Early Reactive A1 Astrocytes Induction by the Neurotoxin 3-Nitropropionic Acid in Rat Brain

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
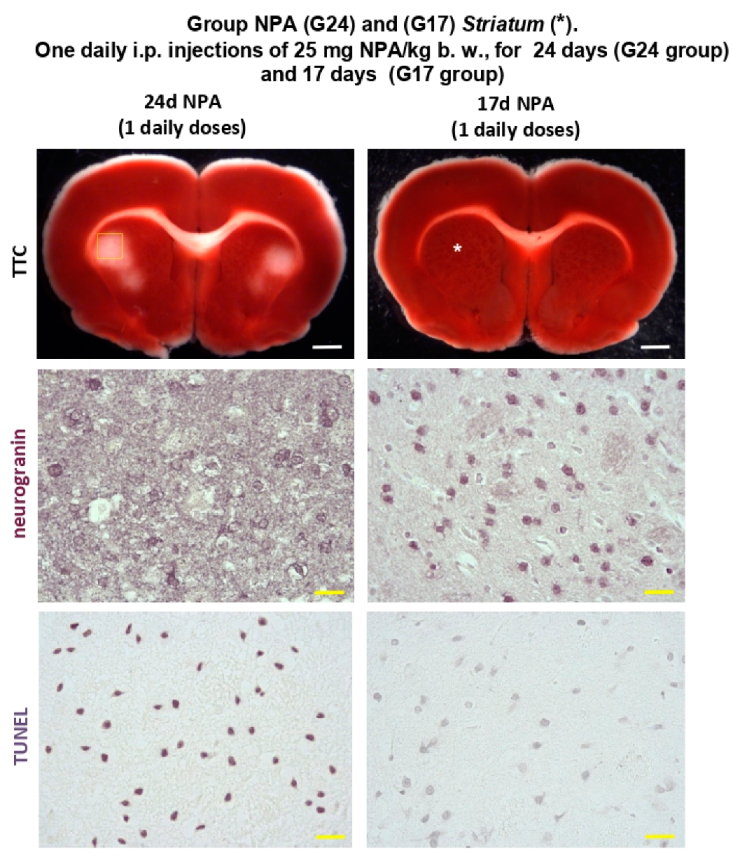
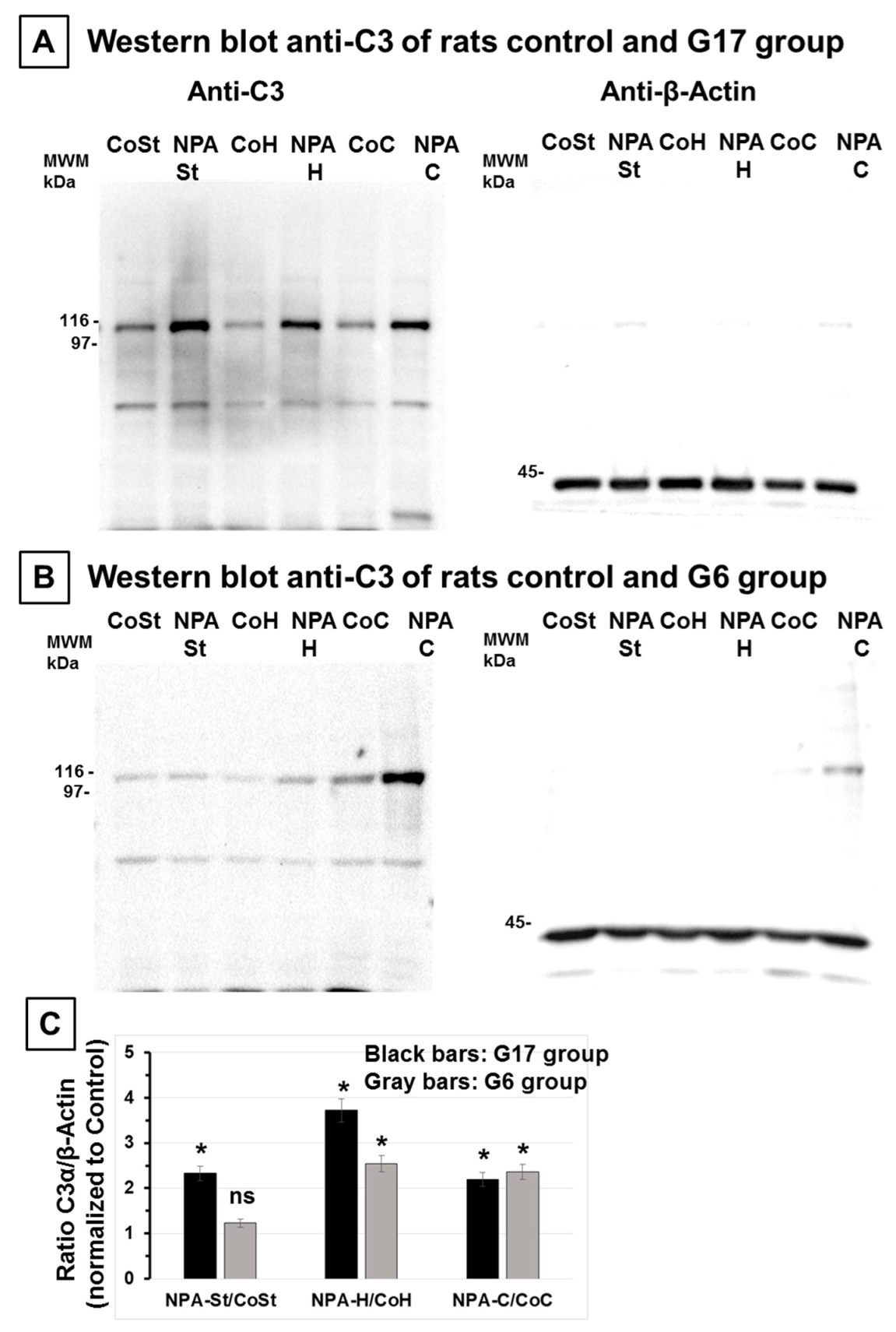
2.1. The Increase of Activated Complement C3 Protein, a Reactive A1 Astrocyte Marker, Precedes Significant NPA-Induced Brain degeneration
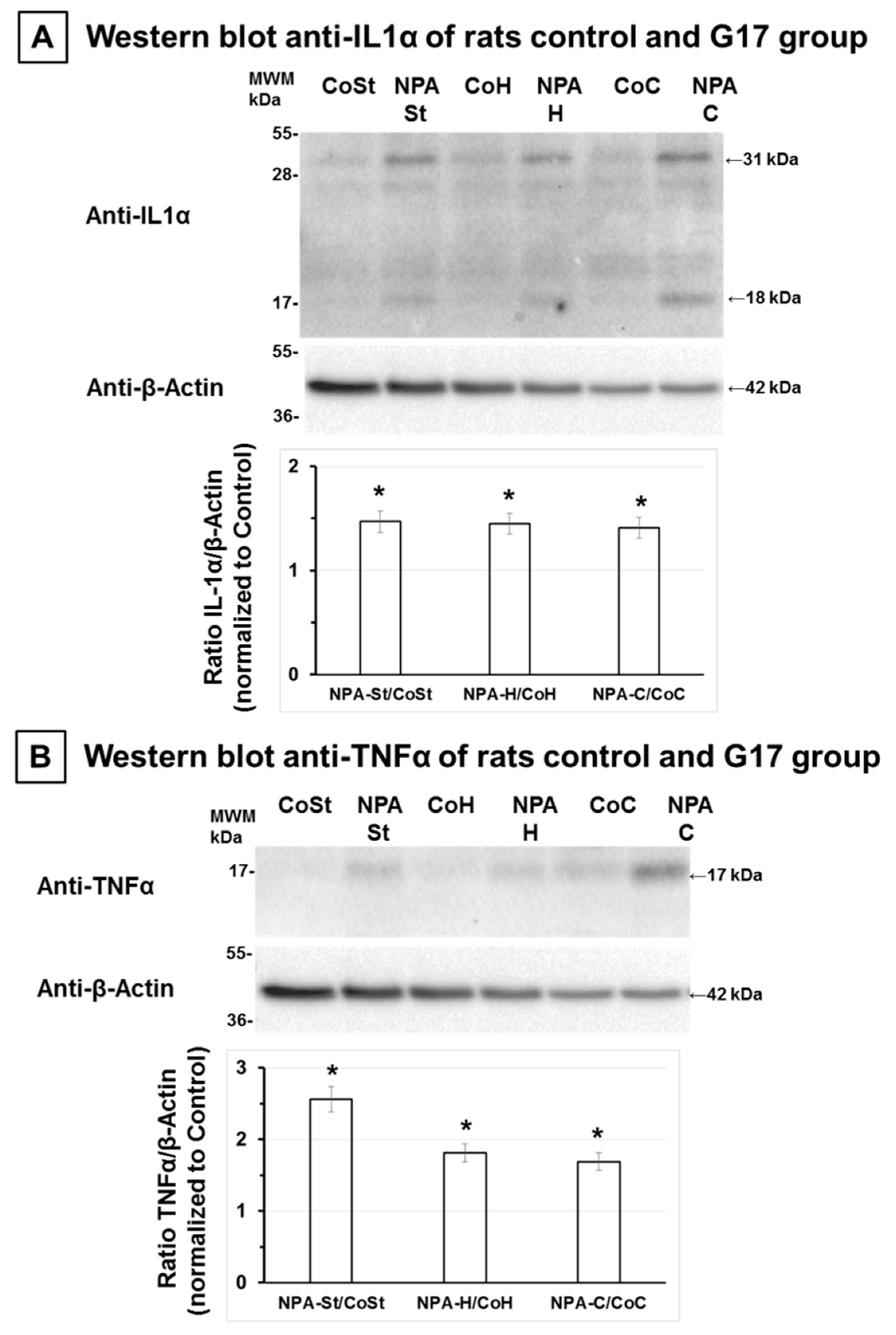
2.2. The Increase of Proinflammatory Cytokines IL-1α and TNFα Also Precedes Significant NPA-Induced Brain Degeneration
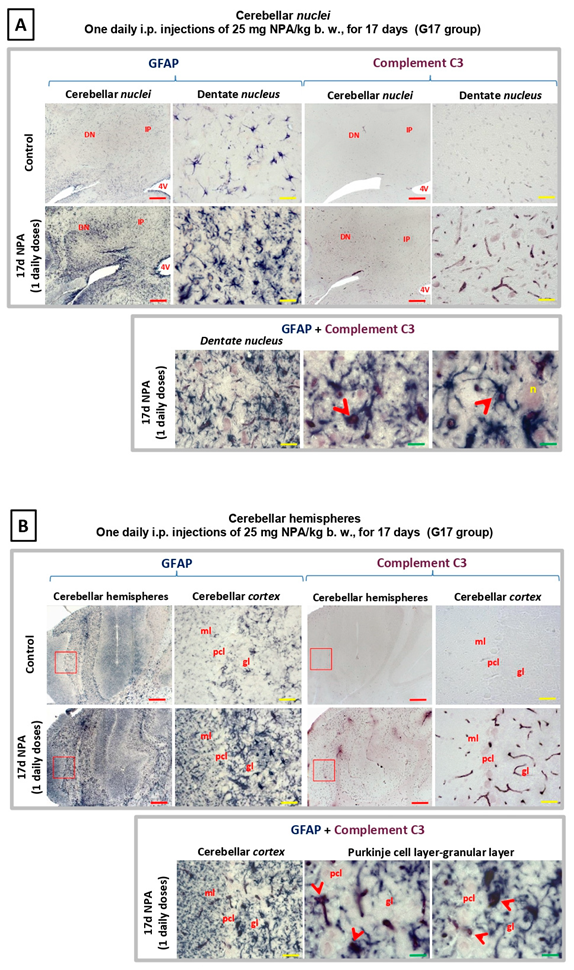
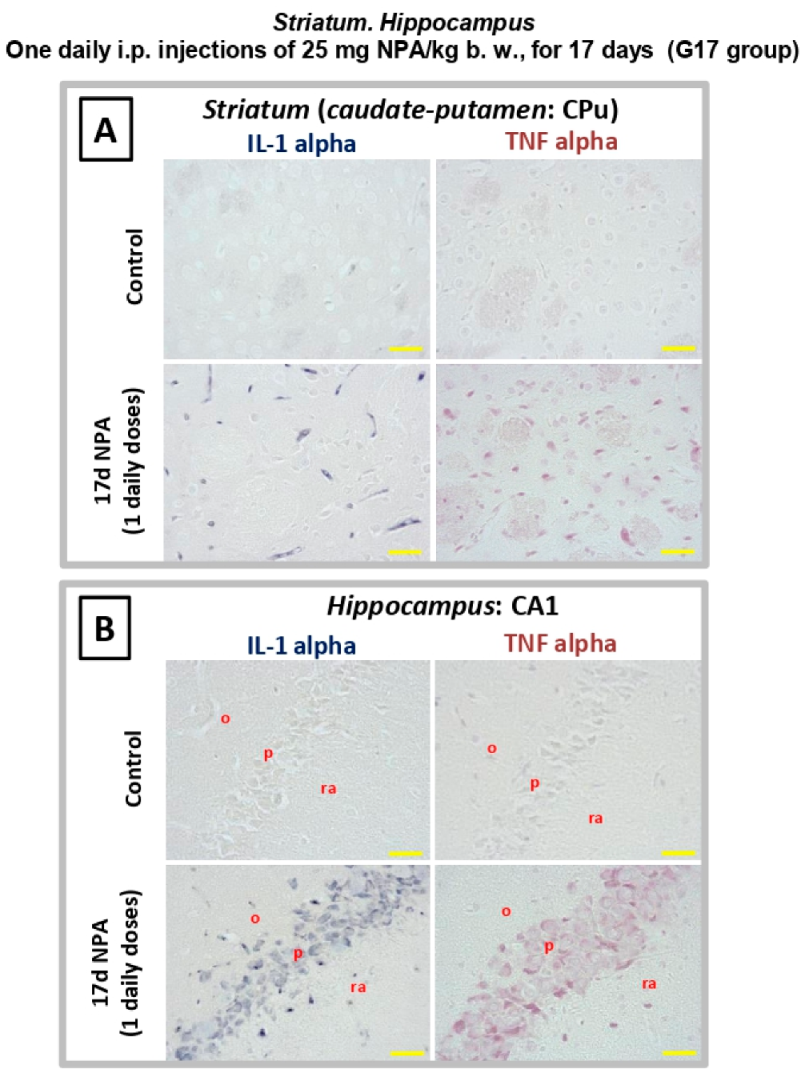
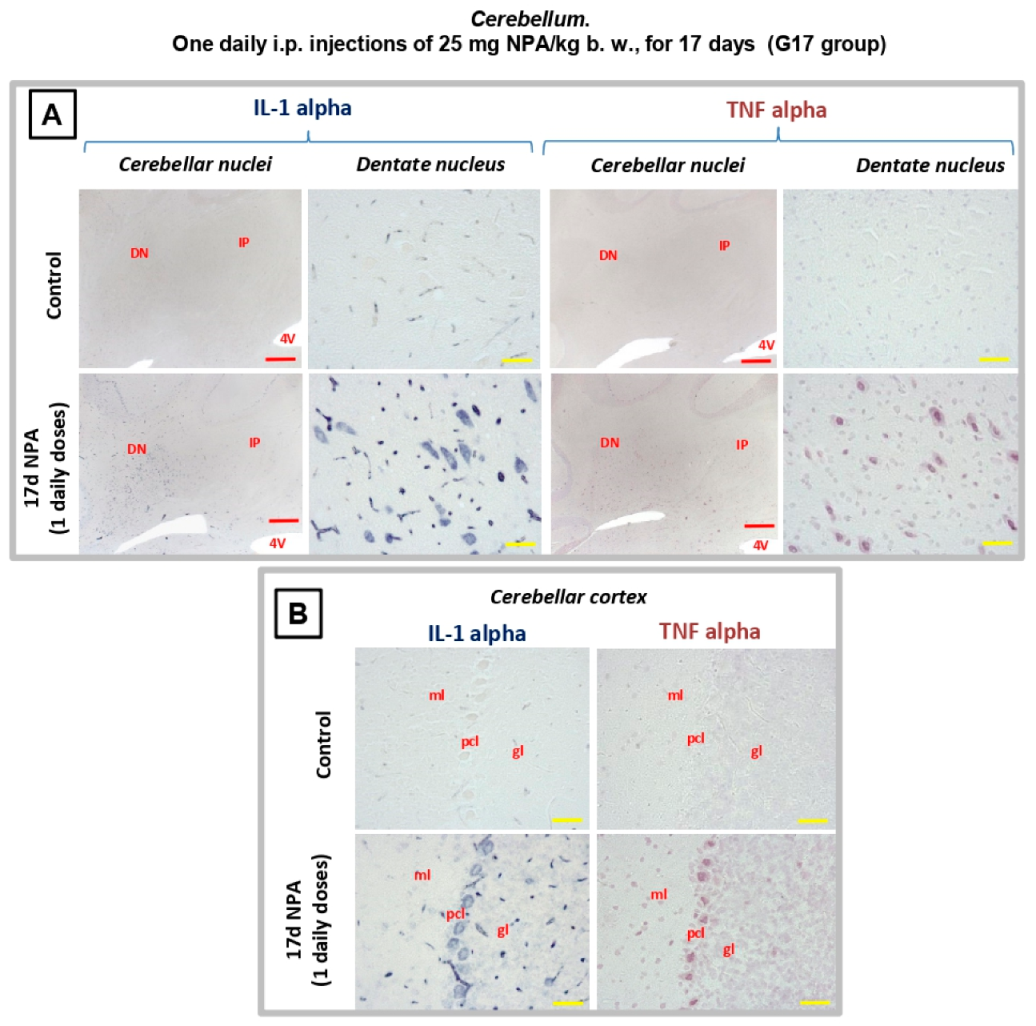
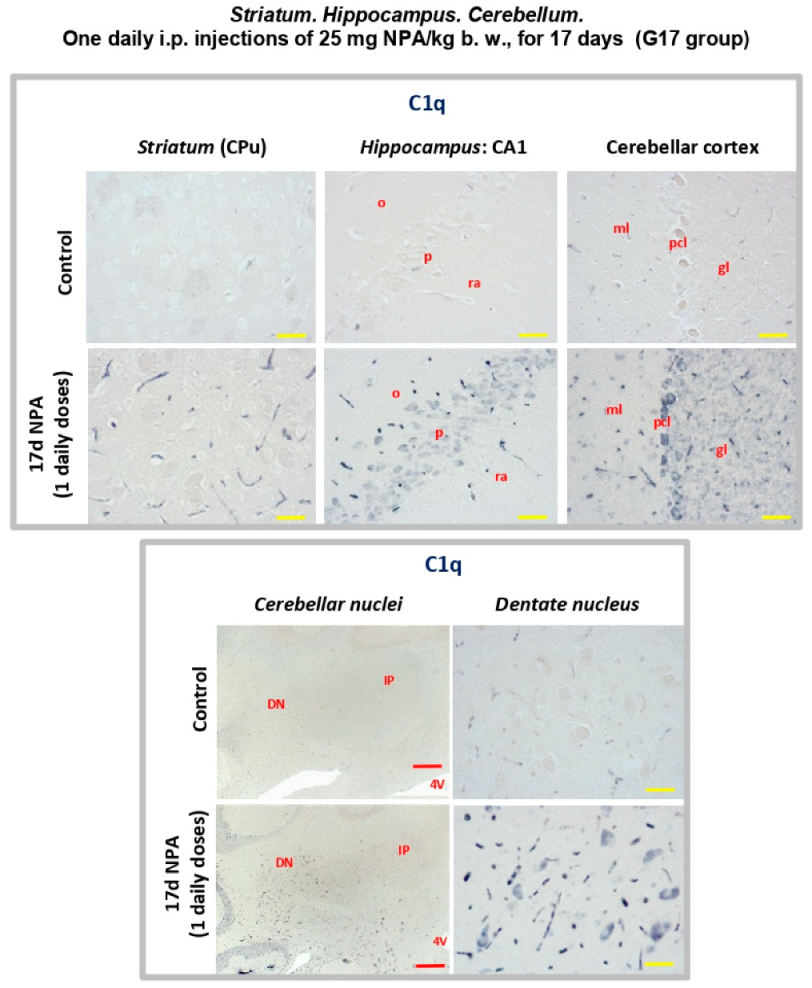
2.3. Immunohistochemical Analysis of the Regionalization and Location of Complement C3 Protein Activation in the Striatum, Hippocampus, and Cerebellum
2.4. Immunohistochemical Analysis of Regionalization and Location of IL1-α and TNFα Cytokines in the Striatum, Hippocampus, and Cerebellum
3. Discussion
4. Materials and Methods
4.1. Animals and Treatments
4.2. Preparation of Rat Brain Slices and TTC Staining
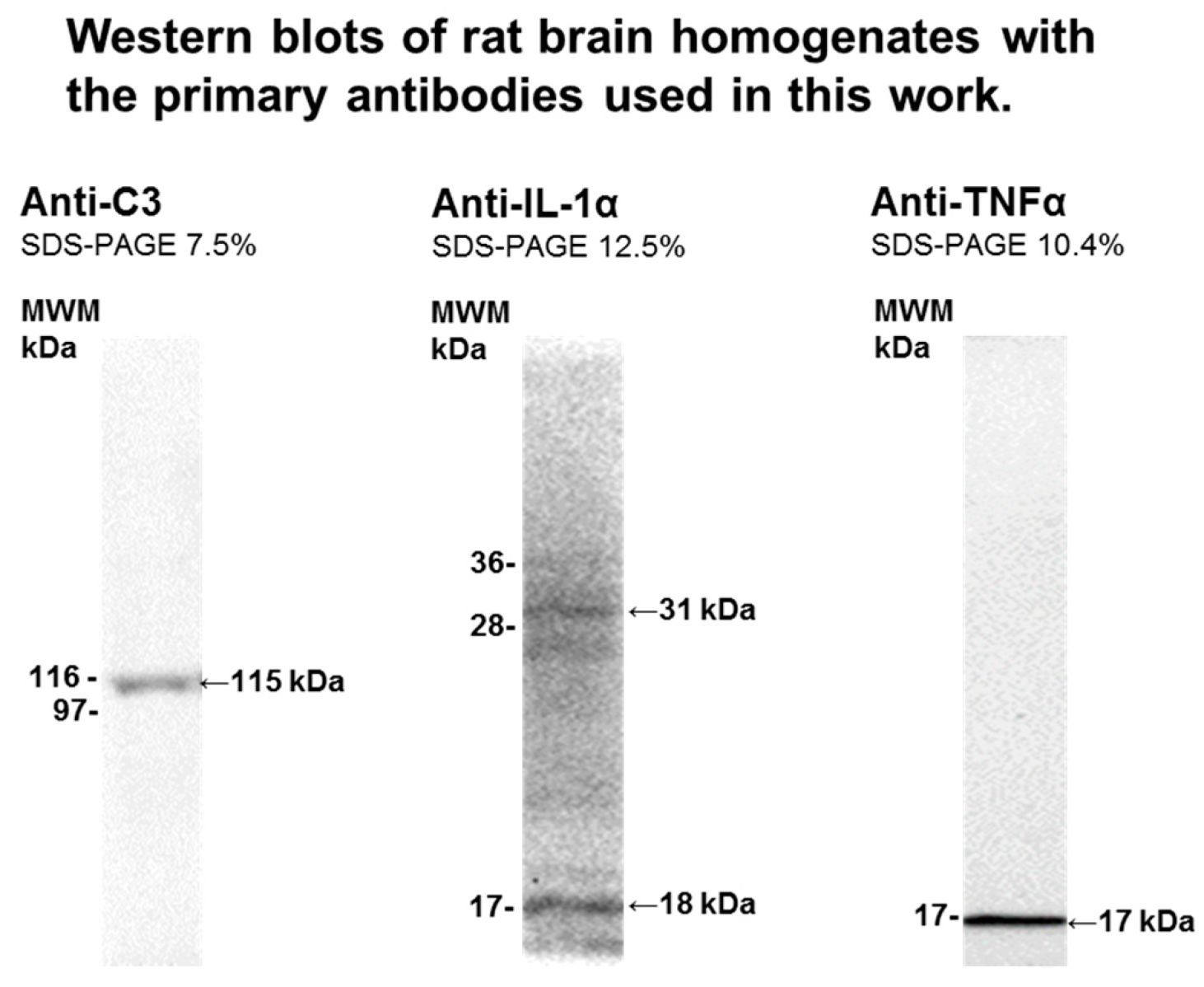
4.3. Brain Samples Homogenization and Western Blotting
4.4. Immunohistochemistry
4.4.1. Glial Fibrillary Acidic Protein (GFAP), Interleukin 1 (IL-1α), and Complement Component 1, Sububcomponent q (C1q-C).
4.4.2. Complement Component 3, (C3), Tumor Necrosis Factor Alpha (TNFα), and Neurogranin.
4.4.3. Terminal Deoxynucleotidyl Transferase-Mediated Deoxyuridine Triphosphate Nick-End Labelling (TUNEL).
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alston, T.A.; Mela, L.; Bright, H.J. 3-Nitropropionate, the toxic substance of Indigofera, is a suicide inactivator of succinate dehydrogenase. Proc. Natl. Acad. Sci. USA 1977, 74, 3767–3771. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Luo, X.; Hu, W. Studies on the epidemiology and etiology of moldy sugarcane poisoning in China. Biomed. Environ. Sci. 1992, 5, 161–177. [Google Scholar]
- Ming, L. Moldy Sugarcane Poisoning—A Case Report with a Brief Review. J. Toxicol. Clin. Toxicol. 1995, 33, 363–367. [Google Scholar] [CrossRef]
- Blumenthal, C.Z. Production of toxic metabolites in Aspergillus niger, Aspergillus oryzae, and Trichoderma reesei: Justification of mycotoxin testing in food grade enzyme preparations derived from the three fungi. Regul. Toxicol. Pharmacol. 2004, 39, 214–228. [Google Scholar] [CrossRef]
- Beal, M.; Brouillet, E.; Jenkins, B.; Ferrante, R.; Kowall, N.; Miller, J.; Storey, E.; Srivastava, R.; Rosen, B.; Hyman, B. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci. 1993, 13, 4181–4192. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F. Neurochemistry and toxin models in Huntingtonʼs disease. Curr. Opin. Neurol. 1994, 7, 542–547. [Google Scholar] [CrossRef]
- Brouillet, E.; Jenkins, B.G.; Hyman, B.T.; Ferrante, R.J.; Kowall, N.W.; Srivastava, R.; Roy, D.S.; Rosen, B.R.; Beal, M.F. Age-Dependent Vulnerability of the Striatum to the Mitochondrial Toxin 3-Nitropropionic Acid. J. Neurochem. 1993, 60, 356–359. [Google Scholar] [CrossRef]
- Brouillet, E.; Condé, F.; Beal, M.F.; Hantraye, P. Replicating Huntington’s disease phenotype in experimental animals. Prog. Neurobiol. 1999, 59, 427–468. [Google Scholar] [CrossRef]
- Blum, D.; Gall, D.; Cuvelier, L.; Schiffmann, S.N. Topological analysis of striatal lesions induced by 3-nitropropionic acid in the Lewis rat. NeuroReport 2001, 12, 1769–1772. [Google Scholar] [CrossRef] [Green Version]
- Lagoa, R.; Lopez-Sanchez, C.; Samhan-Arias, A.K.; Gañan, C.M.; Garcia-Martinez, V.; Gutierrez-Merino, C. Kaempferol protects against rat striatal degeneration induced by 3-nitropropionic acid. J. Neurochem. 2009, 111, 473–487. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P., Jr. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef]
- Ho, A.; Sahakian, B.J.; Brown, R.G.; Barker, R.A.; Hodges, J.; Ane, M.-N.; Snowden, J.; Thompson, J.; Esmonde, T.; Gentry, R.; et al. Profile of cognitive progression in early Huntington’s disease. Neurology 2003, 61, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- Phillips, W.; Shannon, K.M.; Barker, R.A. The current clinical management of Huntington’s disease. Mov. Disord. 2008, 23, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Browne, S.E.; Ferrante, R.J.; Beal, M.F. Oxidative stress in Huntington’s disease. Brain Pathol. 1999, 9, 147–163. [Google Scholar] [CrossRef]
- Menze, E.; Esmat, A.; Tadros, M.G.; Abdel-Naim, A.B.; Khalifa, A.E. Genistein Improves 3-NPA-Induced Memory Impairment in Ovariectomized Rats: Impact of Its Antioxidant, Anti-Inflammatory and Acetylcholinesterase Modulatory Properties. PLoS ONE 2015, 10, e0117223. [Google Scholar] [CrossRef] [Green Version]
- Tsang, T.M.; Haselden, J.N.; Holmes, E. Metabonomic Characterization of the 3-Nitropropionic Acid Rat Model of Huntington’s Disease. Neurochem. Res. 2009, 34, 1261–1271. [Google Scholar] [CrossRef]
- Peraza, A.V.; Guzmán, D.C.; Brizuela, N.O.; Herrera, M.O.; Olguin, H.J.; Silva, M.L.; Tapia, B.J.; Mejía, G.B. Riboflavin and pyridoxine restore dopamine levels and reduce oxidative stress in brain of rats. BMC Neurosci. 2018, 19, 71. [Google Scholar] [CrossRef]
- Singh-Bains, M.K.; Mehrabi, N.F.; Sehji, T.; Austria, M.D.; Tan, A.Y.S.; Tippett, L.J.; Dragunow, M.; Waldvogel, H.J.; Faull, R. Cerebellar degeneration correlates with motor symptoms in Huntington disease. Ann. Neurol. 2019, 85, 396–405. [Google Scholar] [CrossRef]
- Nasr, P.; Gursahani, H.I.; Pang, Z.; Bondada, V.; Lee, J.; Hadley, R.W.; Geddes, J. Influence of cytosolic and mitochondrial Ca2+, ATP, mitochondrial membrane potential, and calpain activity on the mechanism of neuron death induced by 3-nitropropionic acid. Neurochem. Int. 2003, 43, 89–99. [Google Scholar] [CrossRef]
- Rosenstock, T.R.; Carvalho, A.C.P.; Jurkiewicz, A.; Frussa-Filho, R.; Smaili, S. Mitochondrial calcium, oxidative stress and apoptosis in a neurodegenerative disease model induced by 3-nitropropionic acid. J. Neurochem. 2004, 88, 1220–1228. [Google Scholar] [CrossRef]
- Brouillet, E.; Jacquard, C.; Bizat, N.; Blum, D. 3-Nitropropionic acid: A mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J. Neurochem. 2005, 95, 1521–1540. [Google Scholar] [CrossRef]
- Gil, J.M.; Rego, A.C. Mechanisms of neurodegeneration in Huntington’s disease. Eur. J. Neurosci. 2008, 27, 2803–2820. [Google Scholar] [CrossRef] [Green Version]
- Zeevalk, G.D.; Derr-Yellin, E.; Nicklas, W.J. NMDA receptor involvement in toxicity to dopamine neurons in vitro caused by the succinate dehydrogenase inhibitor 3-nitropropionic acid. J. Neurochem. 1995, 64, 455–458. [Google Scholar] [CrossRef]
- Greene, J.G.; Sheu, S.-S.; Gross, R.A.; Greenamyre, J.T. 3-Nitropropionic acid exacerbates N-methyl-d-aspartate toxicity in striatal culture by multiple mechanisms. Neuroscience 1998, 84, 503–510. [Google Scholar] [CrossRef]
- Calabresi, P.; Gubellini, P.; Picconi, B.; Centonze, D.; Pisani, A.; Bonsi, P.; Greengard, P.; Hipskind, R.A.; Borrelli, E.; Bernardi, G. Inhibition of Mitochondrial Complex II Induces a Long-Term Potentiation of NMDA-Mediated Synaptic Excitation in the Striatum Requiring Endogenous Dopamine. J. Neurosci. 2001, 21, 5110–5120. [Google Scholar] [CrossRef] [PubMed]
- Bizat, N.; Hermel, J.-M.; Boyer, F.; Jacquard, C.; Creminon, C.; Ouary, S.; Escartin, C.; Hantraye, P.; Krajewski, S.; Brouillet, E. Calpain Is a Major Cell Death Effector in Selective Striatal Degeneration Induced In Vivo by 3-Nitropropionate: Implications for Huntington’s Disease. J. Neurosci. 2003, 23, 5020–5030. [Google Scholar] [CrossRef] [Green Version]
- Crespo-Biel, N.; Camins, A.; Pelegrí, C.; Vilaplana, J.; Pallàs, M.; Canudas, A. 3-Nitropropionic acid activates calpain/cdk5 pathway in rat striatum. Neurosci. Lett. 2007, 421, 77–81. [Google Scholar] [CrossRef]
- Fu, Y. 3-Nitropropionic acid produces indirect excitotoxic damage to rat striatum. Neurotoxicol. Teratol. 1995, 17, 333–339. [Google Scholar] [CrossRef]
- Stewart, V.C.; Sharpe, M.A.; Clark, J.B.; Heales, S.J.R. Astrocyte-Derived Nitric Oxide Causes Both Reversible and Irreversible Damage to the Neuronal Mitochondrial Respiratory Chain. J. Neurochem. 2002, 75, 694–700. [Google Scholar] [CrossRef] [Green Version]
- Ryu, J.K.; Nagai, A.; Kim, J.; Lee, M.C.; McLarnon, J.G.; Kim, S.U. Microglial activation and cell death induced by the mitochondrial toxin 3-nitropropionic acid: In vitro and in vivo studies. Neurobiol. Dis. 2003, 12, 121–132. [Google Scholar] [CrossRef]
- Chakraborty, J.; Singh, R.; Dutta, D.; Naskar, A.; Rajamma, U.; Mohanakumar, K.P. Quercetin Improves Behavioral Deficiencies, Restores Astrocytes and Microglia, and Reduces Serotonin Metabolism in 3-Nitropropionic Acid-Induced Rat Model of Huntington’s Disease. CNS Neurosci. Ther. 2013, 20, 10–19. [Google Scholar] [CrossRef]
- Jin, X.; Riew, T.-R.; Kim, H.L.; Choi, J.-H.; Lee, M.-Y. Morphological characterization of NG2 glia and their association with neuroglial cells in the 3-nitropropionic acid–lesioned striatum of rat. Sci. Rep. 2018, 8, 5942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niccolini, F.; Politis, M. Neuroimaging in Huntington’s disease. World J. Radiol. 2014, 6, 301–312. [Google Scholar] [CrossRef]
- Hong, H.; Kim, B.; Im, H.-I. Pathophysiological Role of Neuroinflammation in Neurodegenerative Diseases and Psychiatric Disorders. Int. Neurourol. J. 2016, 20 (Suppl. 1), S2–S7. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor Necrosis Factor-α Induces Neurotoxicity via Glutamate Release from Hemichannels of Activated Microglia in an Autocrine Manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef] [Green Version]
- Crotti, A.; Glass, C.K. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015, 36, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Nishino, H.; Kumazaki, M.; Fukuda, A.; Fujimoto, I.; Shimano, Y.; Hida, H.; Sakurai, T.; Deshpande, S.B.; Shimizu, H.; Morikawa, S.; et al. Acute 3-nitropropionic acid intoxication induces striatal astrocytic cell death and dysfunction of the blood-brain barrier: Involvement of dopamine toxicity. Neurosci. Res. 1997, 27, 343–355. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [Green Version]
- Singhrao, S.; Neal, J.; Morgan, B.P.; Gasque, P. Increased Complement Biosynthesis By Microglia and Complement Activation on Neurons in Huntington’s Disease. Exp. Neurol. 1999, 159, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Färber, K.; Cheung, G.; Mitchell, D.; Wallis, R.; Weihe, E.; Schwaeble, W.; Kettenmann, H. C1q, the recognition subcomponent of the classical pathway of complement, drives microglial activation. J. Neurosci. Res. 2009, 87, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, L.; Gates, D.J.; Richfield, E.K.; O’Brien, T.F.; Schweitzer, J.B.; Steindler, D.A. DNA End Labeling (TUNEL) in Huntington’s Disease and Other Neuropathological Conditions. Exp. Neurol. 1995, 133, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Suzuki, K.; Mori, N.; Yasuhara, O. Occurrence of complement protein C3 in dying pyramidal neurons in rat hippocampus after systemic administration of kainic acid. Neurosci. Lett. 2006, 409, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Encinas, E.; Aguilar-Morante, D.; Morales-Garcia, J.A.; Gine, E.; Sanz-SanCristobal, M.; Santos, A.; Perez-Castillo, A. Complement component 3 (C3) expression in the hippocampus after excitotoxic injury: Role of C/EBPβ. J. Neuroinflamm. 2016, 13, 276. [Google Scholar] [CrossRef] [Green Version]
- Boje, K.M.; Arora, P.K. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992, 587, 250–256. [Google Scholar] [CrossRef]
- Murray, K.N.; Parry-Jones, A.R.; Allan, S.M. Interleukin-1 and acute brain injury. Front. Cell. Neurosci. 2015, 9, 1662–5102. [Google Scholar] [CrossRef] [Green Version]
- Marinelli, C.; Di Liddo, R.; Facci, L.; Bertalot, T.; Conconi, M.; Zusso, M.; Skaper, S.; Giusti, P. Ligand engagement of Toll-like receptors regulates their expression in cortical microglia and astrocytes. J. Neuroinflamm. 2015, 12, 244. [Google Scholar] [CrossRef]
- Ouary, S.; Bizat, N.; Altairac, S.; Ménétrat, H.; Mittoux, V.; Condé, F.; Hantraye, P.; Brouillet, E. Major strain differences in response to chronic systemic administration of the mitochondrial toxin 3-nitropropionic acid in rats: Implications for neuroprotection studies. Neuroscience 2000, 97, 521–530. [Google Scholar] [CrossRef]
- Sun, F.; López-Sánchez, C.; Martin-Romero, F.J.; Luis, L.; Gutierrez-Merino, C.; Garcia-Martinez, V. Transfemoral selective “intraluminal wiring” technique for transient middle cerebral artery occlusion in rats. J. Neurosci. Methods 2005, 149, 82–89. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez-Sanchez, C.; Garcia-Martinez, V.; Poejo, J.; Garcia-Lopez, V.; Salazar, J.; Gutierrez-Merino, C. Early Reactive A1 Astrocytes Induction by the Neurotoxin 3-Nitropropionic Acid in Rat Brain. Int. J. Mol. Sci. 2020, 21, 3609. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103609
Lopez-Sanchez C, Garcia-Martinez V, Poejo J, Garcia-Lopez V, Salazar J, Gutierrez-Merino C. Early Reactive A1 Astrocytes Induction by the Neurotoxin 3-Nitropropionic Acid in Rat Brain. International Journal of Molecular Sciences. 2020; 21(10):3609. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103609
Chicago/Turabian StyleLopez-Sanchez, Carmen, Virginio Garcia-Martinez, Joana Poejo, Virginio Garcia-Lopez, Jairo Salazar, and Carlos Gutierrez-Merino. 2020. "Early Reactive A1 Astrocytes Induction by the Neurotoxin 3-Nitropropionic Acid in Rat Brain" International Journal of Molecular Sciences 21, no. 10: 3609. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103609