.; Stoudemire, J.; Lammers, P. Developing therapeutic microRNAs for cancer. Gene Ther. 2011, 18, 1121–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yu, B.; Ren, W.; Mo, X.; Zhou, C.; He, H.; Jia, H.; Wang, L.; Jacob, S.T.; Lee, R.J.; et al. Enhanced hepatic delivery of siRNA and microRNA using oleic acid based lipid nanoparticle formulations. J. Control. Release 2013, 172, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Crawford, M.; Yu, B.; Mao, Y.; Nana-Sinkam, S.P.; Lee, L.J. MicroRNA delivery by cationic lipoplexes for lung cancer therapy. Mol. Pharm. 2011, 8, 1381–1389. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Crawford, M.; Mao, Y.; Lee, R.J.; Davis, I.C.; Elton, T.S.; Lee, L.J.; Nana-Sinkam, S.P. Therapeutic Delivery of MicroRNA-29b by Cationic Lipoplexes for Lung Cancer. Mol. Ther. Nucleic Acids 2013, 2, e84. [Google Scholar] [CrossRef]
- Lee, H.Y.; Mohammed, K.A.; Kaye, F.; Sharma, P.; Moudgil, B.M.; Clapp, W.L.; Nasreen, N. Targeted delivery of let-7a microRNA encapsulated ephrin-A1 conjugated liposomal nanoparticles inhibit tumor growth in lung cancer. Int. J. Nanomed. 2013, 8, 4481–4494. [Google Scholar]
- Shi, S.; Han, L.; Deng, L.; Zhang, Y.; Shen, H.; Gong, T.; Zhang, Z.; Sun, X. Dual drugs (microRNA-34a and paclitaxel)-loaded functional solid lipid nanoparticles for synergistic cancer cell suppression. J. Control. Release 2014, 194, 228–237. [Google Scholar] [CrossRef]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.; Kim, S.A.; Yang, Y.G.; Yoo, H.; Lim, S.J.; Mok, H. Long chain microRNA conjugates in calcium phosphate nanoparticles for efficient formulation and delivery. Arch. Pharm. Res. 2015, 38, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.F.; Weirauch, U.; Thomas, M.; Grünweller, A.; Hartmann, R.K.; Aigner, A. MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 2011, 71, 5214–5224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, H.L.; Lee, H.J.; Uto, K.; Ebara, M.; Kim, W.J.; Aoyagi, T.; Park, I.K. Simultaneous Drug and Gene Delivery from the Biodegradable Poly(ε-caprolactone) Nanofibers for the Treatment of Liver Cancer. J. Nanosci. Nanotechnol. 2015, 15, 7971–7975. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.Y.; Choe, J.W.; Pu, K.; Devulapally, R.; Bachawal, S.; Machtaler, S.; Chowdhury, S.M.; Luong, R.; Tian, L.; Khuri-Yakub, B.; et al. Ultrasound-guided delivery of microRNA loaded nanoparticles into cancer. J. Control. Release 2015, 203, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.B.; Saltzman, W.M.; Slack, F.J. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, E1695–E1704. [Google Scholar] [CrossRef] [Green Version]
- Cosco, D.; Cilurzo, F.; Maiuolo, J.; Federico, C.; Di Martino, M.T.; Cristiano, M.C.; Tassone, P.; Fresta, M.; Paolino, D. Delivery of miR-34a by chitosan/PLGA nanoplexes for the anticancer treatment of multiple myeloma. Sci. Rep. 2015, 5, 17579. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Pan, L.; Xiang, B.; Zhu, H.; Wu, Y.; Chen, M.; Guan, P.; Zou, X.; Valencia, C.A.; Dong, B.; et al. Potential role of exosome-associated microRNA panels and in vivo environment to predict drug resistance for patients with multiple myeloma. Oncotarget 2016, 7, 30876–30891. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Liu, C.J.; Avet-Loiseau, H.; Park, J.; Shi, J.; Campigotto, F.; Salem, K.Z.; Huynh, D.; Glavey, S.V.; Rivotto, B.; et al. Prognostic role of circulating exosomal miRNAs in multiple myeloma. Blood 2017, 129, 2429–2436. [Google Scholar] [CrossRef]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Tian, J.; Chen, X. Effect of surface properties on liposomal siRNA delivery. Biomaterials 2016, 79, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Anwer, K.; Meaney, C.; Kao, G.; Hussain, N.; Shelvin, R.; Earls, R.M.; Leonard, P.; Quezada, A.; Rolland, A.P.; Sullivan, S.M. Cationic lipid-based delivery system for systemic cancer gene therapy. Cancer Gene Ther. 2000, 7, 1156–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthiah, M.; Park, I.K.; Cho, C.S. Nanoparticle-mediated delivery of therapeutic genes: Focus on miRNA therapeutics. Expert Opin. Drug Deliv. 2013, 10, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.M.; Hall, L.L.; Ayyalapu, A.R.; Irion, V.R.; Nantz, M.H.; Hecker, J.G. Stability of mRNA/cationic lipid lipoplexes in human and rat cerebrospinal fluid: Methods and evidence for nonviral mRNA gene delivery to the central nervous system. Hum. Gene Ther. 2003, 143, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Welch, C.; Chen, Y.; Stallings, R.L. MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene 2007, 26, 5017–5022. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, C.M.; Jiang, Z.Z.; Yu, X.J.; Fan, C.G.; Xu, F.F.; Zhang, Q.; Li, L.I.; Li, R.F.; Sun, W.S.; et al. MicroRNA-34c targets TGFB-induced factor homeobox 2, represses cell proliferation and induces apoptosis in hepatitis B virus-related hepatocellular carcinoma. Oncol. Lett. 2015, 10, 3095–3102. [Google Scholar] [CrossRef] [Green Version]
- Krzeszinski, J.Y.; Wei, W.; Huynh, H.; Jin, Z.; Wang, X.; Chang, T.C.; Xie, X.J.; He, L.; Mangala, L.S.; Lopez-Berestein, G.; et al. miR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2. Nature 2014, 512, 431–435. [Google Scholar] [CrossRef]
- Srinivasachari, S.; Zhang, G.D. Novel cationic polymers and glycodendrimers for gene delivery. Pap. Am. Chem. 2004, 227, 1212–1220. [Google Scholar]
- Erbacher, P.; Zou, S.; Bettinger, T.; Steffan, A.M.; Remy, J.S. Chitosan-based vector/DNA complexes for gene delivery: Biophysical characteristics and transfection ability. Pharm. Res. 1998, 15, 1332–1339. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.; Zhang, Y.; Pan, Y.; Zhao, J.; Ren, L.; Liao, M.; Hu, Z.; Kong, L.; Wang, J. A novel PEGylation of chitosan nanoparticles for gene delivery. Biotechnol. Appl. Biochem. 2007, 46, 197–204. [Google Scholar]
- Fernandez-Piñeiro, I.; Badiola, I.; Sanchez, A. Nanocarriers for microRNA delivery in cancer medicine. Biotechnol. Adv. 2017, 35, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Devalliere, J.; Chang, W.G.; Andrejecsk, J.W.; Abrahimi, P.; Cheng, C.J.; Jane-wit, D.; Saltzman, W.M.; Pober, J.S. Sustained delivery of proangiogenic microRNA-132 by nanoparticle transfection improves endothelial cell transplantation. FASEB J. 2014, 28, 908–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turturici, G.; Tinnirello, R.; Sconzo, G.; Geraci, F. Extracellular membrane vesicles as a mechanism of cell-to-cell communication: Advantages and disadvantages. Am. J. Physiol. Cell Physiol. 2014, 306, C621–C633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daßler-Plenker, J.; Küttner, V.; Egeblad, M. Communication in tiny packages: Exosomes as means of tumor-stroma communication. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188340. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L.; Boyiadzis, M. Response commentary: Exosomes vs microvesicles in hematological malignancies. Leukemia 2017, 31, 2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, S.; Palanisamy, V. Horizontal transfer of RNAs: Exosomes as mediators of intercellular communication. Wiley Interdiscip. Rev. RNA 2012, 3, 286–293. [Google Scholar] [CrossRef] [Green Version]
- Squadrito, M.L.; Baer, C.; Burdet, F.; Maderna, C.; Gilfillan, G.D.; Lyle, R.; Ibberson, M.; De Palma, M. Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell Rep. 2014, 8, 1432–1446. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Powers, J.T.; Sacco, A.; Glavey, S.V.; Huynh, D.; Reagan, M.R.; Salem, K.Z.; Moschetta, M.; Shi, J.; Mishima, Y.; et al. The LIN28B/let-7 axis is a novel therapeutic pathway in multiple myeloma. Leukemia 2017, 31, 853–860. [Google Scholar] [CrossRef] [Green Version]
- Spizzo, R.; Nicoloso, M.S.; Croce, C.M.; Calin, G.A. SnapShot: MicroRNAs in Cancer. Cell 2009, 137, 586–586. [Google Scholar] [CrossRef] [Green Version]
- Büssing, I.; Slack, F.J.; Grosshans, H. let-7 microRNAs in development, stem cells and cancer. Trends Mol. Med. 2008, 14, 400–409. [Google Scholar] [CrossRef]
- Krutilina, R.; Sun, W.; Sethuraman, A.; Brown, M.; Seagroves, T.N.; Pfeffer, L.M.; Ignatova, T.; Fan, M. MicroRNA-18a inhibits hypoxia-inducible factor 1α activity and lung metastasis in basal breast cancers. Breast Cancer Res. 2014, 16, R78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Rocco, G.; Baldari, S.; Toietta, G. Exosomes and other extracellular vesicles-mediated microRNA delivery for cancer therapy. Transl. Cancer Res. 2017, 6, S1321–S1330. [Google Scholar] [CrossRef] [Green Version]
- Geraldo, M.V.; Yamashita, A.S.; Kimura, E.T. MicroRNA miR-146b-5p regulates signal transduction of TGF-β by repressing SMAD4 in thyroid cancer. Oncogene 2012, 31, 1910–1922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [Green Version]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The Potential for microRNA Therapeutics and Clinical Research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Yoon-Koo, K.; Brenner, A.J.; Sachdev, J.C.; Ejadi, S.; Borad, M.J.; Kim, T.Y.; Lim, H.Y.; Park, K.; Becerra, C.; et al. MRX34, a liposomal miR-34 mimic, in patients with advanced solid tumors: Final dose-escalation results from a first-in-human phase I trial of microRNA therapy. J. Clin. Oncol. 2016, 34, 2508. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020. [Google Scholar] [CrossRef]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2015, 108, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.D.; Li, X.M.; Gu, J.W.; Sun, X.C. MiR-155 regulates lymphoma cell proliferation and apoptosis through targeting SOCS3/JAK-STAT3 signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 5153–5159. [Google Scholar] [PubMed]
- Deng, S.; Zhang, Y.; Wang, Y.; Lu, X.; Jiang, Q. MicroRNA-92 regulates vascular smooth muscle cell function by targeting KLF4 during vascular restenosis and injury. Int. J. Clin. Exp. Pathol. 2019, 12, 4253–4262. [Google Scholar] [PubMed]
- Zhen, L.; Li, J.; Zhang, M.; Yang, K. MiR-10b decreases sensitivity of glioblastoma cells to radiation by targeting AKT. J. Biol. Res. 2016, 23, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, D.; Nandi, S.; Bhattacharjee, S. Combination therapy to checkmate Glioblastoma: Clinical challenges and advances. Clin. Transl. Med. 2018, 7, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Zandwijk, N.; McDiarmid, J.; Brahmbhatt, H.; Reid, G. Response to “An innovative mesothelioma treatment based on mir-16 mimic loaded EGFR targeted minicells (TargomiRs)”. Transl. Lung Cancer Res. 2018, 7, S60–S61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, G.; Kao, S.C.; Pavlakis, N.; Brahmbhatt, H.; MacDiarmid, J.; Clarke, S.; Boyer, M.; van Zandwijk, N. Clinical development of TargomiRs, a miRNA mimic-based treatment for patients with recurrent thoracic cancer. Epigenomics 2016, 8, 1079–1085. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Ju, S.; Chu, H.; Shen, X.; Zhang, Y.; Luo, X.; Cong, H. The potential function of microRNAs as biomarkers and therapeutic targets in multiple myeloma. Oncol. Lett. 2018, 15, 6094–6106. [Google Scholar] [CrossRef]
- Zarone, M.R.; Misso, G.; Grimaldi, A.; Zappavigna, S.; Russo, M.; Amler, E.; Di Martino, M.T.; Amodio, N.; Tagliaferri, P.; Tassone, P.; et al. Evidence of novel miR-34a-based therapeutic approaches for multiple myeloma treatment. Sci. Rep. 2017, 7, 17949. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.J.; Chu, Z.B.; Hu, Y.; Lin, J.; Wang, Z.; Jiang, M.; Chen, M.; Wang, X.; Kang, Y.; Zhou, Y.; et al. Targeting the miR-221-222/PUMA/BAK/BAX pathway abrogates dexamethasone resistance in multiple myeloma. Cancer Res. 2015, 75, 4384–4397. [Google Scholar] [CrossRef] [Green Version]
- Gullà, A.; Di Martino, M.T.; Gallo Cantafio, M.E.; Morelli, E.; Amodio, N.; Botta, C.; Pitari, M.R.; Lio, S.G.; Britti, D.; Stamato, M.A.; et al. A 13 mer LNA-i-miR-221 Inhibitor Restores Drug Sensitivity in Melphalan-Refractory Multiple Myeloma Cells. Clin. Cancer Res. 2016, 22, 1222–1233. [Google Scholar]
- Jagannathan, S.; Vad, N.; Vallabhapurapu, S.; Vallabhapurapu, S.; Anderson, K.C.; Driscoll, J.J. MiR-29b replacement inhibits proteasomes and disrupts aggresome+autophagosome formation to enhance the antimyeloma benefit of bortezomib. Leukemia 2015, 29, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, C.; Ju, S.; Wang, Y.; Wang, H.; Zhong, R. Myeloma cell adhesion to bone marrow stromal cells confers drug resistance by microRNA-21 up-regulation. Leuk. Lymphoma 2011, 52, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Haider, S.; Jagannathan, S.; Anaissie, E.; Driscoll, J.J. MicroRNA theragnostics for the clinical management of multiple myeloma. Leukemia 2014, 28, 732–738. [Google Scholar] [CrossRef] [Green Version]
- Esposito, C.L.; Cerchia, L.; Catuogno, S.; De Vita, G.; Dassie, J.P.; Santamaria, G.; Swiderski, P.; Condorelli, G.; Giangrande, P.H.; de Franciscis, V. Multifunctional aptamer-miRNA conjugates for targeted cancer therapy. Mol Ther. 2014, 22, 1151–1163. [Google Scholar] [CrossRef] [Green Version]
- Ganju, A.; Khan, S.; Hafeez, B.B.; Behrman, S.W.; Yallapu, M.M.; Chauhan, S.C.; Jaggi, M. miRNA nanotherapeutics for cancer. Drug Discov. Today 2017, 22, 424–432. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| miRNA | Target | Function in MM | Deregulation in MM | Reference |
|---|---|---|---|---|
| miR-21 | PTEN | Proliferation and survival in vitro and in vivo | Overexpressed | [15,35] |
| Rho-B | ||||
| BTG2 | ||||
| AKT | ||||
| miR-106b-25 cluster | PCAF | Cell viability, colony formation | Overexpressed | [32,49] |
| p38 | ||||
| MAPK | ||||
| miR-181a/b | BCL-2 | Cell proliferation, apoptosis | Overexpressed | [32,50] |
| NOVA1 | ||||
| PCAF | ||||
| miR-15a/16-1 cluster | Bcl-2 | Proliferation, apoptosis, angiogenesis | Downregulated | [12,32,33] |
| Cyclin D1 | ||||
| PI3K | ||||
| MAPK | ||||
| VEGF | ||||
| miR-17-92 cluster | SOCS-1 | MM cells drug resistance, poor prognosis | Overexpressed | [32,36] |
| BIM | ||||
| miR-34 family | c-MYC | Cell cycle, apoptosis, tumor growth in vivo | Downregulated | [39,40] |
| CDK6 | ||||
| c-MET | ||||
| Bcl-2 | ||||
| Notch1 | ||||
| miR-29b | HDAC | Cell proliferation, apoptosis, migration | Downregulated | [41,42,43] |
| DNMT3B | ||||
| MCL-1 | ||||
| CDK-6 | ||||
| AKT | ||||
| Sp1 | ||||
| miR-125a-5p | p53 | Cell growth, apoptosis, migration | Overexpressed | [44] |
| p21 | ||||
| BAX | ||||
| MDM2 | ||||
| miR-125b-5p | IRF4 | Cell growth, apoptosis, autophagy | Downregulated | [47] |
| Carrier Type | Delivery System | Targeted miRNA | Cancer Type | Reference |
|---|---|---|---|---|
| Lipid-based Carriers | DOTMA | miR-122 | Liver cancer Lung cancer | [65] |
| miR-133b | [66] | |||
| miR-29b | [67] | |||
| DOTAP | let-7a miR | Lung cancer Mesothelioma | [68] | |
| DDAB | miR-34a | Lung cancer Melanoma | [69] | |
| SNALPs | miR-34a | Multiple Myeloma | [70] | |
| Cationic Polymer-Based Carriers | PEI | miR-34a | Prostate cancer | [71] |
| miR-145 | HCC | [72] | ||
| miR-33a | Colon cancer | [73] | ||
| PLGA | miR-122 | Colon cancer | [74] | |
| miR-155 | Lymphoma | [75] | ||
| Chitosan/PLGA | miR-34a | Multiple Myeloma | [76] | |
| Exososome | miR-16-5p | Multiple Myeloma | [77] | |
| miR-15a-5p | ||||
| miR-20a-5p | ||||
| miR17-5p | ||||
| let-7b | Multiple Myeloma | [78] | ||
| miR-18a | ||||
| miR-27b-3p | Multiple Myeloma | [52] | ||
| miR-214-3p | ||||
| miR-146b | Glioma | [79] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desantis, V.; Saltarella, I.; Lamanuzzi, A.; Melaccio, A.; Solimando, A.G.; Mariggiò, M.A.; Racanelli, V.; Paradiso, A.; Vacca, A.; Frassanito, M.A. MicroRNAs-Based Nano-Strategies as New Therapeutic Approach in Multiple Myeloma to Overcome Disease Progression and Drug Resistance. Int. J. Mol. Sci. 2020, 21, 3084. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093084
Desantis V, Saltarella I, Lamanuzzi A, Melaccio A, Solimando AG, Mariggiò MA, Racanelli V, Paradiso A, Vacca A, Frassanito MA. MicroRNAs-Based Nano-Strategies as New Therapeutic Approach in Multiple Myeloma to Overcome Disease Progression and Drug Resistance. International Journal of Molecular Sciences. 2020; 21(9):3084. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093084
Chicago/Turabian StyleDesantis, Vanessa, Ilaria Saltarella, Aurelia Lamanuzzi, Assunta Melaccio, Antonio Giovanni Solimando, Maria Addolorata Mariggiò, Vito Racanelli, Angelo Paradiso, Angelo Vacca, and Maria Antonia Frassanito. 2020. "MicroRNAs-Based Nano-Strategies as New Therapeutic Approach in Multiple Myeloma to Overcome Disease Progression and Drug Resistance" International Journal of Molecular Sciences 21, no. 9: 3084. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093084