The Role of Immune Checkpoints after Cellular Therapy

1
Department of Hematology, Oncology and Rheumatology, University Hospital Bonn, 53127 Bonn, Germany
2
UKIM 5, Hematology and Oncology, Medical University Innsbruck, 6020 Innsbruck, Austria
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(10), 3650; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103650
Submission received: 27 April 2020
/
Revised: 18 May 2020
/
Accepted: 18 May 2020
/
Published: 21 May 2020
(This article belongs to the Special Issue Microenvironment in Cancer Progression and Metastases: Crosstalk between Cancer Cells and Microenvironment)
Abstract
:Cellular therapies utilize the powerful force of the human immune system to target malignant cells. Allogeneic hematopoietic stem cell transplantation (allo-HCT) is the most established cellular therapy, but chimeric antigen receptor (CAR) T cell therapies have gained attention in recent years. While in allo-HCT an entirely novel allogeneic immune system facilitates a so-called Graft-versus-tumor, respectively, Graft-versus-leukemia (GvT/GvL) effect against high-risk hematologic malignancies, in CAR T cell therapies genetically modified autologous T cells specifically attack target molecules on malignant cells. These therapies have achieved high success rates, offering potential cures in otherwise detrimental diseases. However, relapse after cellular therapy remains a serious clinical obstacle. Checkpoint Inhibition (CI), which was recently designated as breakthrough in cancer treatment and consequently awarded with the Nobel prize in 2018, is a different way to increase anti-tumor immunity. Here, inhibitory immune checkpoints are blocked on immune cells in order to restore the immunological force against malignant diseases. Disease relapse after CAR T cell therapy or allo-HCT has been linked to up-regulation of immune checkpoints that render cancer cells resistant to the cell-mediated anti-cancer immune effects. Thus, enhancing immune cell function after cellular therapies using CI is an important treatment option that might re-activate the anti-cancer effect upon cell therapy. In this review, we will summarize current data on this topic with the focus on immune checkpoints after cellular therapy for malignant diseases and balance efficacy versus potential side effects.
1. Introduction
The human immune system is one of the most powerful weapons in the treatment armamentarium against cancer. Many different immune cell types play distinct roles in the concert of cellular anti-tumor immunity [1]. However, divers mechanisms evading this anti-tumor immune response have been recognized, including a weakened cellular immune system [2,3,4,5,6]. Cellular therapies have been established to overcome this hurdle. They either aim to replace the dysfunctional immune system with a new one from a healthy donor in case of allogeneic stem cell transplantation (allo-HCT), or to add highly specific genetically modified autologous T cells in CAR T cell therapy. Despite these potent treatment options, disease relapse remains a major concern and further options for increasing anti-tumor immunity are under intense investigation. Inhibitory immune checkpoints play a pivotal role in limiting the hyperactivation of the immune response. In the setting of malignant disease, these mechanisms limit an effective anti-tumor immune response. Blocking antibodies targeting inhibitory immune checkpoints have been successfully implemented into the standard treatment algorithms of various malignancies for almost a decade. However, the effect of immune checkpoints in relapse after cellular therapies, such as allogeneic stem cell transplantation (allo-HCT) and CAR T cell therapy has yet to be fully explored. Additionally, the role of checkpoint inhibition (CI) after cell therapies is under current investigation. This review aims to elucidate the role of immune checkpoints after cellular therapies, as well as ways to modulate the immune function after cell therapy with CI for potential prevention or treatment of relapse. Next to pre-clinical and early clinical data elucidating the relationship between immune checkpoints and relapse after cell therapy, analysis of efficacy and responses and the view on toxicities of CI after cellular therapy are crucial points of this review.
1.1. Allogeneic Hematopoietic Stem Cell Transplantation (Allo-HCT)
Already in the late 1950s, Thomas et al. [7] conducted the oldest successfully realized cell therapy by performing the first allogeneic hematopoietic stem cell transplantation (allo-HCT). Initially, allo-HCT was considered to be a therapeutic approach for alleviating the toxic side-effects of high dosage radiation and chemotherapy by replacing the patient’s bone marrow with a new hematopoietic system from a healthy donor [7]. Since then, allo-HCT has become a potentially curative treatment option for various high-risk hematological malignancies as well as benign bone marrow diseases. Eventually, it became more apparent that the efficacy of allo-HCT in malignant diseases was owed to immunologically active cells of the donor against cancer cells of the host, the so-called Graft-versus-leukemia (GvL) effect [8]. However, already early observations affirmed that immunologically active donor cells also cause one of the major and potentially lethal complications of allo-HCT, the Graft-versus-Host Disease (GvHD) [9]. While the outcome after allo-HCT has been greatly improved, especially regarding the prevention and treatment of GvHD and infections, disease relapse is still a major complication after allo-HCT. While the GvL effect is considered to be mainly responsible for the cure of the underlying disease, the loss of this donor-mediated immunological anti-tumor effect is considered to be a major factor for relapse after transplant [10,11]. The restoration of the GvL effect while minimizing the complications of GvHD is a main challenge in relapse after allo-HCT [12]. Donor lymphocyte infusions (DLI) [13] are one possibility for restoring the GvL effect by boosting the host’s immune system. However, with only limited success with current treatment options in relapsed patients after allo-HCT ways to restore the GvL effect are urgently needed. One tempting approach to achieve this goal is to combine allo-HCT with CI.
1.2. Chimeric Antigen Receptor T Cells (Cars)
With the recent implementation of CARs, entirely novel cellular treatment options in various malignancies have arisen. CARs are autologous genetically engineered T cells expressing a chimeric antigen receptor that is specific for a tumor-associated antigen. CARs consist of an extracellular single chain variable fragment (scFv), a transmembrane domain, and an intracellular signaling domain. The CAR technology was first developed in the 1980s and further modified before entering clinical practice [14,15,16,17,18,19,20,21,22]. First generation CARs were called “T-bodies”, which were solely built of a scFv with the CD3ζ intracellular signaling domain to induce T cell activation. Due to initially limited antitumor activity, one (2nd generation) or two (3rd generation) costimulatory molecule domains, respectively (e.g., CD28, 4-1BB (CD137) or OX-40), were added to the CD3ζ intracellular signaling domain. These costimulatory domains enhance activation, function and persistence stimulating their proliferation and cytokine release. Second generation CD19-CARs have been approved by the Food and Drug Administration (FDA) in 2017 and by the European Medicines Agency (EMA) in 2018 for patients with relapsed/refractory diffuse large B cell lymphoma (DLBCL), acute lymphatic leukemia (ALL < 25 years), and primary mediastinal B cell lymphoma (PMBCL), respectively. Significant side effects of CAR T cell therapy are cytokine release syndrome (CRS) and immune cell associated neurotoxicity syndrome (ICANS). While these side effects can have severe manifestations sometimes requiring intensive care treatment, they are mostly well manageable and temporary [23,24]. Long-term toxicities are rare, but the short follow-up time until now must be considered. However, disease relapse is still a major issue after CAR T cell therapy [25]. Antigen loss, exhaustion of CAR T cells, and the expression of immune checkpoints have been linked to relapse [26]. Therefore, a combination of CARs with CI to enhance their anti-tumor efficacy in expectation of higher cure rates is a feasible option.
1.3. Immune Checkpoint Inhibitors (CI)
The inhibition of immune checkpoints, such as programmed death-1 (PD-1) or cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), have led to a breakthrough in cancer treatment by releasing the break on cellular anti-tumor immunity [3,27,28,29,30]. Checkpoint inhibition has evolved to be a well-established and very successful treatment option in various solid tumors, resulting in improved overall survival and even long-term tumor control in some cases [31,32,33,34]. CTLA-4 and PD-1 are central immune checkpoints that negatively regulate T cell immune function [29,35,36,37].
In 1996, James P. Allison et al. provided first evidence that anti-CTLA-4 inhibitory therapy improves anti-tumor immune responses in a pre-clinical murine cancer model [5]. Tasuku Honjo and colleagues showed that the engagement of PD-1 and its ligand PD-L1 (programmed death- ligand 1) on tumor cells lead to a potent inhibition of T cell receptor-mediated lymphocyte proliferation and cytokine secretion. This interaction could be reversed by application of anti-PD-L1 monoclonal-antibody (mAb), providing a promising treatment strategy for specific tumor immunotherapy [3]. The seminal findings of both researchers were awarded with the Nobel Prize for medicine in 2018 as a consequence of the ground-breaking nature of these findings for today’s cancer medicine.
Since ipilimumab’s FDA approval in 2011 for metastatic melanoma, five additional CI, including PD-1 and PD-L1 inhibitors (nivolumab, pembrolizumab, atezolizumab, avelumab, and durvalumab), were approved for numerous advanced solid malignancies and relapsed Hodgkin lymphoma. Alternative novel immune checkpoints in the tumor microenvironment have been detected as potential targets (e.g., T-cell immunoglobulin mucin-3 (TIM-3)/galectin-9, lymphocyte-activation gene 3 (LAG-3) or T cell immunoglobulin and ITIM domains (TIGIT)) in order to enhance the benefit from CI especially in patients with resistance to PD-1/PD-L1/CTLA-4 mAbs. Immune-related toxicities can, in some cases, be life-threatening, but are mostly well manageable when patients are properly monitored and early intervention is initiated [38]. While combinations of CI with other agents, such as conventional chemotherapeutics, have already been approved in some diseases, the role of CI after cellular therapy is still under investigation and it will be discussed throughout this review.
2. Immune Checkpoints and Checkpoint Inhibition after Allo-HCT
Multiple factors facilitating the loss of the GvL effect and, consequently, allowing disease relapse after allo-HCT have been described [39,40]. One reason for the lack or reduction of allo-immune T cell function might be due to the exhaustion of the T cells [41]. An exhausted T cell phenotype is associated with increased expression of inhibitory checkpoints (PD-1, TIM-3, and others) [42]. Several studies have been published analyzing the expression of inhibitory checkpoints in the context of disease relapse after allo-HCT. During the early post-transplantation phase, PD-1 was shown to be ubiquitously overexpressed on T cells, but interestingly without being a reliable predictive marker of potential disease relapse [43]. Hutten and colleagues found that high co-expression of PD-1, TIGIT, and killer cell lectin-like receptor subfamily member 1 (KLRG-1) on minor histocompatibility antigen (MiHA)-reactive CD8+ T cells correlates with disease relapse after allo-HCT [44]. In comparison to relapse-free patients after HLA-matched allo-HCT, a higher proportion of CD8+ T cells that accumulate in the bone marrow and express inhibitory molecules, like CTLA-4, PD-1, and TIM-3, were detected in patients with relapse. Curiously, this was not the case in HLA-haploidentical transplanted patients, possibly due to a higher degree of HLA-mismatch leading to an aggravated pro-inflammatory milieu in these patients [45]. Additionally, these T cells showed impaired immune function and an increase in the expression of CD80, CD86, PD-L1, and Galectin-9 was observed on leukemic blasts [45]. In-depth analyses of the immune signature of leukemic blasts during relapse after allo-HCT confirmed these findings and revealed, amongst others, an up-regulation of inhibitory checkpoints driving leukemia escape [46]. Further phenotypical and functional analyses of patient cells also display an important role of the PD-1/PD-L1 interaction in relapse after allo-HCT [47]. Kong et al. identified circulating CD8+ PD-1hiTIM-3+cells with reduced production of IL-2, TNF-α, and IFN-γ. These cells could already be identified after allo-HCT, but before diagnosis of overt leukemia relapse and thereby might possibly serve as a screening tool [48]. Elevated PD-1 expression on CD4+ T cells early after allo-HCT also correlates with increased mortality [49]. In addition to T cells, natural killer (NK) cells play a fundamental role in providing the GvL effect after allo-HCT [50]. Hattori et al. investigated the role of TIGIT in bone marrow samples after allo-HCT, which was—when highly expressed—associated with a decreased incidence of aGvHD, while overall survival (OS) and progression free survival (PFS) were poor [51].
These findings fueled the hypothesis that the inhibition of inhibitory immune checkpoints might be feasible in the prevention and treatment of disease relapse after allo-HCT. Preclinical murine studies showed that PD-1 blockade after allo-HCT can refuel the GvL effect [52,53,54]. In a murine GvHD model, the up-regulation of PD-1 on dysfunctional T cells during GvHD correlated with a loss of the GvL effect, while PD-L1 blockade was able to restore it [52]. The GvL effect of adoptively transferred genetically modified T cells expressing a T cell receptor against leukemia-associated antigen could also be enhanced by PD-L1 blockade in a murine model [53]. Additionally, it was shown that the PD-1/PD-L1 pathway engages in the compartmentalization of cytotoxic T cells in different tissue environments after allo-HCT, leading to tumor escape. The restoration of tumor control could be achieved by the application of CI [52,54].
In line with these results, early case reports regarding PD-1 blockade in patients with Hodgkin’s Disease (HD) that had relapsed after allo-HCT were published [55,56,57,58,59,60,61]. In those reports, all of the patients responded and none of them suffered from the induction of acute GvHD (aGvHD). Singh and colleagues firstly reported the induction of severe and eventually fatal skin and liver GvHD in a young patient with HD evolving one week after first pembrolizumab dose [62]. Haverkos et al. [63] conducted a multicenter retrospective study with 31 lymphoma patients (29 HD, one transformed follicular lymphoma (FL) and one with FL+ HD), who were treated with anti-PD-1 mAb after relapse following allo-HCT to better characterize the risks and benefits of PD-1 blockade after allo-HCT. Twenty-eight of those were treated with nivolumab and three with pembrolizumab. The overall response rate (ORR) was 77%, median PFS 591 days, and median OS was not reached because 21 of 31 patients were still alive at study termination at a median follow-up of 400 days. However, 17 patients developed GvHD after anti-PD-1 treatment, including eight patients with severe GvHD (six aGvHD and two cGvHD). In the same year (2017), Herbaux et al. published a retrospective analysis of nivolumab mono-therapy in 20 relapsed HD patients after allo-HCT, which again supported the efficacy of nivolumab [64]. ORR was 95% translating into a one-year-PFS of 58.2% and an OS of 78.7% after a median follow-up or 370 days. Six patients (30%), though, developed aGvHD after nivolumab initiation, of which five were reported as severe GvHD. There is only limited data regarding efficacy in the post-transplantation setting with CI in relapsed diseases other than HD. In a case of relapsed anaplastic large cell lymphoma after allo-HCT, low-dose pembrolizumab resulted in CR with tolerable side effects (liver GvHD) [65]. Albring et al. [66] published a case study of three patients with relapsed acute myeloid leukemia (AML) after allo-HCT, who received post-relapse treatment with nivolumab. Two patients responded and side effects, namely GvHD, could be well controlled. In a retrospective German multi-center study, 21 patients were examined, who had received off-label therapy with CI (nivolumab, ipilimumab, or the combination of both mAbs) alone or in combination with DLI for treatment of disease relapse after allo-HCT, (n = 15 after 1st; n = 5 after 2nd, and n = 1 after 3rd) [67]. Twelve patients suffered from relapsed AML or myelodysplastic syndrome (MDS), two from ALL, five from Non-Hodgkin-Lymphoma (NHL) and two from myelofibrosis (MF). ORR was 43% with three complete remissions (CR) and six partial remissions (PR). One patient had stable disease (SD) and 10 patients progressive disease (PD). ORR was 40% in patients receiving nivolumab, 80% when nivolumab was combined with DLI, and 20% in patients receiving ipilimumab. The development of aGvHD III-IV or moderate/severe cGvHD was seen in 29% of the patients. Especially patients receiving the combination of CI with DLI were at very high risk of GvHD development. Further immune-related toxicities were rare. When compared to ipilimumab, Davids and colleagues observed in a phase 1/1b study with nivolumab more severe GvHD and immune-related adverse events (irAEs), even when the lowest dose (0.5 mg/kg) was applied (median time 21 months after allo-HCT). Furthermore, shorter time from allo-HCT until application of CI was significantly associated with a higher risk of development of GvHD [68].
Kline et al. [69] examine pembrolizumab in a prospective, still recruiting clinical trial for the treatment of relapsed disease following allo-HCT (NCT02981914). In an early report, they presented eight patients with AML and three with lymphoma. Patients with AML showed discrete response to pembrolizumab (2 SD, 2 PD). irAEs were observed in 63% (any grade), which were well manageable. The first clinical trial using CTLA-4 blockade after allo-HCT (ipilimumab was administered at doses up to 3 mg/kg) demonstrated an acceptable safety profile [70]. Notably, the response to ipilimumab for the treatment of relapse after allogeneic transplantation is dose-dependent [71], as no objective responses were seen at a dose of 3 mg per kilogram body weight, whereas the best responses were seen among 22 included patients receiving 10 mg/kg of ipilimumab (7 CR/PR, 6 SD), including three patients with leukemia cutis. After 27 months median follow-up, OS and PFS were 54% and 32%, respectively. GvHD, which was steroid-sensitive, appeared in 14%. However, severe irAEs, of which one was fatal, were observed in six patients [71].
Additionally, the combinatory use of lenalidomide and ipilimumab after allo-HCT has shown good tumor control and significant increase of ICOS+ CD4+ FoxP3− T cells, indicating a synergistic effect of these two agents. ORR was good (70%) and no severe irAEs or GvHD were induced [72]. Table 1 summarizes relevant studies regarding CI after allo-HCT. In further currently ongoing clinical trials, mono or dual CI therapy with PD-1 and CTLA-4 inhibition after allo-HCT in high risk relapsed/refractory (r/r) AML or MDS, but also the combination of one checkpoint inhibitor with hypomethylating agents after allo-HCT are currently being evaluated and the results are eagerly awaited.
3. Immune Checkpoints and Checkpoint Inhibition after Cars
The recognition and interaction of CAR T cells with the corresponding target cell leads to their activation and proliferation, to the recruitment of further immune cells, and to the release of pro-inflammatory cytokines, and thereby eliminates tumor cells. Besides the great antitumor effect induced by CAR T cells, unneglectable significant relapse rates still remain an unresolved clinical problem [18,73,74,75,76,77]. Several mechanisms of failure of CAR T cell therapy have been uncovered, including poor CAR T cell persistence, low intrinsic CAR T cell fitness, antigen loss and escape, trogocytosis, as well as an inhibitory tumor microenvironment [25,26,78,79,80,81]. The assessment of immune checkpoints after CAR T cell therapy revealed an up-regulation of PD-1 that was seen on CD4+ and CD8+ anti-CD19-CAR T cells after treatment and PD-1 expression was higher on CAR-T cells than on non-CAR T cells [82,83,84]. Gene signature analyses of patients from the ZUMA-1 trial additionally showed an increase in gene expression of PD-1, LAG-3, and CTLA-4 after CD19-specific CAR T cell treatment [85]. In melanoma patients that were treated with anti-GD2 CAR T cells the CAR T cells showed an up-regulation of PD-1 and PD-L1, which was associated with a limited persistence of the CAR T cells [86]. In the tumor microenvironment of glioblastoma, patients up-regulation of PD-L1 in the microenvironment was seen after receiving anti-epidermal growth factor receptor (EGFRvIII) CAR T cell treatment and associated with incomplete responses [87]. In murine studies, the up-regulation of PD-1, LAG-3, TIM-3, and 2B4 was associated with impaired anti-mesothelin CAR T cell function and the inhibition of PD-L1 could restore CAR T cell function in a xenograft mouse model [88]. The extracellular domain of PD-1 was fused to intracellular co-stimulatory domains to resist suppression by PD-L1 (e.g., CD28). This chimera led to enhanced PD-L1 dependent cytotoxic T cell stimulation with increased cytokine secretion [89]. In adoptive transfer studies in Her-2 transgenic recipient mice, it was shown that the combination of Her-2 targeted CAR T cell therapy with anti-PD-1 mAb significantly reduced tumor mass and prolonged survival of the tumor bearing mice. Combinatory use of gene-modified Her-2 T cells with anti-PD-1 mAb was well tolerated without any signs of autoimmunity in recipient mice [90]. The triple-downregulation of PD-1, TIM-3, and LAG-3 in anti-Her2 CAR T cells using short hairpin RNA cluster resulted in enhanced tumor control in mice [91]. CRISPR/Cas9 (clustered regularly interspaced short palindromic re-peats associated with Cas9 endonuclease)- based gene editing was used to knockout PD-1 on primary T cells, leading to enhanced cytotoxicity [92]. Combined CRISPR/Cas9- based gene editing of anti-CD19 CAR T cells with mediated disruption of PD-1 resulted in augmented killing efficacy of the CRISPR/Cas9-edited CAR T cells in vitro and potently cleared PD-L1+ tumor xenografts in vivo [93]. Approaches with CRISPR/Cas9 modulated allogeneic CAR T cells deficient in TCR and HLA class I, as well as PD-1, showed potent in vivo anti-tumor efficacy with reduced alloreactivity not causing GvHD [94,95]. Anti-CAIX CAR T cells that secrete PD-L1 antibodies in murine renal cell carcinoma [96] or anti-CD19 CAR T cells secreting PD-1-blocking single-chain variable fragments (scFv) [97] showed improved anti-tumor activity of CAR T cells that was superior to conventional CAR-T cells in vivo. The treatment with anti-PD-1 scFv-producing CAR-T cells appeared to have an increased concentration of PD-1 scFv in tumor tissue, but not in sera [98]. These results suggest a possibility to decrease adverse events that are caused by systemically applied PD-1 blockade. In xenograft AML models, a significant up-regulation of TIM-3 on CAR T cells was shown in relapsed mice in comparison to CAR T cells that were isolated from mice in remission after CAR T cell therapy [99]. Refueling the immunological response of the CAR adding PD-1 or TIM-3 blocking antibodies to the same mouse model showed improved response rates. Interestingly, this synergistic effect even further increased in the presence of both checkpoint inhibitors combined [99].
With these and other encouraging preclinical results, several initial case series combining checkpoint inhibition with CAR T cell therapy were published. One report of fourteen heavily pretreated patients with r/r B-cell ALL and B lymphoblastic lymphoma with early CAR T cell loss or insufficient response to anti-CD19 CAR T cell therapy received PD-1 blockade no sooner than fourteen days after CAR T cell infusion [100]. At least partial responses were reported in the majority of the patients without unexpected or fatal toxicities. In other cases of pembrolizumab (2 mg/kg) on day 26 [101] or nivolumab (3 mg/kg) treatment on day 11 [102] for progressive DLBCL after anti-CD19 CAR T cell therapy rapid responses were seen and further assessment showed a drastically increased CAR T cell number after PD-1 blockade when compared to patients not receiving CI. A retrospective study analyzed eleven patients with r/r lymphoma who received nivolumab (3 mg/kg) on day 3 after anti-CD19 CAR T cell infusion [103]. ORR was 81% and, therefore, not superior to patients that received anti-CD19 CAR T cell treatment alone. However, due to differing patient characteristics a direct comparison is not possible. Importantly, CI related toxicities in combination with CAR T cell therapy were again well manageable [103]. In an initial report of the phase 1b multi-center open-label PORTIA trial (NCT03630159), four patients with refractory DLBCL, receiving pembrolizumab on day 21 after anti-CD19 CAR T cells have shown, so far, no irAEs nor severe CRS/ICANS [104]. Early results of the phase 1 of ZUMA-6 trial (NCT02926833), in which 12 patients with r/r DLBCL received anti-CD19 CAR T cells followed by 4 infusions of atezolizumab, no worsening of CAR T cell related adverse events was noted following CI [105]. Encouraging OR of 90% as well as an increased CAR T cell expansion and persistence as compared to anti-CD19 CAR T cell therapy alone support phase 2 of ZUMA-6 [105]. Another approach to target tumor antigens in addition to disrupting the PD-1/PD-L1 interaction is the use of bicistronic CAR T cells (AUTO3) targeting the two antigens CD19 and CD22, followed by anti-PD-1 consolidation courses (NCT03287817) in patients with r/r DLBCL. Early data have shown early efficacy with the lowest CARs dose (50 × 106 cells) and pembrolizumab 200 mg (given every three weeks) with an ORR of 57% and modest side effects [106].
In a phase I clinical trial (NCT03399448), CRISPR/Cas9 edited T cells with triple gene depletion including the PD-1 locus were infused after lymphodepleting chemotherapy into three patients with advanced refractory cancer (multiple myeloma, n = 2; liposarcoma, n = 1). The overall tolerance was good, with only mild toxicity and without the appearance of CRS. The best clinical responses were stable disease in the two myeloma patients, but eventually all three patients showed progress [107].
A growing number of CARs have been designed to target solid tumors, e.g., directed against EGFRvIII in glioblastoma showing prolonged CAR T cell persistence and the improvement of anti-glioma-activity with PD-1 knockout in a xenograft mouse model [108]. The combinatory use of EGFRvIII-CAR and pembrolizumab in patients with newly diagnosed 0-6-Methylguanine DNA-methyltransferase (MGMT)–unmethylated glioblastoma is currently being evaluated in a clinical trial (NCT03726515).
Encouraging synergistic effects have been described in early reports with the limited clinical data of treatment strategies combining CI with CAR T cells available (see Table 2). These strongly support further clinical trials utilizing immune checkpoints to optimize CAR T cell therapies. Table 3 illustrates the current overview with ongoing studies. The overall toxicity with this approach was not severely enhanced in the available, yet small, studies. However, special attention needs to be granted to the occurrence of autoimmune-related adverse events also in this setting [109].
4. Conclusions
Immunotherapy of cancer has revolutionized the treatment landscape in oncology. Strategies targeting immune checkpoints are also entering late clinical testing in hematology (i.e., TIM-3 mAb in MDS/AML) or are already approved (PD-1 blocker in cHL). In addition, allo-HCT and CAR T cells are established cellular therapies in hematology; however, both of them are also characterized by high relapse rates. One cause of relapse after cell therapy is an impaired T cell function accompanied with increased expression of inhibitory immune checkpoints. The currently available data shows the up-regulation of inhibitory immune checkpoints after cell therapy. Higher levels of immune checkpoints, such as PD-1/PD-L1, CTLA-4, TIM-3, LAG-3, or TIGIT, appear to be associated with increased relapse rates and reduced survival. Thus, it is tempting to speculate that the combination of cellular therapy with immune checkpoint blockade is a rational approach for improving outcome after cellular therapy. Preclinical data supported this hypothesis, which led to the individual clinical application and initiation of clinical studies of CI after allo-HCT and CAR T cell therapy. First results with PD-1 or/and CTLA-4 blockade indicate best responses in relapsed HD, but also other hematological malignancies, such as acute leukemia and NHL, show adequate responses to CI after allo-HCT. On the one hand, immune-related adverse events after CI in the setting of allo-HCT appear mostly well manageable, but GvHD induction after CI is often lethal or confers high morbidity burden, even though the underlying disease may be pushed back. On the other hand, a combination of CI with CAR T cell therapy appears mostly safe, while auto-immune related toxicities must be closely considered. Early reports fuel the hope of synergistic effects with this combinatory strategy. Additionally, genetically modified CRISPR/Cas9-based CAR T cells with specific checkpoint-knock-outs show good anti-tumor-efficacy without bearing uncontrollable toxicity. Figure 1 provides an overview of the mechanism of tumor escape, restored anti-tumor immunity after blocked signaling of inhibitory immune checkpoints as well as therapy-mediated toxicity.
To completely answer the question which immune checkpoint in cellular therapy is dominant with respect to resistance evolvement and, thus, representing the most appropriate therapy target warrants further investigations. This might not only be the PD-1/PD-L1 axis or CTLA-4, but also involves other candidates, such as TIM-3/galectin-9, TIGIT, or LAG-3. When considering a combination of checkpoint inhibition with cellular therapies, many aspects regarding their optimal use have to be taken into account to minimize the toxic side effects while guaranteeing optimal efficacy. Especially the identification of the appropriate patient population as well as optimal dosing, sequencing and therapy duration schedules have to be established in future studies, as they are inevitable to ensure the safety and efficacy of this approach.
Author Contributions
F.S.: literature review; data interpretation; manuscript drafting/revising; D.W.: literature review; data interpretation; coordination and supervision; manuscript drafting/revising; T.A.W.H.: literature review; data interpretation; coordination and supervision; manuscript drafting/revising. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| aGvHD | acute Graft-versus-Host Disease |
| allo-HCT | allogeneic hematopoietic stem cell transplantation |
| ALL | acute lymphocytic leukemia |
| AML | acute myeloid leukemia |
| CAIX | carbonic anhydrase IX |
| CAR | chimeric antigen receptor |
| CD | cluster of differentiation |
| cGvHD | chronic Graft-versus-Host Disease |
| CI | checkpoint inhibition/inhibitor |
| CR | complete remission |
| CRS | cytokine release syndrome |
| CRISPR/Cas9 | clustered regularly interspaced short palindromic repeats associated with Cas9 endonuclease |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein-4 |
| DLBCL | diffuse large B-cell lymphoma |
| DLI | donor lymphocyte infusion |
| EGFR | epidermal growth factor receptor |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration |
| FL | follicular lymphoma |
| FOXP3 GD2 | Forkhead-box-protein P3 disialoganglioside |
| GvHD | Graft-versus-Host Disease |
| GvL | Graft-versus-leukemia |
| GvT | Graft-versus-tumor |
| HD | Hodgkin’s disease |
| (c)HL | (classical) Hodgkin lymphoma |
| ICANS | immune cell associated neurotoxicity syndrome |
| ICOS | inducible T cell costimulator |
| irAEs | immune-related adverse events |
| KLRG-1 | killer cell lectin-like receptor subfamily member 1 |
| LAG-3 | lymphocyte-activation gene 3 |
| mAbs | monoclonal antibody |
| MDS | myelodysplastic syndrome |
| MGMT | 0-6-Methylguanine DNA-methyltransferase |
| MiHA | minor histocompatibility antigen |
| MF | myelofibrosis |
| NHL | Non-Hodgkin-Lymphoma |
| ORR | overall response rate |
| OS | overall survival |
| PD-1 | programmed death-1 |
| PD-L1 | programmed death- ligand 1 |
| PFS | progression free survival |
| PMBCL | primary mediastinal B cell lymphoma |
| PR | partial remission |
| RNA | ribonucleotide acid |
| scFv | single chain variable fragment |
| SD | stable disease |
| TIGIT | T cell immunoglobulin and ITIM domains |
| TIM-3 | T-cell immunoglobulin mucin-3 |
References
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [Green Version]
- Zang, X.; Allison, J.P. The B7 Family and Cancer Therapy: Costimulation and Coinhibition. Clin. Cancer Res. 2007, 13, 5271–5279. [Google Scholar] [CrossRef] [Green Version]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [Green Version]
- Kusmartsev, S.; Gabrilovich, D.I. Role Of Immature Myeloid Cells in Mechanisms of Immune Evasion in Cancer. Cancer Immunol. Immunother. 2006, 55, 237–245. [Google Scholar] [CrossRef]
- Thomas, E.D.; Lochte, H.L.; Lu, W.C.; Ferrebee, J.W. Intravenous Infusion of Bone Marrow in Patients Receiving Radiation and Chemotherapy. New Engl. J. Med. 1957, 257, 491–496. [Google Scholar] [CrossRef]
- Weiden, P.L.; Flournoy, N.; Thomas, E.D.; Prentice, R.; Fefer, A.; Buckner, C.D.; Storb, R. Antileukemic Effect of Graft-versus-Host Disease in Human Recipients of Allogeneic-Marrow Grafts. New Engl. J. Med. 1979, 300, 1068–1073. [Google Scholar] [CrossRef]
- Billingham, R.E. The biology of graft-versus-host reactions. Harvey Lect. 1966, 62, 21–78. [Google Scholar]
- Horowitz, M.M.; Gale, R.P.; Sondel, P.M.; Goldman, J.M.; Kersey, J.; Kolb, H.J.; A Rimm, A.; Ringden, O.; Rozman, C.; Speck, B. Graft-versus-leukemia reactions after bone marrow transplantation. Blood 1990, 75, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.J.; Savani, B.N.; Mohty, M.; Gorin, N.C.; Labopin, M.; Ruggeri, A.; Schmid, C.; Baron, F.; Esteve, J.; Giebel, S.; et al. Post-remission strategies for the prevention of relapse following allogeneic hematopoietic cell transplantation for high-risk acute myeloid leukemia: expert review from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Bone Marrow Transplant. 2019, 54, 519–530. [Google Scholar] [CrossRef]
- Parmar, S.; Ritchie, D.S. Allogeneic transplantation as anticancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 38–45. [Google Scholar] [CrossRef]
- Kolb, H.J.; Schattenberg, A.V.; Goldman, J.M.; Hertenstein, B.; Jacobsen, N.; Arcese, W.; Ljungman, P.; Ferrant, A.; Verdonck, L.; Niederwieser, D.; et al. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood 1995, 86, 2041–2050. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Eshhar, Z.; Gross, G.; Waks, T.; Lustgarten, J.; Bach, N.; Ratner, A.; Treisman, J.; Schindler, D.G. T-Bodies: Chimeric T-Cell Receptors with Antibody-Type Specificity. Methods 1995, 8, 133–142. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. New Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Locke, F.L.; Ghobadi, A.; A Jacobson, C.; Miklos, D.B.; Lekakis, L.J.; O Oluwole, O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Grupp, S.A.; Maude, S.L.; Rives, S.; Baruchel, A.; Boyer, M.W.; Bittencourt, H.; Bader, P.; Büchner, J.; Laetsch, T.W.; Stefanski, H.; et al. Updated Analysis of the Efficacy and Safety of Tisagenlecleucel in Pediatric and Young Adult Patients with Relapsed/Refractory (r/r) Acute Lymphoblastic Leukemia. Blood 2018, 132, 895. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.; Borchmann, P.; Jaeger, U.; Waller, E.K.; Holte, H.; McGuirk, J.P.; Jaglowski, S.; Tobinai, K.; et al. Sustained Disease Control for Adult Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma: An Updated Analysis of Juliet, a Global Pivotal Phase 2 Trial of Tisagenlecleucel. Blood 2018, 132, 1684. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. New Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Fitzgerald, J.C.; Weiss, S.L.; Maude, S.L.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; Shaw, P.; Berg, R.A.; June, C.H.; Porter, D.L.; et al. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit. Care Med. 2017, 45, e124–e131. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef]
- Cheng, J.; Zhao, L.; Zhang, Y.; Qin, Y.; Guan, Y.; Zhang, T.; Liu, C.; Zhou, J. Understanding the Mechanisms of Resistance to CAR T-Cell Therapy in Malignancies. Front. Oncol. 2019, 9, 1237. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Haanen, J.B. Immune checkpoint inhibitors for the treatment of cancer. Ann. Oncol. 2015, 26, vii7. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. New Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, E.; Ricciuti, B.; Gainor, J.; Kehl, K.; Kravets, S.; Dahlberg, S.; Nishino, M.; Sholl, L.; Adeni, A.; Subegdjo, S.; et al. Outcomes to first-line pembrolizumab in patients with non-small-cell lung cancer and very high PD-L1 expression. Ann. Oncol. 2019, 30, 1653–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonia, S.J.; Borghaei, H.; Ramalingam, S.S.; Horn, L.; Carpeño, J.D.C.; Pluzanski, A.; A Burgio, M.; Garassino, M.; Chow, L.Q.M.; Gettinger, S.; et al. Four-year survival with nivolumab in patients with previously treated advanced non-small-cell lung cancer: a pooled analysis. Lancet Oncol. 2019, 20, 1395–1408. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazieres, J.; Hermes, B.; Şenler, F.Ç.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non–Small-Cell Lung Cancer. New Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Hanahan, D.; A Weinberg, R. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Haanen, J.B.A.G.; Carbonnel, F.; Robert, C.; Kerr, K.M.; Peters, S.; Larkin, J.; Jordan, K. on behalf of the ESMO Guidelines Committee Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28, iv119–iv142. [Google Scholar] [CrossRef]
- Zeiser, R.; Vago, L. Mechanisms of immune escape after allogeneic hematopoietic cell transplantation. Blood 2019, 133, 1290–1297. [Google Scholar] [CrossRef]
- Blazar, B.R.; Hill, G.R.; Murphy, W.J. Dissecting the biology of allogeneic HSCT to enhance the GvT effect whilst minimizing GvHD. Nat. Rev. Clin. Oncol. 2020, 2020, 1–18. [Google Scholar] [CrossRef]
- Falkenburg, J.F.; Jedema, I. Graft versus tumor effects and why people relapse. Hematology 2017, 2017, 693–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Tian, X.; Cordes, S.; Chen, J.; Cantilena, C.R.; Bradley, C.; Panjwani, R.; Chinian, F.; Keyvanfar, K.; Battiwalla, M.; et al. Over-expression of PD-1 Does Not Predict Leukemic Relapse after Allogeneic Stem Cell Transplantation. Boil. Blood Marrow Transplant. 2019, 25, 216–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutten, T.; Norde, W.J.; Woestenenk, R.; Wang, R.C.; Maas, F.; Kester, M.; Falkenburg, J.F.; Berglund, S.; Luznik, L.; Jansen, J.H.; et al. Increased Coexpression of PD-1, TIGIT, and KLRG-1 on Tumor-Reactive CD8+ T Cells During Relapse after Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2018, 24, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noviello, M.; Manfredi, F.; Ruggiero, E.; Perini, T.; Oliveira, G.; Cortesi, F.; De Simone, P.; Toffalori, C.; Gambacorta, V.; Greco, R.; et al. Bone marrow central memory and memory stem T-cell exhaustion in AML patients relapsing after HSCT. Nat. Commun. 2019, 10, 1065. [Google Scholar] [CrossRef]
- Toffalori, C.; Zito, L.; Gambacorta, V.; Riba, M.; Oliveira, G.; Bucci, G.; Barcella, M.; Spinelli, O.; Greco, R.; Crucitti, L.; et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat. Med. 2019, 25, 603–611. [Google Scholar] [CrossRef]
- Norde, W.J.; Maas, F.; Hobo, W.; Korman, A.; Quigley, M.; Kester, M.G.; Hebeda, K.; Falkenburg, J.H.; Schaap, N.; de Witte, T.M.; et al. PD-1/PD-L1 interactions contribute to functional T-cell impairment in patients who relapse with cancer after allogeneic stem cell transplantation. Cancer Res. 2011, 71, 5111–5122. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Zhu, L.; Zeng, H.; Schell, T.; Zheng, H. PD-1(hi)TIM-3(+) T cells associate with and predict leukemia relapse in AML patients post allogeneic stem cell transplantation. Blood Cancer J. 2015, 5, e330. [Google Scholar] [CrossRef] [Green Version]
- Schade, H.; Sen, S.; Neff, C.P.; Freed, B.M.; Gao, D.; Gutman, J.A.; Palmer, B.E. Programmed Death 1 Expression on CD4 + T Cells Predicts Mortality after Allogeneic Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2016, 22, 2172–2179. [Google Scholar] [CrossRef]
- Hattori, N.; Nakamaki, T. Natural Killer Immunotherapy for Minimal Residual Disease Eradication Following Allogeneic Hematopoietic Stem Cell Transplantation in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2019, 20, 2057. [Google Scholar] [CrossRef] [Green Version]
- Hattori, N.; Kawaguchi, Y.; Sasaki, Y.; Shimada, S.; Murai, S.; Abe, M.; Baba, Y.; Watanuki, M.; Fujiwara, S.; Arai, N.; et al. Monitoring TIGIT/DNAM-1 and PVR/PVRL2 Immune Checkpoint Expression Levels in Allogeneic Stem Cell Transplantation for Acute Myeloid Leukemia. Biol. Blood Marrow Transplant. 2019, 25, 861–867. [Google Scholar] [CrossRef]
- Michonneau, D.; Sagoo, P.; Breart, B.; Garcia, Z.; Celli, S.; Bousso, P. The PD-1 Axis Enforces an Anatomical Segregation of CTL Activity that Creates Tumor Niches after Allogeneic Hematopoietic Stem Cell Transplantation. Immunity 2016, 44, 143–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koestner, W.; Hapke, M.; Herbst, J.; Klein, C.; Welte, K.; Fruehauf, J.; Flatley, A.; Vignali, D.A.; Hardtke-Wolenski, M.; Jaeckel, E.; et al. PD-L1 blockade effectively restores strong graft-versus-leukemia effects without graft-versus-host disease after delayed adoptive transfer of T-cell receptor gene-engineered allogeneic CD8+ T cells. Blood 2011, 117, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Asakura, S.; Hashimoto, D.; Takashima, S.; Sugiyama, H.; Maeda, Y.; Akashi, K.; Tanimoto, M.; Teshima, T. Alloantigen expression on non-hematopoietic cells reduces graft-versus-leukemia effects in mice. J. Clin. Investig. 2010, 120, 2370–2378. [Google Scholar] [CrossRef] [PubMed]
- Angenendt, L.; Schliemann, C.; Lutz, M.; Rebber, E.; Schulze, A.B.; Weckesser, M.; Stegger, L.; Schäfers, M.; Groth, C.; Kessler, T.; et al. Nivolumab in a patient with refractory Hodgkin’s lymphoma after allogeneic stem cell transplantation. Bone Marrow Transplant. 2016, 51, 443–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Yared, J.; Hardy, N.; Singh, Z.; Hajj, S.; Badros, A.Z.; Kocoglu, M.; Yanovich, S.; A Sausville, E.; Ujjani, C.; Ruehle, K.; et al. Major clinical response to nivolumab in relapsed/refractory Hodgkin lymphoma after allogeneic stem cell transplantation. Bone Marrow Transplant. 2016, 51, 850–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villasboas, J.C.; Ansell, S.M.; Witzig, T.E. Targeting the PD-1 pathway in patients with relapsed classic Hodgkin lymphoma following allogeneic stem cell transplant is safe and effective. Oncotarget 2016, 7, 13260–13264. [Google Scholar] [CrossRef] [Green Version]
- Shad, A.; Huo, J.; Darcy, C.; Abu-Ghosh, A.; Esposito, G.; Holuba, M.-J.; Robey, N.; Cooke, K.R.; Symons, H.J.; Chen, A.R.; et al. Tolerance and effectiveness of nivolumab after pediatric T-cell replete, haploidentical, bone marrow transplantation: A case report. Pediatr. Blood Cancer 2017, 64, e26257. [Google Scholar] [CrossRef]
- Onizuka, M.; Kojima, M.; Matsui, K.; Machida, S.; Toyosaki, M.; Aoyama, Y.; Kawai, H.; Amaki, J.; Hara, R.; Ichiki, A.; et al. Successful treatment with low-dose nivolumab in refractory Hodgkin lymphoma after allogeneic stem cell transplantation. Int. J. Hematol. 2017, 106, 141–145. [Google Scholar] [CrossRef]
- Covut, F.; Pinto, R.; Cooper, B.W.; Tomlinson, B.; Metheny, L.; Malek, E.; Lazarus, H.M.; De Lima, M.; Caimi, P.F. Nivolumab before and after allogeneic hematopoietic cell transplantation. Bone Marrow Transplant. 2017, 52, 1054–1056. [Google Scholar] [CrossRef] [Green Version]
- El Cheikh, J.; Massoud, R.; Abudalle, I.; Haffar, B.; Mahfouz, R.; A Kharfan-Dabaja, M.; Jisr, T.; Mougharbel, A.; Ibrahim, A.; Bazarbachi, A. Nivolumab salvage therapy before or after allogeneic stem cell transplantation in Hodgkin lymphoma. Bone Marrow Transplant. 2017, 52, 1074–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Porrata, L.F.; Aljitawi, O.; Lin, T.; Shune, L.; Ganguly, S.; McGuirk, J.P.; Abhyankar, S. Fatal GvHD induced by PD-1 inhibitor pembrolizumab in a patient with Hodgkin’s lymphoma. Bone Marrow Transplant. 2016, 51, 1268–1270. [Google Scholar] [CrossRef] [PubMed]
- Haverkos, B.M.; Abbott, D.; Hamadani, M.; Armand, P.; Flowers, M.E.; Merryman, R.; Kamdar, M.; Kanate, A.S.; Saad, A.; Mehta, A.; et al. PD-1 blockade for relapsed lymphoma post–allogeneic hematopoietic cell transplant: high response rate but frequent GVHD. Blood 2017, 130, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Herbaux, C.; Gauthier, J.; Brice, P.; Drumez, E.; Ysebaert, L.; Doyen, H.; Fornecker, L.; Bouabdallah, K.; Manson, G.; Ghesquières, H.; et al. Efficacy and tolerability of nivolumab after allogeneic transplantation for relapsed Hodgkin lymphoma. Blood 2017, 129, 2471–2478. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.S.Y.; Khong, P.-L.; Kwong, Y. Pembrolizumab for relapsed anaplastic large cell lymphoma after allogeneic haematopoietic stem cell transplantation: efficacy and safety. Ann. Hematol. 2016, 95, 1913–1915. [Google Scholar] [CrossRef]
- Albring, J.C.; Inselmann, S.; Sauer, T.; Schliemann, C.; Altvater, B.; Kailayangiri, S.; Rössig, C.; Hartmann, W.; Knorrenschild, J.R.; Sohlbach, K.; et al. PD-1 checkpoint blockade in patients with relapsed AML after allogeneic stem cell transplantation. Bone Marrow Transplant. 2017, 52, 317–320. [Google Scholar] [CrossRef]
- Holderried, T.A.W.; Fraccaroli, A.; Schumacher, M.; Heine, A.; Brossart, P.; Stelljes, M.; Klobuch, S.; Kröger, N.; Apostolova, P.; Finke, J.; et al. The role of checkpoint blockade after allogeneic stem cell transplantation in diseases other than Hodgkin’s Lymphoma. Bone Marrow Transplant. 2019, 54, 1662–1667. [Google Scholar] [CrossRef]
- Davids, M.S.; Kim, H.T.; Costello, C.L.; Herrera, A.F.; Locke, F.L.; Maegawa, R.O.; Savell, A.; Mazzeo, M.; Avigan, D.E.; Chen, Y.-B.; et al. A Phase I/Ib Study of Nivolumab for Relapsed Hematologic Malignancies after Allogeneic Hematopoietic Cell Transplantation (alloHCT). Blood 2018, 132, 705. [Google Scholar] [CrossRef]
- Kline, J.; Liu, H.; Michael, T.; Artz, A.S.; Godfrey, J.; Curran, E.K.; Stock, W.; Smith, S.M.; Bishop, M.R. Pembrolizumab for the Treatment of Disease Relapse Following Allogeneic Hematopoietic Cell Transplantation. Blood 2018, 132, 3415. [Google Scholar] [CrossRef]
- Bashey, A.; Medina, B.; Corringham, S.; Pasek, M.; Carrier, E.; Vrooman, L.; Lowy, I.; Solomon, S.R.; Morris, L.E.; Holland, H.K.; et al. CTLA4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoietic cell transplantation. Blood 2009, 113, 1581–1588. [Google Scholar] [CrossRef]
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.-B.; McSweeney, P.; et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. New Engl. J. Med. 2016, 375, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Khouri, I.F.; Scutti, J.A.B.; Milton, D.R.; Nicholas, C.; Pineda, M.; Jabbour, E.J.; Bassett, R.L.; Yadav, S.S.; Vence, L.M.; Allison, J.P.; et al. Durable Responses with Ipilimumab Plus Lenalidomide after Allogeneic and Autologous Stem Cell Transplantation for Patients with Lymphoid Malignancies. Blood 2018, 132, 4585. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; A Feldman, S.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Gardner, R.; Wu, D.; Cherian, S.; Fang, M.; Hanafi, L.-A.; Finney, O.; Smithers, H.; Jensen, M.C.; Riddell, S.R.; Maloney, D.G.; et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 2016, 127, 2406–2410. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Geyer, M.B.; Brentjens, R.J. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood 2016, 127, 3312–3320. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef]
- Shah, N.N.; Fry, T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 372–385. [Google Scholar] [CrossRef]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; Van Der Stegen, S.J.C.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochenderfer, J.N.; Dudley, M.E.; Carpenter, R.O.; Kassim, S.H.; Rose, J.J.; Telford, W.G.; Hakim, F.T.; Halverson, D.C.; Fowler, D.H.; Hardy, N.M.; et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013, 122, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brudno, J.N.; Somerville, R.P.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J. Clin. Oncol. 2016, 34, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Rossi, J.; Turcan, S.; Danan, C.; Locke, F.L.; Neelapu, S.S.; Miklos, D.B.; Bartlett, N.L.; Jacobson, C.A.; Braunschweig, I.; et al. Characterization of anti-CD19 chimeric antigen receptor (CAR) T cell-mediated tumor microenvironment immune gene profile in a multicenter trial (ZUMA-1) with axicabtagene ciloleucel (axi-cel, KTE-C19). J. Clin. Oncol. 2017, 35, 3025. [Google Scholar] [CrossRef]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol. Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Moon, E.K.; Ranganathan, R.; Eruslanov, E.; Kim, S.; Newick, K.; O’Brien, S.; Lo, A.; Liu, X.; Zhao, Y.; Albelda, S.M. Blockade of Programmed Death 1 Augments the Ability of Human T Cells Engineered to Target NY-ESO-1 to Control Tumor Growth after Adoptive Transfer. Clin. Cancer Res. 2016, 22, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Prosser, M.E.; E Brown, C.; Shami, A.F.; Forman, S.J.; Jensen, M.C. Tumor PD-L1 co-stimulates primary human CD8+ cytotoxic T cells modified to express a PD1:CD28 chimeric receptor. Mol. Immunol. 2012, 51, 263–272. [Google Scholar] [CrossRef]
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.; Darcy, P.K. Anti-PD-1 Antibody Therapy Potently Enhances the Eradication of Established Tumors By Gene-Modified T Cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef] [Green Version]
- Zou, F.; Lu, L.; Liu, J.; Xia, B.; Zhang, W.; Hu, Q.; Liu, W.; Zhang, Y.; Lin, Y.; Jing, S.; et al. Engineered triple inhibitory receptor resistance improves anti-tumor CAR-T cell performance via CD56. Nat. Commun. 2019, 10, 4109–4114. [Google Scholar] [CrossRef]
- Su, S.; Hu, B.; Shao, J.; Shen, B.; Du, J.; Du, Y.; Zhou, J.; Yu, L.; Zhang, L.; Chen, F.; et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci. Rep. 2016, 6, 20070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Cheng, C.; Cheng, A.; Zhang, X.; Li, N.; Xia, C.; Wei, X.; Liu, X.; Wang, H. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017, 27, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [Green Version]
- Suarez, E.; Chang, D.-K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; Van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Nakajima, M.; Sakoda, Y.; Adachi, K.; Nagano, H.; Tamada, K. Improved survival of chimeric antigen receptor-engineered T (CAR-T) and tumor-specific T cells caused by anti-programmed cell death protein 1 single-chain variable fragment-producing CAR-T cells. Cancer Sci. 2019, 110, 3079–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenderian, S.S.; Ruella, M.; Shestova, O.; Klichinsky, M.; Kim, M.; Porter, D.L.; June, C.H.; Gill, S. Identification of PD1 and TIM3 As Checkpoints That Limit Chimeric Antigen Receptor T Cell Efficacy in Leukemia. Boil. Blood Marrow Transplant. 2016, 22, S19–S21. [Google Scholar] [CrossRef]
- Li, A.M.; E Hucks, G.; Dinofia, A.M.; Seif, A.E.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Fasano, C.; McBride, B.; Gonzalez, V.; et al. Checkpoint Inhibitors Augment CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy in Relapsed B-Cell Acute Lymphoblastic Leukemia. Blood 2018, 132, 556. [Google Scholar] [CrossRef]
- Chong, E.A.; Melenhorst, J.J.; Lacey, S.F.; Ambrose, D.E.; Gonzalez, V.; Levine, B.L.; June, C.H.; Schuster, S.J. PD-1 blockade modulates chimeric antigen receptor (CAR)–modified T cells: refueling the CAR. Blood 2017, 129, 1039–1041. [Google Scholar] [CrossRef] [Green Version]
- Hill, B.T.; Roberts, Z.J.; Xue, A.; Rossi, J.M.; Smith, M.R. Rapid tumor regression from PD-1 inhibition after anti-CD19 chimeric antigen receptor T-cell therapy in refractory diffuse large B-cell lymphoma. Bone Marrow Transplant. 2019, 2019, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Lu, W.; Sun, R.; Jin, X.; Cheng, L.; He, X.; Wang, L.; Yuan, T.; Lyu, C.; Zhao, M. Anti-CD19 Chimeric Antigen Receptor T Cells in Combination With Nivolumab Are Safe and Effective Against Relapsed/Refractory B-Cell Non-hodgkin Lymphoma. Front. Oncol. 2019, 9, 767. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, U.; Worel, N.; McGuirk, J.P.; Riedell, P.A.; Fleury, I.; Borchmann, P.; Chu, J.; Abdelhady, A.M.; Forcina, A.; Pacaud, L.B.; et al. Portia: A Phase 1b Study Evaluating Safety and Efficacy of Tisagenlecleucel and Pembrolizumab in Patients with Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Blood 2019, 134, 5325–5353. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Locke, F.L.; Miklos, D.B.; Herrera, A.F.; Westin, J.R.; Lee, J.; Rossi, J.M.; Sun, J.; Zheng, L.; Avanzi, M.P.; et al. End of Phase 1 Results from Zuma-6: Axicabtagene Ciloleucel (Axi-Cel) in Combination with Atezolizumab for the Treatment of Patients with Refractory Diffuse Large B Cell Lymphoma. Biol. Blood Marrow Transplant. 2019, 25, S173. [Google Scholar] [CrossRef]
- Ardeshna, K.M.; Marzolini, M.A.V.; Norman, J.; Al-Hajj, M.; Thomas, S.; Faulkner, J.; Kotsopoulou, E.; Pule, M.; Peddareddigari, V.G.R.; Khokhar, N.Z.; et al. Phase 1/2 Study of AUTO3 the First Bicistronic Chimeric Antigen Receptor (CAR) Targeting CD19 and CD22 Followed By an Anti-PD1 in Patients with Relapsed/Refractory (r/r) Diffuse Large B Cell Lymphoma (DLBCL): Results of Cohort 1 and 2 of the Alexander Study. Blood 2019, 134, 246. [Google Scholar]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; You, Y.; Shen, Z.; Shi, L. EGFRvIII-CAR-T Cells with PD-1 Knockout Have Improved Anti-Glioma Activity. Pathol. Oncol. Res. 2020, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kambhampati, S.; Gray, L.; Fakhri, B.; Lo, M.; Vu, K.; Arora, S.; Kaplan, L.; Ai, W.Z.; Andreadis, C. Immune-related Adverse Events Associated With Checkpoint Inhibition in the Setting of CAR T Cell Therapy: A Case Series. Clin. Lymphoma Myeloma Leuk. 2020, 20, e118–e123. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
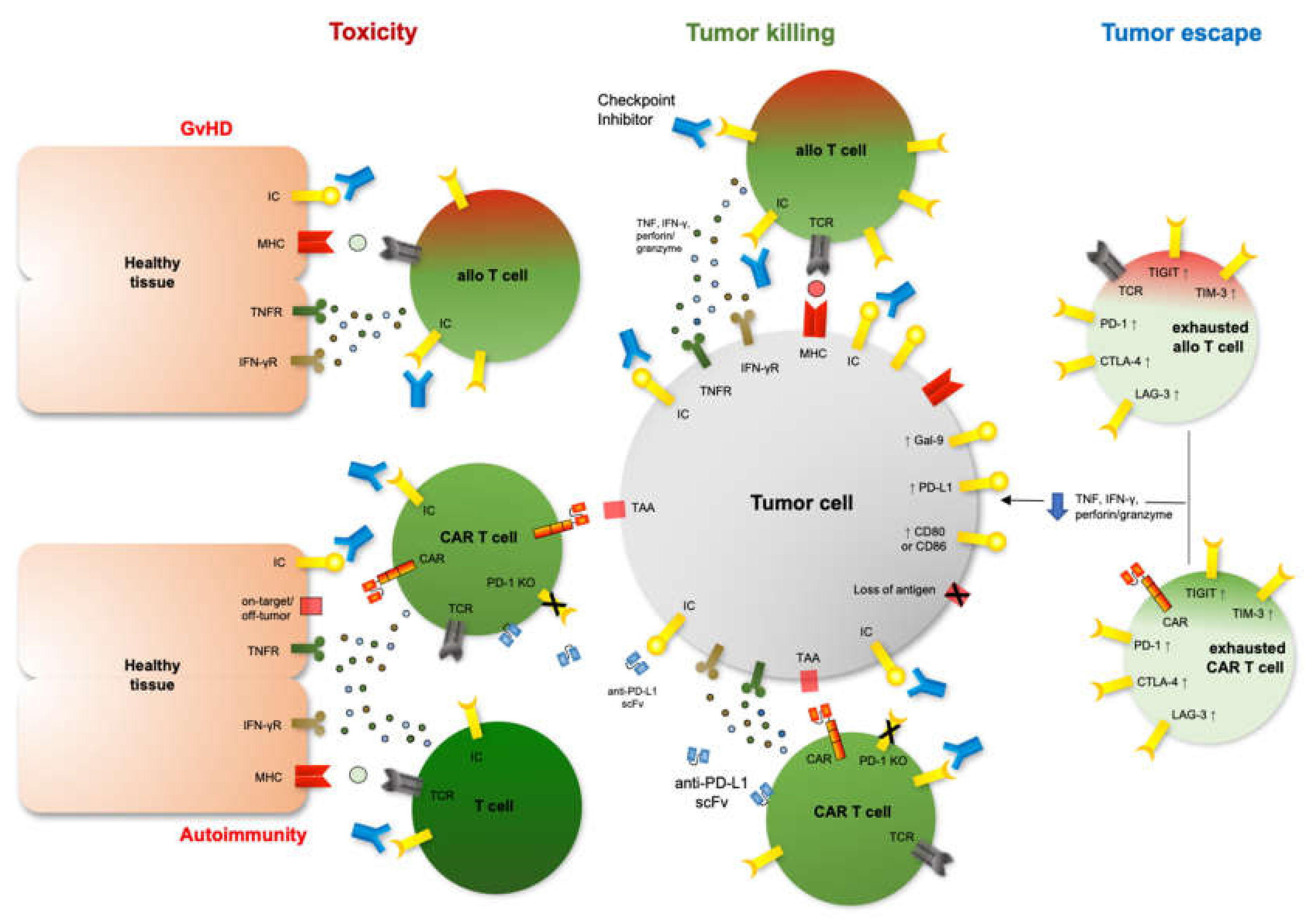
Mechanisms of tumor escape, restored anti-tumor immunity after blocked signaling of inhibitory immune checkpoints as well as therapy-mediated toxicity. Right: Up-regulation of inhibitory immune checkpoints (IC) like programmed death-1 (PD-1), cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), T-cell immunoglobulin mucin-3 (TIM-3), lymphocyte-activation gene 3 (LAG-3) or T-cell immunoglobulin and ITIM domains (TIGIT) on exhausted T cells after cell therapy leads to impaired tumor recognition and killing. Center: Therapeutic strategies modifying inhibitory ICs, i.e., checkpoint inhibitors or CAR T cells expressing anti-PD-L1 single chain variable fragments (scFv) or harboring a PD-1 knockout (KO), can restore anti-tumor immunity after cellular therapy. Left: Increased immune function after blocked inhibitory immune checkpoints may amplify undesired toxic side effects on healthy tissue after cell therapy such as Graft-versus-Host-Disease (GvHD), immune-related adverse events (irAEs) or increased on-target/off-tumor activity.
Figure 1.
Mechanisms of tumor escape, restored anti-tumor immunity after blocked signaling of inhibitory immune checkpoints as well as therapy-mediated toxicity. Right: Up-regulation of inhibitory immune checkpoints (IC) like programmed death-1 (PD-1), cytotoxic T-lymphocyte-associated protein-4 (CTLA-4), T-cell immunoglobulin mucin-3 (TIM-3), lymphocyte-activation gene 3 (LAG-3) or T-cell immunoglobulin and ITIM domains (TIGIT) on exhausted T cells after cell therapy leads to impaired tumor recognition and killing. Center: Therapeutic strategies modifying inhibitory ICs, i.e., checkpoint inhibitors or CAR T cells expressing anti-PD-L1 single chain variable fragments (scFv) or harboring a PD-1 knockout (KO), can restore anti-tumor immunity after cellular therapy. Left: Increased immune function after blocked inhibitory immune checkpoints may amplify undesired toxic side effects on healthy tissue after cell therapy such as Graft-versus-Host-Disease (GvHD), immune-related adverse events (irAEs) or increased on-target/off-tumor activity.


{kind=link}
Table 1.
Overview of relevant studies targeting immune checkpoints after allogeneic hematopoietic stem cell transplantation.
Table 1.
Overview of relevant studies targeting immune checkpoints after allogeneic hematopoietic stem cell transplantation.
| n | Disease | Characteristics | Intervention | Response | irAEs (Grade) | GvHD (Grade) | Ref. |
|---|---|---|---|---|---|---|---|
31 | r/r HL/FL | retrospective multi-center study HL, n = 29; transformed FL, n = 1; FL + HL, n = 1 85% received ≥ 1 salvage therapy post allo-HCT before anti-PD-1 mAb prior GvHD NOS (19/31) | nivolumab (q2w, 3 mg/kg) n = 28 pembrolizumab (q3w, 200 mg) n = 3 first application 26 mo. after allo-HCT (median) | ORR/CR/PR/SD/PD: 77/50/27/10/13% OS: N/A PFS: 591 days | N/A | aGvHD (N/A, n = 6), overlap (N/A n = 4), cGvHD (N/A, n = 7) In 16/17 GvHD onset after 1–2 doses of CI 8 GvHD related deaths | [63] |
| 20 | r/r HL | retrospective multi-center study HL, n = 20 65% received ≥ 1 salvage therapy post allo-HCT prior aGvHD (10/20), cGvHD (3/20) | nivolumab (q2w, 3 mg/kg) first application 23 mo. after allo-HCT (median) | ORR/CR/PR/PD: 95/42/52/5% OS/PFS: 79/58% | cerebellar ataxia (II, n = 1) hepatitis (II, n = 7) | aGVHD (I, n = 1; III, n = 3; IV, n = 2) In 6/20 GvHD onset after 1 dose of CI 2 GvHD related deaths | [64] |
| 21 | r/r HM | retrospective multi-center study AML/MDS, n = 12; ALL, n = 2; NHL, n = 5; MF, n = 2 relapse after 1st allo-HCT, n = 15; 2nd, n = 5; 3rd, n = 1 prior aGvHD (13/21), cGvHD (7/21) | nivolumab (0.5 mg/kg, 3 mg/kg, 40 mg or 200 mg absolute), n = 5 nivolumab + DLI (0.5 mg/kg, 1 mg/kg, or 40 mg absolute), n = 5 nivo + ipilimumab (both 3 mg/kg), n = 1 ipilimumab (3 mg/kg, or 10 mg/kg), n = 10 first application 4.5 mo. after allo-HCT (median) | ORR/CR/PR/SD/PD: 43/14/29/4/48% ORR (nivolumab): 40% ORR (nivo + DLI): 80% ORR (ipilimumuab): 20% OS: 79 days PFS: N/A | N/A | 10/21 (any grade) 6/21 aGvHD III/IV or moderate/severe cGvHD; 5/5 GvHD in patients with nivo + DLI In 6/20 GvHD onset after 1 dose of CI 4 GvHD related deaths | [67] |
| 11 | r/r AML r/r (N)HL | phase I prospective single-center study AML/MDS, n = 8, lymphoma (DLBCL, HL), n = 3 no prior aGvHD > I or cGvHD | pembrolizumab (200 mg q3w) first application after allo-HCT N/A | ORR/CR/PR/SD/PD: 28/28/0/28/43% OS/PFS: N/A | 7/11 (any grade) pneumonitis (III-IV, n = 2), hyperthyrodism (III, n = 1), rash (II, n = 1) onset after 1-2 cycles of CI | none | [69] |
| 29 | r/r hematologic and solid malignancies | phase I multi-center study HL, n = 14; Myeloma, n = 6; AML, n = 2; CML, n = 2; CLL, n = 2; NHL, n = 1; breast cancer, n = 1; renal cell cancer, n = 1 no ongoing GvHD, no prior III/IV GvHD | ipilimumab (+ DLI if progressive after CI) single dose 0.1 mg/kg, n = 4 single dose 0.33 mg/kg, n = 3 single dose 0.66 mg/kg, n = 4 single dose 1 mg/kg, n = 3 single dose 3 mg/kg, n = 15 application after allo-HCT N/A | CR: 2/29 (cHL) PR: 1/29 (NHL) OS/PFS: N/A | polyarthropathy (III, n = 1); hyperthyrodism (I-II, n = 1); dyspnea on exertion (N/A); pneumonitis (IV, n = 1) | no grade III-IV time of GvHD onset after CI N/A GvHD related deaths N/A | [70] |
| 28 | r/r HM | phase I/Ib multicenter study AML, n = 12; HL, n = 7; NHL, n = 4; MDS, n = 2; MM, n = 1; MPN, n = 1; ALL, n = 1 72% received ≥ 1 salvage therapy post allo-HCT before anti-PD-1 mAb no ongoing GvHD, no prior III/IV aGvHD | ipilimumab (q3w, 4 courses) 3 mg/kg; n = 6 3 mg escalated to 10 mg/kg; n = 7 10 mg/kg; n = 15 first application 56 mo. after allo-HCT (median) | ORR/CR/PR/SD/PD 23/9/27/41% (10 mg/kg) no response with 3 mg/kg OS/PFS: N/A | 6/28 (any grade) pneumonitis (II-IV, n = 3) colitis (III, n = 1) ITP (II, n = 1) diarrhea (II, n = 1) death (n = 1) | with 10 mg/kg ipilimumab aGvHD gut (N/A, n = 1) cGvHD liver (N/A, n = 3) time of GvHD onset after CI N/A GvHD related deaths N/A | [71] |
| 28 | r/r HM | phase I/Ib multi-center study AML, n = 11; MDS n = 7; HL, n = 5; NHL, n = 3; MPN, n = 1; CLL, n = 1 64% received ≥ 1 salvage therapy post allo-HCT | nivolumab (q2w) 1 mg/kg (initial dose), n = 6 0.5 mg/kg (dose de-escalation), n = 8 0.5 mg/kg (initial dose), n = 14 3 mg/kg (dose escalation not realized due to toxicity) first application 21 mo. after allo-HCT (median) | with 1 mg/kg nivolumab CR/PR: 17/33% with 0.5 mg/kg nivolumab ORR/CR/PR/SD/PD 16/8/8/47/37% 6 mo. PFS/OS: 39/61% | with 1 mg/kg nivolumab sepsis/fatal ARDS (n = 1) fatal APS (n = 1) pneumonitis (III, n = 1) transaminitis (III, n = 1) bilirubinemia (III, n = 1) | with 1 mg/kg nivolumab aGvHD liver, gut (III, n = 2) with 0.5 mg/kg nivolumab new onset/worsening of GvHD (n = 10) aGvHD (N/A, n = 1) cGvHD (N/A, n = 7) aGvHD+ cGvHD (N/A, n = 2) time of GvHD onset after CI N/A GvHD related deaths N/A | [68] |
| 10 | r/r lymphoma | phase II multi-center study FL, n = 2; MCL, n = 3; THL, n = 1; DLBCL, n = 1; CLL, n = 2; ALCL, n = 1 no ongoing GvHD at study entry 44% had prior extensive cGvHD | lenalidomide (10 mg/day x 21d) + ipilimumab (3 mg/kg single-dose) 1st anti-CTLA-4 3d after completed 1st cycle of len repetition of len-cycle after 30d 2nd anti-CTLA-4 dose as before first application 29 mo. after allo- HCT (median) | ORR/CR/PR/SD 77/44/33/22% OS/PFS: N/A 4 mo RFS: 100% 12 mo RFS: 56% | hypothyrodism (II, n = 1) | cGvHD liver, mouth (N/A, n = 1) time of GvHD onset after CI N/A GvHD related deaths N/A | [72] |
irAES: immune-related adverse events, a/cGvHD: acute/chronic Graft-versus-Host Disease, allo-HCT: allogeneic hematopoietic stem cell transplantation, r/r: relapsed and refractory, HL: Hodgkin’s lymphoma, FL: follicular lymphoma, mAb: monoclonal antibody, AML: acute myeloid leukemia, MDS: myelodysplastic syndrome, MF: myelofibrosis, NHL: Non-Hodgkin lymphoma, DLBCL: diffuse large B-cell lymphoma, HM: hematologic malignancy, CML: chronic myeloid leukemia, CLL: chronic lymphocytic leukemia, MPN: myeloid proliferative neoplasm, DLI: donor lymphocyte infusion, ITP: idiopathic thrombocytopenic purpura, MCL: mantle cell lymphoma, THL: triple hit lymphoma, ALCL: anaplastic large T-cell lymphoma, ARDS: acute respiratory deficiency syndrome, APS: antiphospholipid syndrome, ORR: overall remission rate, CR: complete remission, PR: partial remission, SD: stable disease, PD: progressive disease, OS: overall survival, PFS: progression free survival, RFS: remission free survival, N/A: not available, NOS: no otherwise specified.
Table 2.
Overview of relevant studies targeting immune checkpoints after CAR T cell therapy.
| n | Disease | Characteristics | Intervention | Response | irAEs (Grade) | CRS/ICANS (Grade) | Ref. |
|---|---|---|---|---|---|---|---|
11 | r/r B-NHL | retrospective single-center study DLBCL (Stage III-IV), n = 10 Burkitt’s lymphoma, n = 1 | CD19 CAR + nivolumab (3 mg/kg single-dose) anti-PD-1 applied on d3 after CAR infusion | ORR/CR/PR/NR: 82/46/36/18% PFS: 6 (1–14 months) | no grade III-IV toxicity | CRS (I, n = 3; II, n = 6) ICANS (N/A, n = 1) | [103] |
| 14 | r/r B-ALL r/r B-NHL | retrospective single-center study B-ALL, n = 13 B-lymphoblastic lymphoma, n = 1 | CD19 CAR + pembrolizumab (200 mg, q3w) CD19 CAR + nivolumab (3 mg/kg, q3w) anti-PD-1 application ≧ 14d after CAR T cell infusion, median time after CAR T cell infusion N/A | ORR/CR/PR/PD: 43/14/29/7% PFS: N/A | pancreatitis (N/A, n = 1) hypothyroidism (N/A, n = 1) urticaria (N/A, n = 1) arthropathy (N/A, n = 1) no grade V toxicities | CRS (N/A, n = 3) | [100] |
| 4 | r/r DLBCL | phase 1b prospective multi-center study (PORTIA) DLBCL, n = 4 | CD19 CAR + pembrolizumab (200 mg, q3w, 6 courses) first anti-PD-1 application on d15 after CAR T cell infusion | N/A | none | CRS (N/A, n = 1) | [104] |
| 12 | r/r DLBCL | phase 1 prospective multi-center study (ZUMA-6) DLBCL, n = 12 | CD19 CAR + atezolizumab (1200 mg, q3w, 4 courses) first anti-PD-L1 application on d21 (cohort 1, n = 3), d14 (cohort 2, n = 3), d1 (cohort 3, n = 6) after CAR infusion | ORR/CR/PR: 90/60/30% PFS: N/A | N/A | CRS (≥ III, n = 3) ICANS (≥ III, n = 6) | [105] |
| 11 | r/r DLBCL | phase 1/2 prospective multi-center study (ALEXANDER) DLBCL NOS, n = 4 DLBCL transformed from FL/MZL, n = 7 | AUTO-3 CD19/CD22 CAR mono (n = 4) AUTO-3 CAR + pembrolizumab (200 mg, q3w) n = 7 first anti-PD-1 application on d14 Cohort 1: 50 × 106 AUTO3, n = 7 Cohort 2: 150 × 106 AUTO3, n = 4 | Cohort 1: 50 × 106 AUTO3 ORR/CR/PR: 57/29/28% PFS: N/A Cohort 2: 150 × 106 AUTO3 N/A | N/A | CRS (I, n = 3) ICANS (III, n = 1) | [106] |
CAR: chimeric antigen receptor, irAES: immune-related adverse events, CRS: cytokine release syndrome, ICANS: immune effector cell-associated neurotoxicity syndrome, r/r: relapsed and refractory, B-NHL: B-cell non-Hodgkin lymphoma, DLBCL: diffuse large B-cell lymphoma, B-ALL: B-cell acute lymphocytic leukemia, FL: follicular lymphoma, MZL: marginal zone B-cell lymphoma, CD: cluster of differentiation, ORR: overall remission rate, CR: complete remission, CRR: complete remission rate, PR: partial remission, PD: progressive disease, NR: no response, PFS: progression free survival, N/A: not available.
Table 3.
Selected ongoing clinical trials targeting immune checkpoints after allogeneic hematopoietic cell transplantation or CAR T cell therapy.
Table 3.
Selected ongoing clinical trials targeting immune checkpoints after allogeneic hematopoietic cell transplantation or CAR T cell therapy.
| Clinical Trial | Phase | Disease | Intervention | Sponsor |
|---|---|---|---|---|
NCT02981914 | I | r/r HL, B-NHL, AML, MDS after allo-HCT | pembrolizumab (q3w, 200 mg) | University of Chicago |
| NCT03286114 | IB | r/r MDS, AML, ALL after allo-HCT | pembrolizumab (q3w, 200 mg) | University of Michigan Rogel Cancer Center |
| NCT04361058 | I | r/r high risk AML, MDS after allo-HCT | nivolumab (q2w, 0.25 mg/kg, 4 courses) | SCRI Development Innovations, LLC |
| NCT02890329 | I | r/r AML(+MRC), MDS after allo-HCT | decitabine + ipilimumab (q4w, dose N/A) priming: decitabine (d1-5 of 28 days) induction: decitabine (d1-5) + ipilimumab (d1); 4 courses maintenance: decitabine (d1-5) + ipilimumab (d1); 4 courses | National Cancer Institute (NCI) |
| NCT03588936 | I | r/r AL, CL, MDS, lymphoma after allo-HCT | tocilizumab (8 mg/kg on day 0 and 29) + nivolumab (q2w, 0.25 or 0.5 mg/kg on day 1; up to 4 courses) | Medical College of Wisconsin |
| NCT03146468 | II | r/r hematologic disease after allo-HCT | nivolumab (q2w, 3 mg/kg) | Melbourne Health |
| NCT01822509 | I/IB | r/r AML, MDS, MPN, ALL, CLL, CML, (N)HL, MM after allo-HCT | nivolumab or ipilimumab induction: nivolumab (q2w, dose N/A, 8 courses) or ipilimumab (q3w, dose N/A; 4 courses) maintenance: nivolumab (q2w, dose N/A, up to a total of 60 weeks) or ipilimumab (q12w, dose N/A; 4 courses) | National Cancer Institute (NCI) |
| NCT03600155 | IB | r/r high risk AML, MDS after allo-HCT | nivolumab or ipilimumab or nivolumab + ipilimumab Arm A: nivolumab (q4w, d1+15, dose N/A 6 courses) ≥ six weeks post allo-HCT Arm B: ipilimumab (q3w, d1, dose N/A, 6 courses) ≥ six weeks post allo-HCT Arm C; nivolumab (q6w, d1,14,28, dose N/A 6 courses) + ipilimumab (q6w, d1, dose N/A, 6 courses) ≥ six weeks post allo-HCT | M.D. Anderson Cancer Center |
| NCT00586391 | I | B-NHL, CLL, ALL | CD19CAR-28-zeta T cells Dose Level 1: 2 × 107 T cells/m2 Dose Level 2: 1 × 108 T cells/m2 Dose Level 3: 2 × 108 T cells/m2 ± ipilimumab (once in week 2 after CAR infusion, dose N/A, only in patients with low/intermediate grade leukemia/ lymphoma) | Baylor College of Medicine |
| NCT03630159 | IB | r/r DLBCL | tisagenlecleucel + pembrolizumab timing and dose N/A | Novartis Pharmaceuticals |
| NCT03630159 | I/II | r/r DLBCL | axicabtagene ciloleucel + atezolizumab timing and dose N/A | Kite, A Gilead Company |
| NCT03287817 | I/II | r/r DLBCL | AUTO-3 (50 × 106 to 900 × 106 CD19/CD22 CAR T cells) ± pembrolizumab timing and dose N/A | Autolus Limited |
| NCT04134325 | I | r/r HL after CAR T cell therapy | pembrolizumab (q3w, 200 mg) or nivolumab (q2w, 240 mg or q4w, 480 mg) | UNC Lineberger Comprehensive Cancer Center |
| NCT02650999 | I/II | r/r DLBCL, FL, MCL after CAR T cell therapy | pembrolizumab (timing N/A, 200 mg) | Abramson Cancer Center of the University of Pennsylvania |
| NCT04205409 | II | r/r CLL, DLBCL, FL, MZL, NHL, MM after CAR T cell therapy | nivolumab (q4w, dose N/A) | University of Washington |
| NCT04337606 | I/II | r/r NHL after CAR T cell therapy | cohort 1: chidamide (q3w, 10 mg on d1-5 and 20 mg on d8,11,15,18) + decitabine (q3w, 10 mg on d1-5) cohort 2: decitabine (q3w, 10 mg on d1-5) + camrelizumab (q3w, 200 mg on d6) | Chinese PLA General Hospital |
CAR: chimeric antigen receptor, allo-HCT: allogeneic hematopoietic stem cell transplantation, r/r: relapsed and refractory, HL: Hodgkin’s lymphoma, B-NHL: B-cell non-Hodgkin lymphoma, AML: acute myeloid leukemia, MDS: myeloid dysplastic syndrome, ALL: acute lymphocytic leukemia, MRC: myelodysplasia-related changes, N/A: not available, AL: acute leukemia, CL: chronic leukemia, MPN: myeloproliferative neoplasia, CLL: chronic lymphocytic leukemia, CML: chronic myeloid leukemia, NHL: Non-Hodgkin-lymphoma, MM: multiple myeloma, DLBCL: diffuse large B-cell lymphoma, FL: follicular lymphoma, MZL: marginal zone B-cell lymphoma.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Schmitz, F.; Wolf, D.; Holderried, T.A.W. The Role of Immune Checkpoints after Cellular Therapy. Int. J. Mol. Sci. 2020, 21, 3650. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103650
AMA Style
Schmitz F, Wolf D, Holderried TAW. The Role of Immune Checkpoints after Cellular Therapy. International Journal of Molecular Sciences. 2020; 21(10):3650. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103650
Chicago/Turabian StyleSchmitz, Friederike, Dominik Wolf, and Tobias A.W. Holderried. 2020. "The Role of Immune Checkpoints after Cellular Therapy" International Journal of Molecular Sciences 21, no. 10: 3650. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103650
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.