Micheliolide Enhances Radiosensitivities of p53-Deficient Non-Small-Cell Lung Cancer via Promoting HIF-1α Degradation

Abstract
:1. Introduction
2. Results
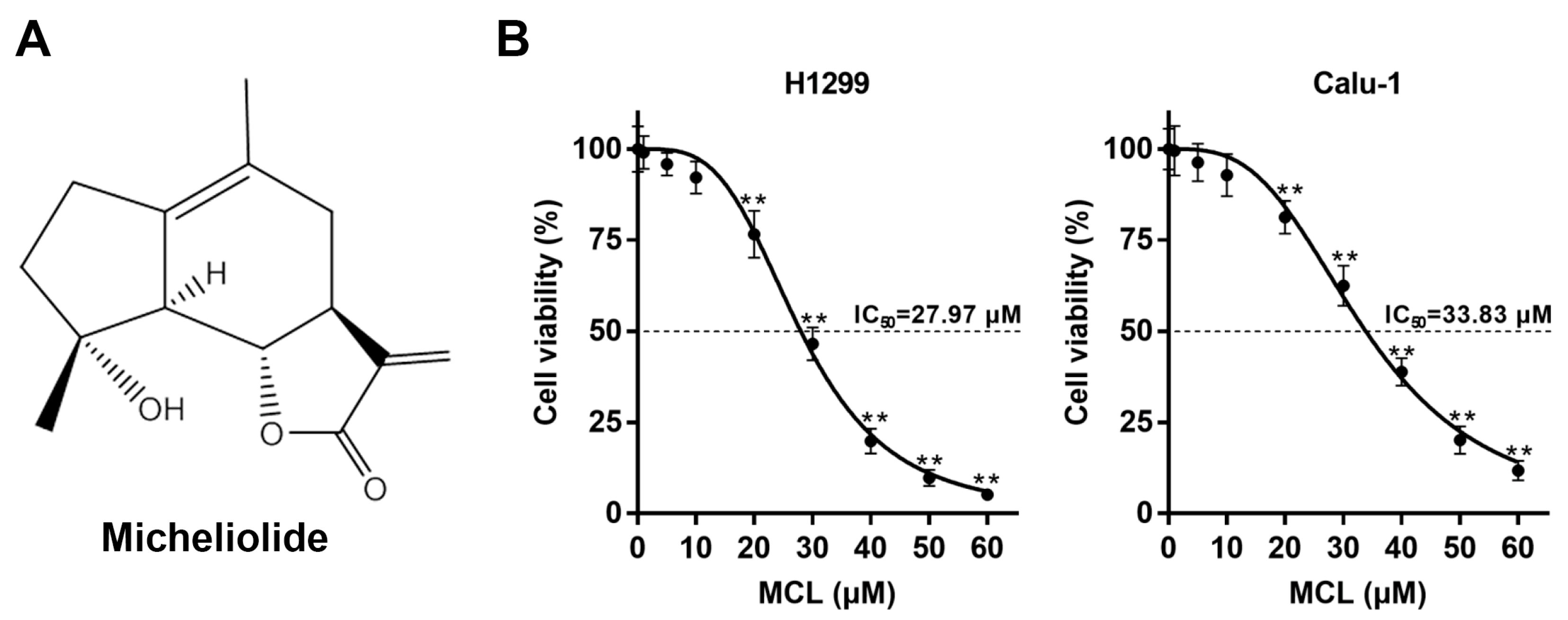
2.1. MCL Inhibits Cell Growth in NSCLC
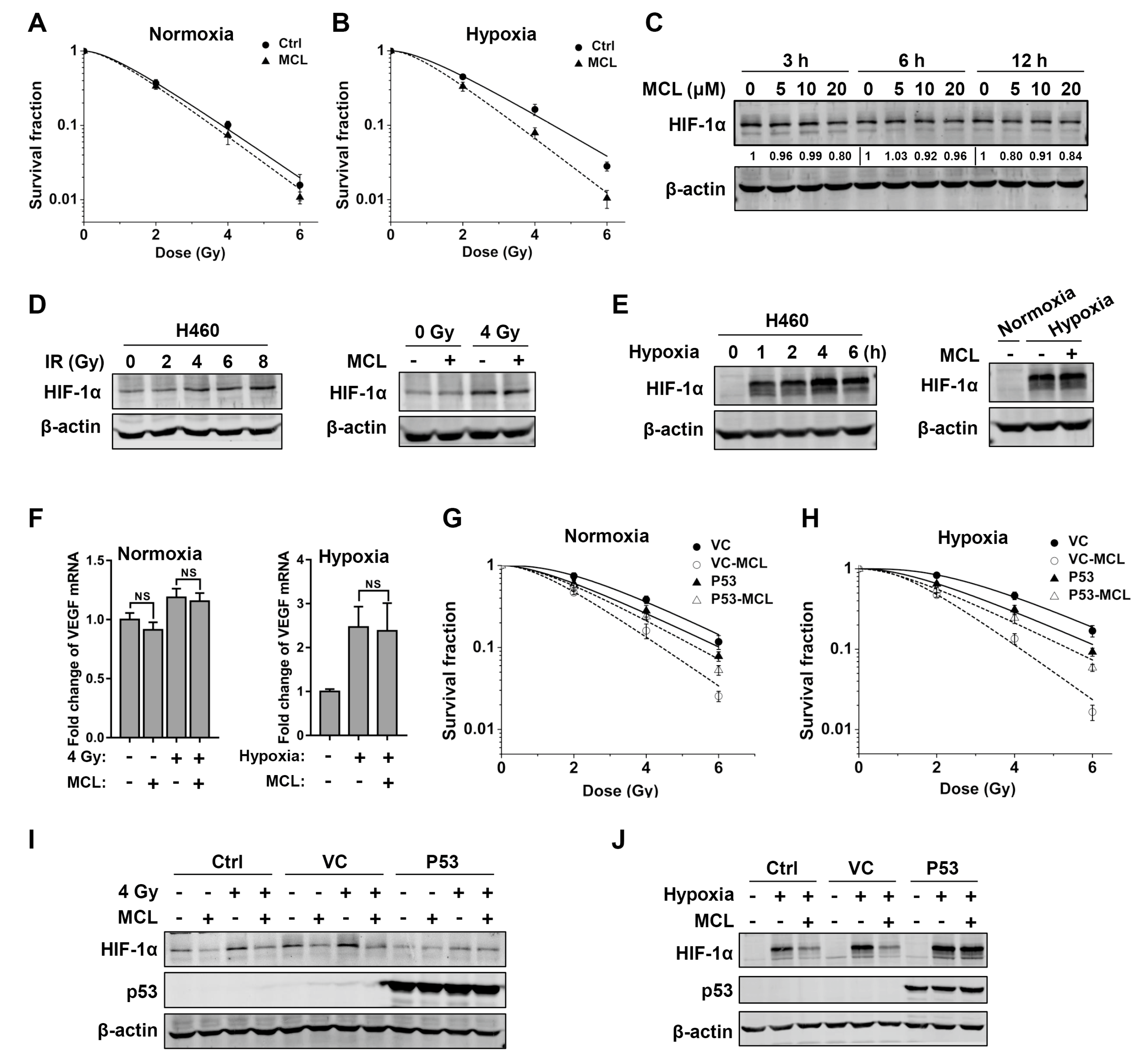
2.2. MCL Sensitizes NSCLC to IR under Both Normoxia and Hypoxia
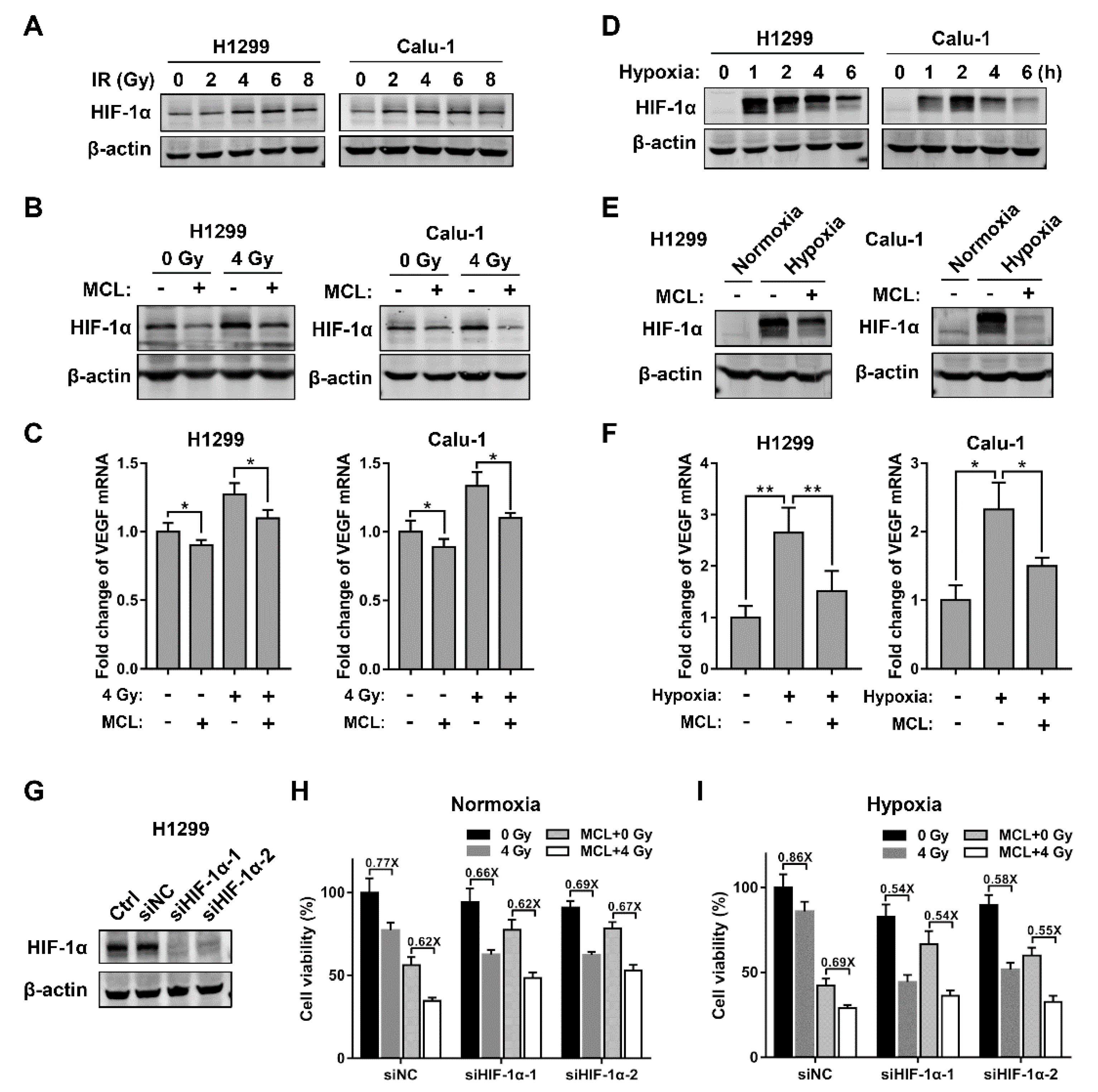
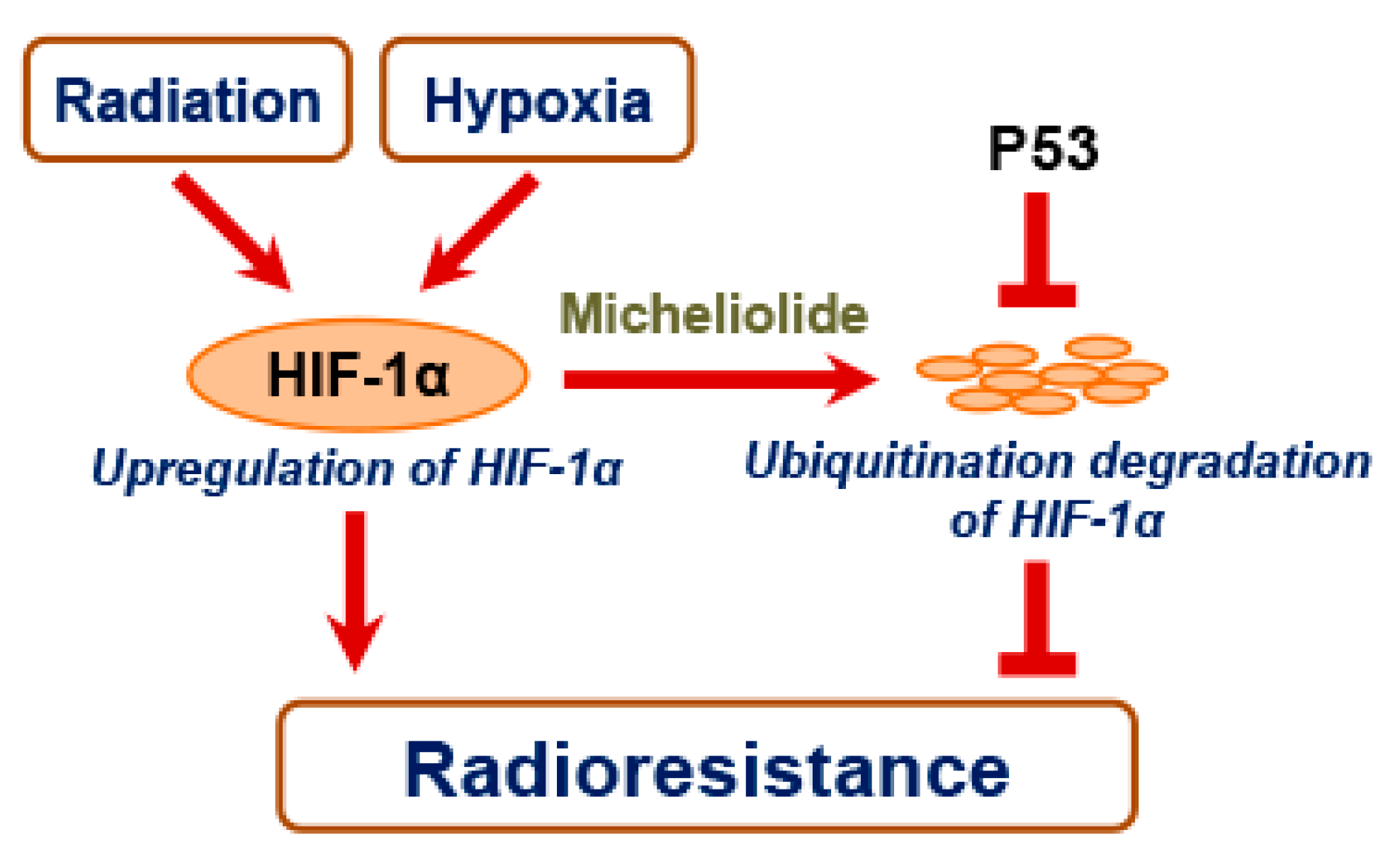
2.3. MCL Inhibits Radiation- and Hypoxia-Induced HIF-1α Expression in NSCLC
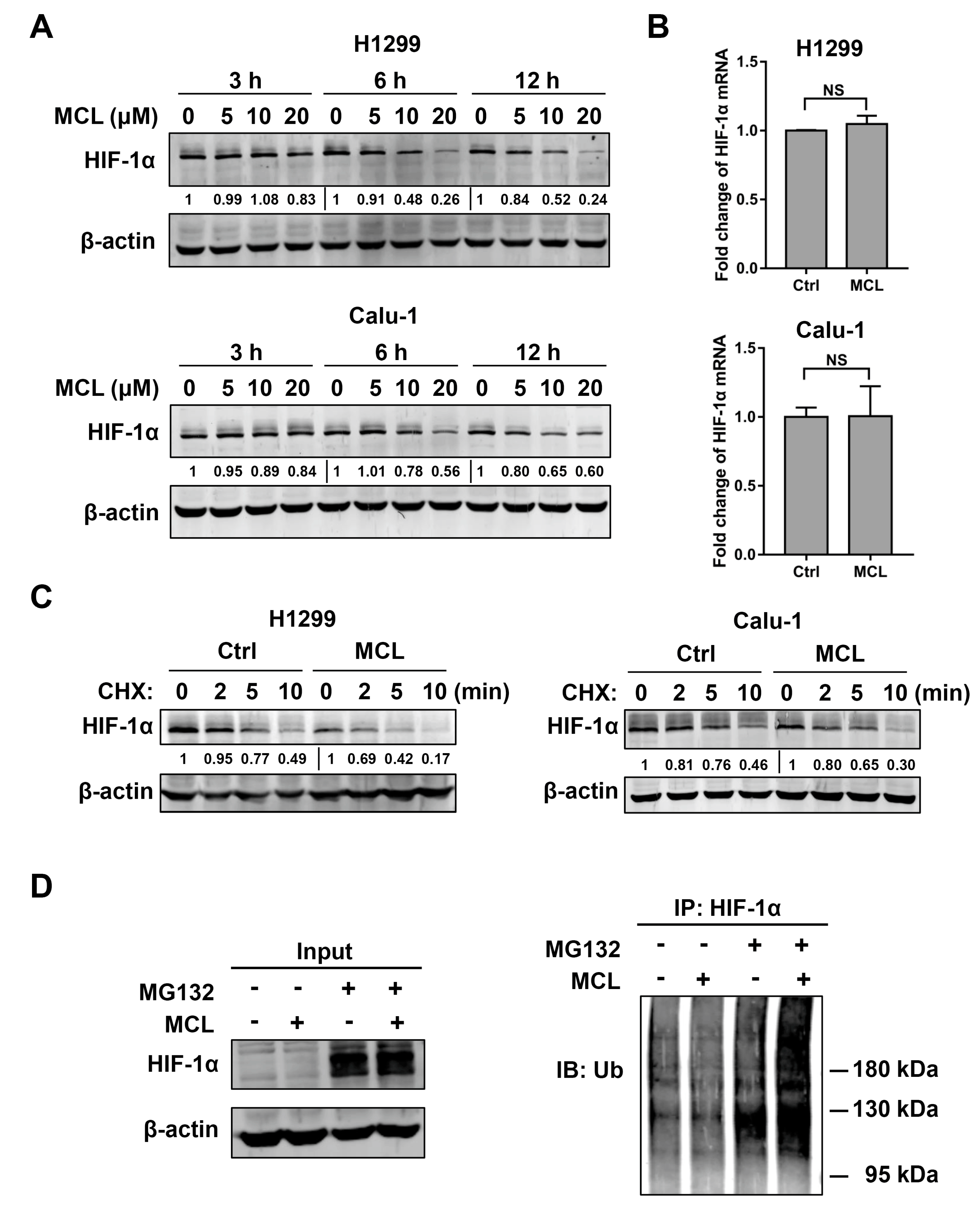
2.4. MCL Promotes the Degradation of HIF-1α
2.5. p53 Attenuates Radiosensitizing Effect of MCL
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Irradiation
4.3. Cell Viability Assay
4.4. Colony Formation Assay
4.5. Western Blot and Immunoprecipitation
4.6. RT-PCR
4.7. Small Interfering RNA Transfection
4.8. Retroviral Infection
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IR MCL PTL DMAMCL | Irradiation Micheliolide Parthenolide Dimethylamino Michael adduct of MCL |
| NSCLC | Non-small-cell lung cancer |
| IC50 DSB HIF-1α | Inhibitory concentration at 50% growth DNA double-strand break Hypoxia-inducible factor-1α |
| VEGF | Vascular endothelial growth factor |
| CHX | Cycloheximide |
| SF2 | Survival fraction at 2 Gy |
| Dq | Quasithreshould dose |
| SERDq NF-κB HDAC | Sensitizer enhancement ratio for Dq Nuclear factor kappa B Histone deacetylase |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meza, R.; Meernik, C.; Jeon, J.; Cote, M.L. Lung cancer incidence trends by gender, race and histology in the United States, 1973–2010. PLoS ONE 2015, 10, e0121323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, M.; Stamatis, G.; Thomas, M. Therapy of localized non-small cell lung cancer (take home messages). Lung Cancer 2001, 33, S47–S49. [Google Scholar] [CrossRef]
- Bristow, R.G.; Alexander, B.; Baumann, M.; Bratman, S.V.; Brown, J.M.; Camphausen, K.; Choyke, P.; Citrin, D.; Contessa, J.N.; Dicker, A. Combining precision radiotherapy with molecular targeting and immunomodulatory agents: a guideline by the American Society for Radiation Oncology. Lancet Oncol. 2018, 19, e240–e251. [Google Scholar] [CrossRef]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437. [Google Scholar] [CrossRef] [PubMed]
- Moeller, B.; Dewhirst, M. HIF-1 and tumour radiosensitivity. Br. J. Cancer 2006, 95, 1. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef]
- Berra, E.; Roux, D.; Richard, D.E.; Pouysségur, J. Hypoxia-inducible factor-1α (HIF-1α) escapes O2-driven proteasomal degradation irrespective of its subcellular localization: nucleus or cytoplasm. EMBO Rep. 2001, 2, 615–620. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.L. Hypoxia-a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38. [Google Scholar] [CrossRef]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721. [Google Scholar] [CrossRef]
- Kim, W.-Y.; Oh, S.H.; Woo, J.-K.; Hong, W.K.; Lee, H.-Y. Targeting heat shock protein 90 overrides the resistance of lung cancer cells by blocking radiation-induced stabilization of hypoxia-inducible factor-1α. Cancer Res. 2009, 69, 1624–1632. [Google Scholar] [CrossRef] [Green Version]
- Pidgeon, G.P.; Barr, M.P.; Harmey, J.H.; Foley, D.A.; Bouchier-Hayes, D.J. Vascular endothelial growth factor (VEGF) upregulates BCL-2 and inhibits apoptosis in human and murine mammary adenocarcinoma cells. Br. J. Cancer 2001, 85, 273. [Google Scholar] [CrossRef] [PubMed]
- Calvaruso, M.; Pucci, G.; Musso, R.; Bravata, V. Nutraceutical Compounds as Sensitizers for Cancer Treatment in Radiation Therapy. Int. J. Mol. Sci. 2019, 20, 5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, L.; Boult, J.; Walker-Samuel, S.; Chung, Y.; Jamin, Y.; Ashcroft, M.; Robinson, S. The HIF-pathway inhibitor NSC-134754 induces metabolic changes and anti-tumour activity while maintaining vascular function. Br. J. Cancer 2012, 106, 1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aebersold, D.M.; Burri, P.; Beer, K.T.; Laissue, J.; Djonov, V.; Greiner, R.H.; Semenza, G.L. Expression of hypoxia-inducible factor-1α: a novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Res. 2001, 61, 2911–2916. [Google Scholar]
- Koukourakis, M.I.; Giatromanolaki, A.; Skarlatos, J.; Corti, L.; Blandamura, S.; Piazza, M.; Gatter, K.C.; Harris, A.L. Hypoxia inducible factor (HIF-1a and HIF-2a) expression in early esophageal cancer and response to photodynamic therapy and radiotherapy. Cancer Res. 2001, 61, 1830–1832. [Google Scholar]
- Hui, E.P.; Chan, A.T.; Pezzella, F.; Turley, H.; To, K.-F.; Poon, T.C.; Zee, B.; Mo, F.; Teo, P.M.; Huang, D.P. Coexpression of hypoxia-inducible factors 1α and 2α, carbonic anhydrase IX, and vascular endothelial growth factor in nasopharyngeal carcinoma and relationship to survival. Clin. Cancer. Res. 2002, 8, 2595–2604. [Google Scholar]
- Moeller, B.J.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell 2004, 5, 429–441. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Kon, T.; Wang, H.; Li, F.; Huang, Q.; Rabbani, Z.N.; Kirkpatrick, J.P.; Vujaskovic, Z.; Dewhirst, M.W.; Li, C.-Y. Enhancement of hypoxia-induced tumor cell death in vitro and radiation therapy in vivo by use of small interfering RNA targeted to hypoxia-inducible factor-1α. Cancer Res. 2004, 64, 8139–8142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viennois, E.; Xiao, B.; Ayyadurai, S.; Wang, L.; Wang, P.G.; Zhang, Q.; Chen, Y.; Merlin, D. Micheliolide, a new sesquiterpene lactone that inhibits intestinal inflammation and colitis-associated cancer. Lab. Invest. 2014, 94, 950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, S.; Guo, J.; Li, Q.; Long, J.; Ma, C.; Ding, Y.; Yan, C.; Li, L.; Wu, Z. Natural product micheliolide (MCL) irreversibly activates pyruvate kinase M2 and suppresses leukemia. J. Med. Chem. 2018, 61, 4155–4164. [Google Scholar] [CrossRef] [PubMed]
- Ghantous, A.; Gali-Muhtasib, H.; Vuorela, H.; Saliba, N.A.; Darwiche, N. What made sesquiterpene lactones reach cancer clinical trials? Drug Discov. Today 2010, 15, 668–678. [Google Scholar] [CrossRef]
- Hehner, S.P.; Heinrich, M.; Bork, P.M.; Vogt, M.; Ratter, F.; Lehmann, V.; Schulze-Osthoff, K.; Dröge, W.; Schmitz, M.L. Sesquiterpene lactones specifically inhibit activation of NF-κB by preventing the degradation of IκB-α and IκB-β. J. Biol. Chem. 1998, 273, 1288–1297. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Clair, D.K.S.; Fang, F.; Warren, G.W.; Rangnekar, V.M.; Crooks, P.A.; Clair, W.H.S. The radiosensitization effect of parthenolide in prostate cancer cells is mediated by nuclear factor-κB inhibition and enhanced by the presence of PTEN. Mol. Cancer Ther. 2007, 6, 2477–2486. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Clair, D.K.S.; Xu, Y.; Crooks, P.A.; Clair, W.H.S. A NADPH oxidase-dependent redox signaling pathway mediates the selective radiosensitization effect of parthenolide in prostate cancer cells. Cancer Res. 2010, 70, 2880–2890. [Google Scholar] [CrossRef] [Green Version]
- Jin, P.; Madieh, S.; Augsburger, L.L. The solution and solid state stability and excipient compatibility of parthenolide in feverfew. AAPS PharmSciTech 2007, 8, 200. [Google Scholar] [CrossRef]
- Zhang, Q.; Lu, Y.; Ding, Y.; Zhai, J.; Ji, Q.; Ma, W.; Yang, M.; Fan, H.; Long, J.; Tong, Z. Guaianolide sesquiterpene lactones, a source to discover agents that selectively inhibit acute myelogenous leukemia stem and progenitor cells. J. Med. Chem. 2012, 55, 8757–8769. [Google Scholar] [CrossRef]
- An, Y.; Guo, W.; Li, L.; Xu, C.; Yang, D.; Wang, S.; Lu, Y.; Zhang, Q.; Zhai, J.; Fan, H.; et al. Micheliolide derivative DMAMCL inhibits glioma cell growth in vitro and in vivo. PLoS ONE 2015, 10, e0116202. [Google Scholar] [CrossRef] [Green Version]
- Unruh, A.; Ressel, A.; Mohamed, H.G.; Johnson, R.S.; Nadrowitz, R.; Richter, E.; Katschinski, D.M.; Wenger, R.H. The hypoxia-inducible factor-1α is a negative factor for tumor therapy. Oncogene 2003, 22, 3213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Liu, X.; Su, L. Parthenolide induces apoptosis via TNFRSF10B and PMAIP1 pathways in human lung cancer cells. J. Exp. Clin. Cancer Res. 2014, 33, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Qiu, L.; Jin, X.; Guo, Z.; Guo, C. Nuclear factor-kappaB inhibition by parthenolide potentiates the efficacy of Taxol in non-small cell lung cancer in vitro and in vivo. Mol. Cancer Res. 2009, 7, 1139–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deraska, P.V.; O’Leary, C.; Reavis, H.D.; Labe, S.; Dinh, T.K.; Lazaro, J.B.; Sweeney, C.; D’Andrea, A.D. NF-kappaB inhibition by dimethylaminoparthenolide radiosensitizes non-small-cell lung carcinoma by blocking DNA double-strand break repair. Cell Death Discov. 2018, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Estabrook, N.C.; Chin-Sinex, H.; Borgmann, A.J.; Dhaemers, R.M.; Shapiro, R.H.; Gilley, D.; Huda, N.; Crooks, P.; Sweeney, C.; Mendonca, M.S. Inhibition of NF-kappaB and DNA double-strand break repair by DMAPT sensitizes non-small-cell lung cancers to X-rays. Free Radic. Biol. Med. 2011, 51, 2249–2258. [Google Scholar] [CrossRef]
- Shanmugam, R.; Kusumanchi, P.; Appaiah, H.; Cheng, L.; Crooks, P.; Neelakantan, S.; Peat, T.; Klaunig, J.; Matthews, W.; Nakshatri, H.; et al. A water soluble parthenolide analog suppresses in vivo tumor growth of two tobacco-associated cancers, lung and bladder cancer, by targeting NF-kappaB and generating reactive oxygen species. Int. J. Cancer 2011, 128, 2481–2494. [Google Scholar] [CrossRef] [Green Version]
- Ackerman, D.; Simon, M.C. Hypoxia, lipids, and cancer: Surviving the harsh tumor microenvironment. Trends Cell Biol. 2014, 24, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Simopoulos, C.; Turley, H.; Talks, K.; Gatter, K.C.; Harris, A.L.; Tumour and Angiogenesis Research Group. Hypoxia-inducible factor (HIF1A and HIF2A), angiogenesis, and chemoradiotherapy outcome of squamous cell head-and-neck cancer. Int. J. Radiat. Oncol. 2002, 53, 1192–1202. [Google Scholar] [CrossRef]
- Bachtiary, B.; Schindl, M.; Pötter, R.; Dreier, B.; Knocke, T.H.; Hainfellner, J.A.; Horvat, R.; Birner, P. Overexpression of hypoxia-inducible factor 1α indicates diminished response to radiotherapy and unfavorable prognosis in patients receiving radical radiotherapy for cervical cancer. Clin. Cancer. Res. 2003, 9, 2234–2240. [Google Scholar]
- Gorski, D.H.; Beckett, M.A.; Jaskowiak, N.T.; Calvin, D.P.; Mauceri, H.J.; Salloum, R.M.; Seetharam, S.; Koons, A.; Hari, D.M.; Kufe, D.W. Blockade of the vascular endothelial growth factor stress response increases the antitumor effects of ionizing radiation. Cancer Res. 1999, 59, 3374–3378. [Google Scholar]
- Lee, C.-G.; Heijn, M.; di Tomaso, E.; Griffon-Etienne, G.; Ancukiewicz, M.; Koike, C.; Park, K.; Ferrara, N.; Jain, R.K.; Suit, H.D. Anti-vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Res. 2000, 60, 5565–5570. [Google Scholar] [PubMed]
- Hess, C.; Vuong, V.; Hegyi, I.; Riesterer, O.; Wood, J.; Fabbro, D.; Glanzmann, C.; Bodis, S.; Pruschy, M. Effect of VEGF receptor inhibitor PTK787/ZK222548 combined with ionizing radiation on endothelial cells and tumour growth. Br. J. Cancer 2001, 85, 2010. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, A.; Oda, K.; Ikeda, Y.; Sone, K.; Fukuda, T.; Inaba, K.; Makii, C.; Enomoto, A.; Hosoya, N.; Tanikawa, M. PI3K/mTOR pathway inhibition overcomes radioresistance via suppression of the HIF1-α/VEGF pathway in endometrial cancer. Gynecol. Oncol. 2015, 138, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.; Simons, J.; Semenza, G. Modulation of HIF-1α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar]
- Fukuda, R.; Hirota, K.; Fan, F.; Do Jung, Y.; Ellis, L.M.; Semenza, G.L. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J. Biol. Chem. 2002, 277, 38205–38211. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-H.; Jeong, J.-W.; Park, J.; Lee, J.-W.; Seo, J.H.; Jung, B.-K.; Bae, M.-K.; Kim, K.-W. Regulation of the HIF-1α stability by histone deacetylases. Oncol. Rep. 2007, 17, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Gopal, Y.V.; Arora, T.S.; Van Dyke, M.W. Parthenolide specifically depletes histone deacetylase 1 protein and induces cell death through ataxia telangiectasia mutated. Chem. Biol. 2007, 14, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; Van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Zhu, S.; Wang, H.; Ding, S. Reprogramming fibroblasts toward cardiomyocytes, neural stem cells and hepatocytes by cell activation and signaling-directed lineage conversion. Nat. Protoc. 2015, 10, 959. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H1299 | Calu-1 | |||||
|---|---|---|---|---|---|---|
| SF2 | Dq | SERDq | SF2 | Dq | SERDq | |
| Ctrl | 0.71 ± 0.06 | 1.95 | - | 0.53 ± 0.06 | 1.24 | - |
| MCL | 0.48 ± 0.07 | 1.20 | 1.62 | 0.33 ± 0.04 | 0.73 | 1.69 |
| H1299 | Calu-1 | |||||
|---|---|---|---|---|---|---|
| SF2 | Dq | SERDq | SF2 | Dq | SERDq | |
| Ctrl | 0.77 ± 0.03 | 2.46 | - | 0.58 ± 0.09 | 1.33 | - |
| MCL | 0.36 ± 0.04 | 0.95 | 2.59 | 0.37 ± 0.06 | 0.73 | 1.82 |
| Normoxia | Hypoxia | |||||
|---|---|---|---|---|---|---|
| SF2 | Dq | SERDq | SF2 | Dq | SERDq | |
| Ctrl | 0.37 ± 0.04 | 0.91 | - | 0.45 ± 0.02 | 1.00 | - |
| MCL | 0.34 ± 0.03 | 0.85 | 1.07 | 0.33 ± 0.05 | 0.83 | 1.21 |
| Normoxia | Hypoxia | |||||
|---|---|---|---|---|---|---|
| SF2 | Dq | SERDq | SF2 | Dq | SERDq | |
| VC | 0.74 ± 0.07 | 2.24 | - | 0.83 ± 0.03 | 2.78 | - |
| VC-MCL | 0.47 ± 0.03 | 1.18 | 1.89 | 0.47 ± 0.03 | 1.33 | 2.10 |
| P53 | 0.60 ± 0.06 | 1.39 | - | 0.65 ± 0.04 | 1.67 | - |
| P53-MCL | 0.56 ± 0.07 | 1.33 | 1.04 | 0.55 ± 0.04 | 1.29 | 1.30 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, P.; Yu, K.N.; Yang, M.; Almahi, W.A.; Nie, L.; Chen, G.; Han, W. Micheliolide Enhances Radiosensitivities of p53-Deficient Non-Small-Cell Lung Cancer via Promoting HIF-1α Degradation. Int. J. Mol. Sci. 2020, 21, 3392. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093392
Kong P, Yu KN, Yang M, Almahi WA, Nie L, Chen G, Han W. Micheliolide Enhances Radiosensitivities of p53-Deficient Non-Small-Cell Lung Cancer via Promoting HIF-1α Degradation. International Journal of Molecular Sciences. 2020; 21(9):3392. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093392
Chicago/Turabian StyleKong, Peizhong, K.N. Yu, Miaomiao Yang, Waleed Abdelbagi Almahi, Lili Nie, Guodong Chen, and Wei Han. 2020. "Micheliolide Enhances Radiosensitivities of p53-Deficient Non-Small-Cell Lung Cancer via Promoting HIF-1α Degradation" International Journal of Molecular Sciences 21, no. 9: 3392. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21093392