Interplay between Type 2 Transglutaminase (TG2), Gliadin Peptide 31-43 and Anti-TG2 Antibodies in Celiac Disease

 , and
, and
Abstract
:1. Introduction
2. Generality on Celiac Disease
3. Gluten Proteins and the Adaptive/Innate Immune Response
4. The Multi-Functionality of TG2
5. TG2 in the Intestine and the Auto-immune Response
Biological Activities of Anti-TG2 Antibodies
6. The Gliadin Peptide 31-43 (P31-43)
6.1. Structural Properties of P31-43
6.2. Entrance of P31-43 Into the Cells: a Role for TG2?
6.3. Biological Activity of P31-43
7. Interplay between TG2 and 31-43
7.1. Modulation of TG2 Activity and Expression by P31-43
7.2. Modulation of P31-43 Effects by TG2 Inhibition
7.3. P31-43 as TG2 Substrate
8. Interplay between Anti-TG2 Antibodies and P31-43
9. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TG2 | type 2 transglutaminase |
| TG | transglutaminase |
| CD | celiac disease |
| ECM P31-43 | extra-cellular matrix α-gliadin peptide 31-43 |
| P57-68 | α-gliadin peptide 57-68 |
| HLA | human leukocytes antigen |
| APC | antigen presenting cell |
| IL15 | interleukin 15 |
| MDC | mono-dansyl-cadaverine |
| CPPs | cell-penetrating peptides |
| EGF | epidermal growth factor |
| PT-gliadin | gliadin peptic-tryptic digest |
| DON | 6-diazo-5-oxo-norleucine |
| INF | interferon |
| CFTR | cystic fibrosis transmembrane conductance regulator |
References
- Sollid, L.M.; Jabri, B. Celiac disease and transglutaminase 2: A model for posttranslational modification of antigens and HLA association in the pathogenesis of auto-immune disorders. Curr. Opin. Immunol. 2011, 23, 732–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, M.V.; Troncone, R.; Auricchio, S. Gliadin peptides as triggers of the proliferative and stress/innate immune response of the celiac small intestinal mucosa. Int. J. Mol. Sci. 2014, 15, 20518–20537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martucciello, S.; Paolella, G.; Esposito, C.; Lepretti, M.; Caputo, I. Anti-type 2 transglutaminase antibodies as modulators of type 2 transglutaminase functions: A possible pathological role in celiac disease. Cell. Mol. Life Sci. 2018, 75, 4107–4124. [Google Scholar] [CrossRef] [PubMed]
- Sollid, L.M. Molecular basis of celiac disease. Annu. Rev. Immunol. 2000, 18, 53–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leffler, D.A.; Green, P.H.R.; Fasano, A. Extraintestinal manifestations of coeliac disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, W.; Ehnis, T.; Bauer, M.; Donner, P.; Volta, U.; Riecken, E.O.; Schuppan, D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 1997, 3, 797–801. [Google Scholar] [CrossRef]
- Leffler, D.A.; Schuppan, D. Update on serologic testing in celiac disease. Am. J. Gastroenterol. 2010, 105, 2520–2524. [Google Scholar] [CrossRef]
- Giersiepen, K.; Lelgemann, M.; Stuhldreher, N.; Ronfani, L.; Husby, S.; Koletzko, S.; Korponay-Szabó, I.R. ESPGHAN Working Group on Coeliac Disease Diagnosis. Accuracy of diagnostic antibody tests for coeliac disease in children: Summary of an evidence report. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 229–241. [Google Scholar] [CrossRef] [Green Version]
- Reunala, T.; Salmi, T.T.; Hervonen, K. Dermatitis herpetiformis: Pathognomonic transglutaminase IgA deposits in the skin and excellent prognosis on a gluten-free diet. Acta Derm. Venereol. 2015, 95, 917–922. [Google Scholar] [CrossRef]
- Collin, P.; Salmi, T.T.; Hervonen, K.; Kaukinen, K.; Reunala, T. Dermatitis herpetiformis: A cutaneous manifestation of coeliac disease. Ann. Med. 2017, 49, 23–31. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Aeschlimann, P.; Strigun, A.; Sanders, D.S.; Woodroofe, N.; Aeschlimann, D. Auto-antibodies in gluten ataxia recognize a novel neuronal transglutaminase. Ann. Neurol. 2008, 64, 332–343. [Google Scholar] [CrossRef]
- Cenit, M.C.; Olivares, M.; Codoñer-Franch, P.; Sanz, Y. Intestinal microbiota and celiac disease: Cause, consequence or co-evolution? Nutrients 2015, 7, 6900–6923. [Google Scholar] [CrossRef] [Green Version]
- Bouziat, R.; Hinterleitner, R.; Brown, J.J.; Stencel-Baerenwald, J.E.; Ikizler, M.; Mayassi, T.; Meisel, M.; Kim, S.M.; Discepolo, V.; Pruijssers, A.J.; et al. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 2017, 356, 44–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, A.; Ajay, R.; Torsten, M. The revival of the battle between David and Goliath in the enteric viruses and microbiota struggle: Potential implication for celiac disease. Microorganisms 2019, 7, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sollid, L.M. The roles of MHC class II genes and post-translational modification in celiac disease. Immunogenetics 2017, 69, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Kårhus, L.L.; Thuesen, B.H.; Skaaby, T.; Rumessen, J.J.; Linneberg, A. The distribution of HLA DQ2 and DQ8 haplotypes and their association with health indicators in a general Danish population. United Eur. Gastroenterol. J. 2018, 6, 866–878. [Google Scholar] [CrossRef] [PubMed]
- Caio, C.; Volta, U.; Sapone, A.; Leffler, D.A.; De Giorgio, R.; Catassi, C.; Alessio, F. Celiac disease: A comprehensive current review. BMC Med. 2019, 17, 142. [Google Scholar] [CrossRef] [Green Version]
- Withoff, S.; Li, Y.; Jonkers, I.; Wijmenga, C. Understanding celiac disease by genomics. Trends Genet. 2016, 32, 295–308. [Google Scholar] [CrossRef]
- Wieser, H. Chemistry of gluten proteins. Food Microbiol. 2007, 24, 115–119. [Google Scholar] [CrossRef]
- Sollid, L.M.; Qiao, S.W.; Anderson, R.P.; Gianfrani, C.; Koning, F. Nomenclature and listing of celiac disease relevant gluten T-cell epitopes restricted by HLA-DQ molecules. Immunogenetics 2012, 64, 455–460. [Google Scholar] [CrossRef] [Green Version]
- Sollid, L.M.; Tye-Din, J.A.; Qiao, S.W.; Anderson, R.P.; Gianfrani, C.; Koning, F. Update 2020: Nomenclature and listing of celiac disease-relevant gluten epitopes recognized by CD4(+) T cells. Immunogenetics 2020, 72, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Molberg, Ø.; Parrot, I.; Hausch, F.; Filiz, F.; Gray, G.M.; Sollid, L.M.; Khosla, C. Structural basis for gluten intolerance in celiac sprue. Science 2002, 297, 2275–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianfrani, C.; Auricchio, S.; Troncone, R. Adaptive and innate immune responses in celiac disease. Immunol. Lett. 2005, 99, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Mamone, G.; Ferranti, P.; Rossi, M.; Roepstorff, P.; Fierro, O.; Malorni, A.; Addeo, F. Identification of a peptide from alpha-gliadin resistant to digestive enzymes: Implications for celiac disease. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 855, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Mamone, G.; Nitride, C.; Picariello, G.; Addeo, F.; Ferranti, P.; Mackie, A. Tracking the fate of pasta (T. Durum semolina) immunogenic proteins by in vitro simulated digestion. J. Agric. Food Chem. 2015, 63, 2660–2667. [Google Scholar] [CrossRef] [PubMed]
- Itzlinger, A.; Branchi, F.; Elli, L.; Schumann, M. Gluten-Free Diet in Celiac Disease—Forever and for All? Nutrients 2018, 10, 1796. [Google Scholar] [CrossRef] [Green Version]
- Lorand, L.; Graham, R.M. Transglutaminases: Crosslinking enzymes with pleiotropic functions. Nat. Rev. Mol. Cell. Biol. 2003, 4, 140–156. [Google Scholar] [CrossRef]
- Duarte, L.; Matte, C.R.; Bizarro, C.V.; Ayub, M.A.Z. Review transglutaminases: Part II-industrial applications in food, biotechnology, textiles and leather products. World J. Microbiol. Biotechnol. 2019, 36, 1–20. [Google Scholar] [CrossRef]
- Muszbek, L.; Bereczky, Z.; Bagoly, Z.; Komáromi, I.; Katona, É. Factor XIII: A coagulation factor with multiple plasmatic and cellular functions. Physiol. Rev. 2011, 91, 931–972. [Google Scholar] [CrossRef] [Green Version]
- Eckert, R.L.; Kaartinen, M.T.; Nurminskaya, M.; Belkin, A.M.; Colak, G.; Johnson, G.V.; Mehta, K. Transglutaminase regulation of cell function. Physiol. Rev. 2014, 94, 383–417. [Google Scholar] [CrossRef] [Green Version]
- Nurminskaya, M.V.; Belkin, A.M. Cellular functions of tissue transglutaminase. Int. Rev. Cell. Mol. Biol. 2012, 294, 1–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemskov, E.A.; Mikhailenko, I.; Hsia, R.C.; Zaritskaya, L.; Belkin, A.M. Unconventional secretion of tissue transglutaminase involves phospholipid-dependent delivery into recycling endosomes. PLoS ONE 2011, 6, e19414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, T.S.; Lin, C.J.; Greenberg, C.S. Role of tissue transglutaminase-2 (TG2)-mediated aminylation in biological processes. Amino Acids 2017, 49, 501–551. [Google Scholar] [CrossRef] [PubMed]
- Stamnaes, J.; Fleckenstein, B.; Sollid, L.M. The propensity for deamidation and transamidation of peptides by transglutaminase 2 is dependent on substrate affinity and reaction conditions. Biochim. Biophys. Acta 2008, 1784, 1804–1811. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cerione, R.A.; Clardy, J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc. Natl. Acad. Sci. USA 2002, 99, 2743–2747. [Google Scholar] [CrossRef] [Green Version]
- Pinkas, D.M.; Strop, P.; Brunger, A.T.; Khosla, C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007, 5, e327. [Google Scholar] [CrossRef]
- Jin, X.; Stamnaes, J.; Klöck, C.; Di Raimondo, T.R.; Sollid, L.M.; Khosla, C. Activation of extracellular transglutaminase 2 by thioredoxin. J. Biol. Chem. 2011, 286, 37866–37873. [Google Scholar] [CrossRef] [Green Version]
- Yi, M.C.; Melkonian, A.V.; Ousey, J.A.; Khosla, C. Endoplasmic reticulum-resident protein 57 (ERp57) oxidatively inactivates human transglutaminase 2. J. Biol. Chem. 2018, 293, 2640–2649. [Google Scholar] [CrossRef] [Green Version]
- Nakaoka, H.; Perez, D.M.; Baek, K.J.; Das, T.; Husain, A.; Misono, K.; Im, M.J.; Graham, R.M. Gh: A GTP-binding protein with transglutaminase activity and receptor signaling function. Science 1994, 264, 1593–1596. [Google Scholar] [CrossRef]
- Lai, T.S.; Lin, C.J.; Wu, Y.T.; Wu, C.J. Tissue transglutaminase (TG2) and mitochondrial function and dysfunction. Front. Biosci. 2017, 22, 1114–1137. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Mehta, K. Tissue transglutaminase, inflammation, and cancer: How intimate is the relationship? Amino Acids 2013, 44, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Lorand, l.; Iismaa, S.E. Transglutaminase diseases: From biochemistry to the bedside. FASEB J. 2019, 33, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Esposito, C.; Paparo, F.; Caputo, I.; Porta, R.; Salvati, V.M.; Mazzarella, G.; Auricchio, S.; Troncone, R. Expression and enzymatic activity of small intestinal tissue transglutaminase in celiac disease. Am. J. Gastroenterol. 2003, 98, 1813–1820. [Google Scholar] [CrossRef]
- Biagi, F.; Campanella, J.; Laforenza, U.; Gastaldi, G.; Tritto, S.; Grazioli, M.; Villanacci, V.; Corazza, G.R. Transglutaminase 2 in the enterocytes is coeliac specific and gluten dependent. Dig. Liver Dis. 2006, 38, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Villanacci, V.; Not, T.; Sblattero, D.; Gaiotto, T.; Chirdo, F.; Galletti, A.; Bassotti, G. Mucosal tissue transglutaminase expression in celiac disease. J. Cell. Mol. Med. 2009, 13, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, L.; Ciacci, C.; Ricciardelli, I.; Vacca, L.; Raia, V.; Rispo, A.; Griffin, M.; Issekutz, T.; Quaratino, S.; Londei, M. Unexpected role of surface transglutaminase type II in celiac disease. Gastroenterology 2005, 129, 1400–1413. [Google Scholar] [CrossRef]
- Sollid, L.M.; Molberg, Ø.; McAdam, S.; Lundin, K.E.A. Auto-antibodies in celiac disease: Tissue transglutaminase–guilt by association? Gut 1997, 41, 851–852. [Google Scholar] [CrossRef] [Green Version]
- Dolcino, M.; Zanoni, G.; Bason, C.; Tinazzi, E.; Boccola, E.; Valletta, E.; Contreas, G.; Lunardi, C.; Puccetti, A. A subset of anti-rotavirus antibodies directed against the viral protein VP7 predicts the onset of celiac disease and induces typical features of the disease in the intestinal epithelial cell line T84. Immunol. Res. 2013, 56, 465–476. [Google Scholar] [CrossRef]
- Stamnaes, J.; Sollid, L.M. Celiac disease: Autoimmunity in response to food antigen. Semin. Immunol. 2015, 27, 343–352. [Google Scholar] [CrossRef]
- HogenEsch, C.E.; Wolters, V.M.; Gerritsen, S.A.; Putter, H.; von Blomberg, B.M.; van Hoogstraten, I.M.W.; Houwen, R.H.J.; van der Lely, N.; Mearin, M.L. Specific celiac disease antibodies in children on a gluten-free diet. Pediatrics 2011, 128, 547–552. [Google Scholar] [CrossRef]
- Kaukinen, K.; Peräaho, M.; Collin, P.; Partanen, J.; Woolley, N.; Kaartinen, T.; Nuutinen, T.; Halttunen, T.; Mäki, M.; Korponay-Szabo, I. Small-bowel mucosal transglutaminase 2-specific IgA deposits in coeliac disease without villous atrophy: A prospective and randomized clinical study. Scand. J. Gastroenterol. 2005, 40, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Ludvigsson, J.F.; Leffler, D.A.; Bai, J.C.; Biagi, F.; Fasano, A.; Green, P.H.; Hadjivassiliou, M.; Kaukinen, K.; Kelly, C.P.; Leonard, J.N.; et al. The Oslo definitions for coeliac disease and related terms. Gut 2013, 62, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Tosco, A.; Aitoro, R.; Auricchio, A.; Ponticelli, D.; Miele, E.; Paparo, F.; Greco, L.; Troncone, R.; Maglio, M. Intestinal anti-tissue transglutaminase antibodies in potential coeliac disease. Clin. Exp. Immunol. 2012, 171, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Maglio, M.; Ziberna, F.; Aitoro, R.; Discepolo, V.; Lania, G.; Bassi, V.; Miele, E.; Not, T.; Troncone, R.; Auricchio, R. Intestinal production of anti-tissue transglutaminase 2 antibodies in patients with diagnosis otherthanC celiac disease. Nutrients 2017, 9, 1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrelli, M.; Maglio, M.; Agnese, M.; Paparo, F.; Gentile, S.; Colicchio, B.; Tosco, A.; Auricchio, R.; Troncone, R. High density of intraepithelial gammadelta lymphocytes and deposits of immunoglobulin (Ig)M anti-tissue transglutaminase antibodies in the jejunum of coeliac patients with IgA deficiency. Clin. Exp. Immunol. 2010, 160, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Caputo, I.; Barone, M.V.; Lepretti, M.; Martucciello, S.; Nista, I.; Troncone, R.; Auricchio, S.; Sblattero, D.; Esposito, C. Celiac anti-tissuetransglutaminaseantibodiesinterfere with the uptake of alphagliadin peptide 31–43 butnot of peptide 57–68 by epithelialcells. Biochim. Biophys. Acta 2010, 1802, 717–727. [Google Scholar] [CrossRef] [Green Version]
- Caputo, I.; Lepretti, M.; Secondo, A.; Martucciello, S.; Paolella, G.; Sblattero, D.; Barone, M.V.; Esposito, C. Anti-tissue transglutaminase antibodies activate intracellular tissue transglutaminase by modulating cytosolic Ca2+ homeostasis. Amino Acids 2013, 44, 251–260. [Google Scholar] [CrossRef]
- Martucciello, S.; Lavric, M.; Toth, B.; Korponay-Szabo, I.; Nadalutti, C.; Myrsky, E.; Rauhavirta, T.; Esposito, C.; Sulic, A.M.; Sblattero, D.; et al. RhoB is associated with the antiangiogenic effects of celiac patient transglutaminase 2-targeted autoantibodies. J. Mol. Med. 2012, 90, 817–826. [Google Scholar] [CrossRef]
- Nadalutti, C.A.; Korponay-Szabo, I.R.; Kaukinen, K.; Griffin, M.; Mäki, M.; Lindfors, K. CeliacdiseasepatientIgAantibodies induce endothelialadhesion and cellpolarizationdefects via extracellulartransglutaminase 2. Cell. Mol. Life Sci. 2014, 71, 1315–1326. [Google Scholar] [CrossRef]
- Kalliokoski, S.; Piqueras, V.O.; Frías, R.; Sulic, A.M.; Määttä, J.A.; Kähkönen, N.; Viiri, K.; Huhtala, H.; Pasternack, A.; Laurila, K.; et al. Transglutaminase 2-specific coeliacdisease auto-antibodies induce morphologicalchanges and signs of inflammation in the small-bowel mucosa of mice. Amino Acids 2017, 49, 529–540. [Google Scholar] [CrossRef] [Green Version]
- Quaglia, S.; Ferrara, F.; De Leo, L.; Ziberna, F.; Vatta, S.; Marchiò, S.; Sblattero, D.; Ventura, A.; Not, T. A functional idiotype/anti-idiotype network is active in genetically gluten-intolerant individuals negative for both celiac disease-related intestinal damage and serum autoantibodies. J. Immunol. 2019, 202, 1079–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemente, M.G.; De Virgiliis, S.; Kang, J.S.; Macatagney, R.; Musu, M.P.; Di Pierro, M.R.; Drago, S.; Congia, M.; Fasano, A. Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function. Gut 2003, 52, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.E.; Sapone, A.; Fasano, A.; Vogel, S.N. Gliadin stimulation of murine macrophage inflammatory gene expression and intestinal permeability are MyD88-dependent: Role of the innate immune response in Celiac disease. J. Immunol. 2006, 176, 2512–2521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, M.V.; Zimmer, K.P. Endocytosis and transcytosis of gliadin peptides. Mol. Cell. Pediatr. 2016, 3, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez Castro, M.F.; Miculán, E.; Herrera, M.G.; Ruera, C.; Perez, F.; Prieto, E.D.; Barrera, E.; Pantano, S.; Carasi, P.; Chirdo, F.G. p31-43 Gliadin peptide formsoligomers and induces NLRP3 inflammasome/caspase 1-dependentmucosaldamage in small intestine. Front. Immunol. 2019, 10, 31. [Google Scholar] [CrossRef] [Green Version]
- Herrera, M.G.; Gómez Castro, M.F.; Prieto, E.; Barrera, E.; Dodero, V.I.; Pantano, S.; Chirdo, F. Structural conformation and self-assembly process of p31–43 gliadin peptide in aqueous solution. Implications for celiac disease. FEBS J. 2020, 287, 2134–2149. [Google Scholar] [CrossRef]
- Iacomino, G.; Fierro, O.; D’Auria, S.; Picariello, G.; Ferranti, P.; Liguori, C.; Addeo, F.; Mamone, G. Structural analysis and Caco-2 cell permeability of the celiac-toxic A-gliadin peptide 31-55. J. Agric. Food Chem. 2013, 61, 1088–1096. [Google Scholar] [CrossRef]
- Barrera, E.; Chirdo, F.; Pantano, S. Commentary: p31–43 gliadin peptide forms oligomers and induces NLRP3 inflammasome/caspase 1-dependent mucosal damage in small intestine. Front. Immunol. 2019, 10, 2792. [Google Scholar] [CrossRef]
- Calvanese, L.; Nanayakkara, M.; Aitoro, R.; Sanseverino, M.; Tornesello, A.L.; Falcigno, L.; D’Auria, G.; Barone, M.V. Structural insights on P31–43, a gliadin peptide able to promote an innate but not an adaptive response in celiac disease. J. Pept. Sci. 2019, 25, e3161. [Google Scholar] [CrossRef]
- Zimmermann, C.; Rudloff, S.; Lochnit, G.; Arampatzi, S.; Maison, W.; Zimmer, K.P. Epithelial transport of immunogenic and toxic gliadin peptides in vitro. PLoS ONE 2014, 9, e113932. [Google Scholar] [CrossRef]
- Barone, M.V.; Nanayakkara, M.; Paolella, G.; Maglio, M.; Vitale, V.; Troiano, R.; Ribecco, M.T.; Lania, G.; Zanzi, D.; Santagata, S.; et al. Gliadin peptide P31–43 localises to endocytic vesicles and interferes with their maturation. PLoS ONE 2010, 5, e12246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer, K.P.; Fischer, I.; Mothes, T.; Weissen-Plenz, G.; Schmitz, M.; Wieser, H.; Büning, J.; Lerch, M.M.; Ciclitira, P.C.; Weber, P.; et al. Endocytotic segregation of gliadin peptide 31–49 in enterocytes. Gut 2010, 59, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Lebreton, C.; Menard, S.; Abed, J.; Moura, I.C.; Coppo, R.; Dugave, C.; Monteiro, R.C.; Fricot, A.; Traore, M.G.; Griffin, M.; et al. Interactions among secretory immunoglobulin A, CD71, and transglutaminase-2 affectpermeabilityofintestinalepithelialcellstogliadinpeptides. Gastroenterology 2012, 143, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Paolella, G.; Lepretti, M.; Martucciello, S.; Nanayakkara, M.; Auricchio, S.; Esposito, C.; Barone, M.V.; Caputo, I. The toxic alpha-gliadin peptide 31-43 enters cells without a surface membrane receptor. Cell. Biol. Int. 2018, 42, 112–120. [Google Scholar] [CrossRef]
- Vilasi, S.; Sirangelo, I.; Irace, G.; Caputo, I.; Barone, M.V.; Esposito, C.; Ragone, R. Interaction of ‘toxic’ and ‘immunogenic’ A-gliadin peptides with a membrane-mimetic environment. J. Mol. Recognit. 2010, 23, 322–328. [Google Scholar] [CrossRef]
- Bechara, C.; Sagan, S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef]
- Franz, J.; Lelle, M.; Peneva, K.; Bonn, M.; Weidner, T. SAP(E)-A cell-penetrating polyproline helix at lipid interfaces. Biochim. Biophys. Acta 2016, 1858, 2028–2034. [Google Scholar] [CrossRef]
- Oba, M.; Nagano, Y.; Kato, T.; Tanaka, M. Secondary structures and cell-penetrating abilities of arginine-rich peptide foldamers. Sci. Rep. 2019, 9, 1349. [Google Scholar] [CrossRef] [Green Version]
- Zhu, P.; Jin, L. Cell penetrating peptides: A promising tool for the cellular uptake of macromolecular drugs. Curr. Protein Pept. Sci. 2018, 19, 211–220. [Google Scholar] [CrossRef]
- Furini, G.; Verderio, E.A. Spotlight on the transglutaminase 2-heparan sulfate interaction. Med. Sci. 2019, 7, 5. [Google Scholar] [CrossRef] [Green Version]
- Maiuri, L.; Ciacci, C.; Ricciardelli, I.; Vacca, L.; Raia, V.; Auricchio, S.; Picard, J.; Osman, M.; Quaratino, S.; Londei, M. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet 2003, 362, 30–37. [Google Scholar] [CrossRef]
- Barone, M.V.; Gimigliano, A.; Castoria, G.; Paolella, G.; Maurano, F.; Paparo, F.; Maglio, M.; Mineo, A.; Miele, E.; Nanayakkara, M.; et al. Growth factor-like activity of gliadin, an alimentary protein: Implications for coeliac disease. Gut 2007, 56, 480–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanayakkara, M.; Lania, G.; Maglio, M.; Discepolo, V.; Sarno, M.; Gaito, A.; Troncone, R.; Auricchio, S.; Auricchio, R.; Barone, M.V. An undigested gliadin peptide activates innate immunity and proliferative signaling in enterocytes: The role in celiac disease. Am. J. Clin. Nutr. 2013, 98, 1123–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caputo, I.; Secondo, A.; Lepretti, M.; Paolella, G.; Auricchio, S.; Barone, M.V.; Esposito, C. Gliadin peptides induce tissue transglutaminase activation and ER-stress through Ca2+ mobilization in Caco-2 cells. PLoS ONE 2012, 7, e45209. [Google Scholar] [CrossRef]
- Chladkova, B.; Kamanova, J.; Palova-Jelinkova, L.; Cinova, J.; Sebo, P.; Tuckova, L. Gliadin fragments promote migration of dendritic cells. J. Cell. Mol. Med. 2011, 15, 938–948. [Google Scholar] [CrossRef] [Green Version]
- Nanayakkara, M.; Lania, G.; Maglio, M.; Kosova, R.; Sarno, M.; Gaito, A.; Discepolo, V.; Troncone, R.; Auricchio, S.; Auricchio, R.; et al. Enterocyte proliferation and signaling are constitutively altered in celiac disease. PLoS ONE 2013, 8, e76006. [Google Scholar] [CrossRef] [Green Version]
- Lania, G.; Nanayakkara, M.; Maglio, M.; Auricchio, R.; Porpora, M.; Conte, M.; De Matteis, M.M.; Rizzo, R.; Luini, A.; Discepolo, V.; et al. Constitutive alterations in vesicular trafficking increase the sensitivity of cells from celiac disease patients to gliadin. Commun. Biol. 2019, 2, 190. [Google Scholar] [CrossRef]
- Luciani, A.; Villella, V.R.; Vasaturo, A.; Giardino, I.; Pettoello-Mantovani, M.; Guido, S.; Cexus, O.N.; Peake, N.; Londei, M.; Quaratino, S.; et al. Lysosomal accumulation of gliadin p31–43 peptide induces oxidative stress and tissue transglutaminase-mediated PPARgamma downregulation in intestinal epithelial cells and coeliac mucosa. Gut 2010, 59, 311–319. [Google Scholar] [CrossRef]
- Paolella, G.; Nanayakkara, M.; Sposito, S.; Lepretti, M.; Auricchio, S.; Esposito, C.; Barone, M.V.; Martucciello, S.; Caputo, I. Constitutive differential features of type 2 transglutaminase in cells derived from celiac patients and from healthy subjects. Int. J. Mol. Sci. 2020, 21, 1231. [Google Scholar] [CrossRef] [Green Version]
- Molberg, O.; McAdam, S.; Lundin, K.E.; Kristiansen, C.; Arentz-Hansen, H.; Kett, K.; Sollid, L.M. T cells from celiac disease lesions recognize gliadin epitopes deamidated in situ by endogenous tissue transglutaminase. Eur. J. Immunol. 2001, 31, 1317–1323. [Google Scholar] [CrossRef]
- Szondy, Z.; Korponay-Szabó, I.; Király, R.; Sarang, Z.; Tsay, G.J. Transglutaminase 2 in human diseases. Biomedicine 2017, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, C.; Paparo, F.; Caputo, I.; Rossi, M.; Maglio, M.; Sblattero, D.; Not, T.; Porta, R.; Auricchio, S.; Marzari, R.; et al. Anti-tissue transglutaminase antibodies from coeliac patients inhibit transglutaminase activity both in vitro and in situ. Gut 2002, 51, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, W.; Trapp, D.; Esslinger, B.; Leidenberger, M.; Piper, J.; Hahn, E.; Schuppan, D. Auto-antibodies of patients with coeliac disease are insufficient to block tissue transglutaminase activity. Gut 2003, 52, 1562–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, M.V.; Caputo, I.; Ribecco, M.T.; Maglio, M.; Marzari, R.; Sblattero, D.; Troncone, R.; Auricchio, S.; Esposito, C. Humoral immune response to tissue transglutaminase is related to epithelial cell proliferation in celiac disease. Gastroenterology 2007, 132, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Király, R.; Vecsei, Z.; Deményi, T.; Korponay-Szabó, I.R.; Fésüs, L. Coeliac auto-antibodies can enhance transamidating and inhibit GTPase activity of tissue transglutaminase: Dependence on reaction environment and enzyme fitness. J. Autoimmun. 2006, 26, 278–287. [Google Scholar] [CrossRef]
- Vincentini, O.; Maialetti, F.; Gonnelli, E.; Silano, M. Gliadin-dependen cytokine production in a bidimensional cellular model of celiac intestinal mucosa. Clin. Exp. Med. 2015, 15, 447–454. [Google Scholar] [CrossRef]
- Rauhavirta, T.; Oittinen, M.; Kivistö, R.; Männistö, P.T.; Garcia-Horsman, J.A.; Wang, Z.; Griffin, M.; Mäki, M.; Kaukinen, K.; Lindfors, K. Are transglutaminase 2 inhibitors able to reduce gliadin-induced toxicity related to celiac disease? A proof-of-concept study. J. Clin. Immunol. 2013, 33, 134–142. [Google Scholar] [CrossRef]
- Feriotto, G.; Calza, R.; Bergamini, C.M.; Griffin, M.; Wang, Z.; Beninati, S.; Ferretti, V.; Marzola, E.; Guerrini, R.; Pagnoni, A.; et al. Involvement of cell surface TG2 in the aggregation of K562 cells triggered by gluten. Amino Acids 2017, 49, 551–565. [Google Scholar] [CrossRef] [Green Version]
- Silano, M.; Vincentini, O.; Luciani, A.; Felli, C.; Caserta, S.; Esposito, S.; Villella, V.R.; Pettoello-Mantovani, M.; Guido, S.; Maiuri, L. Early tissue transglutaminase-mediated response underlies K562(S)-cell gliadin-dependent agglutination. Pediatr. Res. 2012, 71, 532–538. [Google Scholar] [CrossRef] [Green Version]
- Gerace, E.; Resta, F.; Landucci, E.; Renzi, D.; Masi, A.; Pellegrini-Giampietro, D.E.; Calabrò, A.; Mannaioni, G. The gliadin peptide 31-43 exacerbates kainate neurotoxicity in epilepsy models. Sci. Rep. 2017, 7, 15146. [Google Scholar] [CrossRef] [Green Version]
- Abadie, V.; Kim, S.M.; Lejeune, T.; Palanski, B.A.; Ernest, J.D.; Tastet, O.; Voisine, J.; Discepolo, V.; Marietta, E.V.; Hawash, M.B.F.; et al. IL-15, gluten and HLA-DQ8 drive tissue destruction in coeliac disease. Nature 2020, 578, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Klöck, C.; Herrera, Z.; Albertelli, M.; Khosla, C. Discovery of potent and specific dihydroisoxazole inhibitors of human transglutaminase 2. J. Med. Chem. 2014, 57, 9042–9064. [Google Scholar] [CrossRef] [Green Version]
- Rauhavirta, T.; Qiao, S.W.; Jiang, Z.; Myrsky, E.; Loponen, J.; Korponay-Szabó, I.R.; Salovaara, H.; Garcia-Horsman, J.A.; Venäläinen, J.; Männistö, P.T.; et al. Epithelial transport and deamidation of gliadin peptides: A role for coeliac disease patient immunoglobulin A. Clin. Exp. Immunol. 2011, 164, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Villella, V.R.; Venerando, A.; Cozza, G.; Esposito, S.; Ferrari, E.; Monzani, R.; Spinella, M.C.; Oikonomou, V.; Renga, G.; Tosco, A.; et al. A pathogenic role for cystic fibrosis trans membrane conductance regulator in celiac disease. EMBO J. 2019, 38, e100101. [Google Scholar] [CrossRef]
- Picarelli, A.; Di Tola, M.; Sabbatella, L.; Anania, M.C.; Di Cello, T.; Greco, R.; Silano, M.; De Vincenzi, M. 31–43 amino acid sequence of the alpha-gliadin induces anti-endomysial antibody production during in vitro challenge. Scand. J. Gastroenterol. 1999, 34, 1099–1102. [Google Scholar] [CrossRef]
- Paolella, G.; Caputo, I.; Marabotti, A.; Lepretti, M.; Salzano, A.M.; Scaloni, A.; Vitale, M.; Zambrano, N.; Sblattero, D.; Esposito, C. Celiac anti-type 2 transglutaminase antibodies induce phosphoproteome modification in intestinal epithelial Caco-2 cells. PLoS ONE 2013, 8, e84403. [Google Scholar] [CrossRef] [Green Version]
- Nanayakkara, M.; Kosova, R.; Lania, G.; Sarno, M.; Gaito, A.; Galatola, M.; Greco, L.; Cuomo, M.; Troncone, R.; Auricchio, S.; et al. A celiac cellular phenotype, with altered LPP sub-cellular distribution, is inducible in controls by the toxic gliadin peptide P31-43. PLoS ONE 2013, 8, e79763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervio, E.; Volta, U.; Verri, M.; Boschi, F.; Pastoris, O.; Granito, A.; Barbara, G.; Parisi, C.; Felicani, C.; Tonini, M.; et al. Sera of patients with celiac disease and neurologic disorders evoke a mitochondrial-dependent apoptosis in vitro. Gastroenterology 2007, 133, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Paolella, G.; Lepretti, M.; Barone, M.V.; Nanayakkara, M.; Di Zenzo, M.; Sblattero, D.; Auricchio, S.; Esposito, C.; Caputo, I. Celiac anti-type 2 transglutaminase antibodies induce differential effects in fibroblasts from celiac disease patients and from healthy subjects. Amino Acids 2017, 49, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Korponay-Szabó, I.R.; Halttunen, T.; Szalai, Z.; Laurila, K.; Király, R.; Kovács, J.B.; Fésüs, L.; Mäki, M. In vivo targeting of intestinal and extraintestinal transglutaminase 2 by coeliac auto-antibodies. Gut 2004, 53, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Moreno, M.L.; Cebolla, Á.; Muñoz-Suano, A.; Carrillo-Carrion, C.; Comino, I.; Pizarro, Á.; León, F.; Rodríguez-Herrera, A.; Sousa, C. Detection of gluten immunogenic peptides in the urine of patients with coeliac disease reveals transgressions in the gluten-free diet and incomplete mucosal healing. Gut 2017, 66, 250–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]


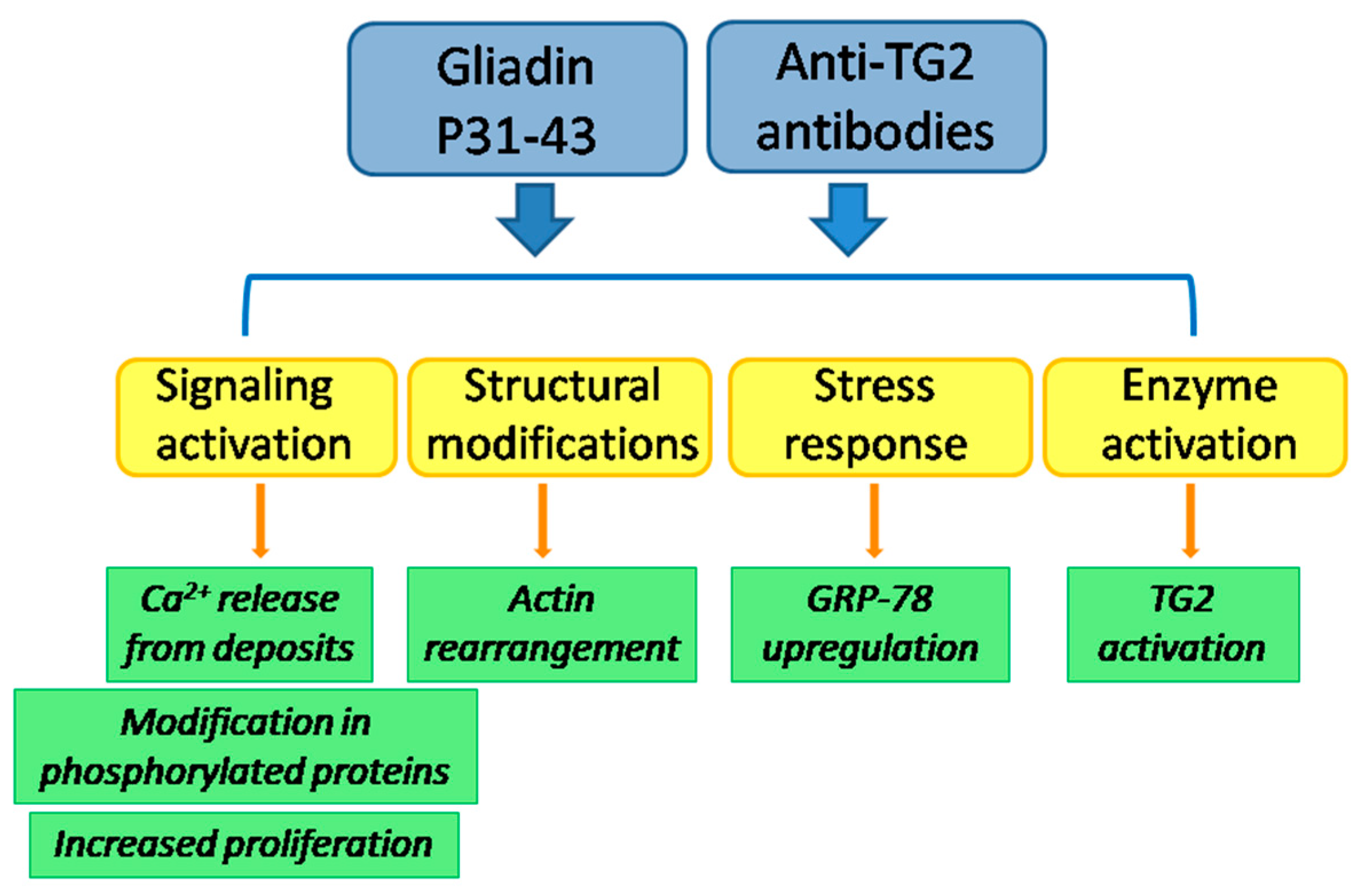{kind=link}
| Biological Process Investigated | In Vitro Model Employed | TG2 Inhibitor or Antibody Employed | Effects Observed | TG2 Activity Involvement? |
|---|---|---|---|---|
| P31-43-induced proliferation | Caco-2 cells | MDC/cystamine [56] R281(unpublished data) | Inhibitors have no effect on P31-43 induced proliferation | TG2 activity seems not to be involved |
| Anti-TG2 antibody CUB7402 [56] | Reduction of proliferation at low doses of antibody | TG2 activity seems not to be involved; antibodies reduce P31-43 uptake | ||
| P57-68-T cell activation after the treatment with P31-43 | CD intestinal biopsies | R283 [46] | Reduction of T cell activation | TG2 activity appears involved |
| P57-68 -induced INF-γ production after the treatment with P31-43 | T84 cells co-culture | Anti-TG2 antibody CUB7402 [96] | CUB7402 prevents INF-γ production | Maybe CUB7402 is reducing P31-43 uptake |
| PPAR-γ down-regulation induced by P31-43 | CD (but not control) intestinal biopsies | Cystamine [92] | Reduction of PPAR-γ down-regulation | TG2 activity appears involved |
| Increased kainate neurotoxicity induced by P31-43 | Mice hippocampal slides | Z-DON [100] | Reduction of kainate cytotoxicity | TG2 activity appears involved |
| P31-43-induced apoptosis | CD intestinal (but not control) intestinal biopsies and T84 cells | R283 and CUB7402 [46] | No effect on apoptosis | TG2 activity seems not to be involved |
| P31-43 endocytosis | Caco-2 cells | MDC/cystamine [56] | No effect on P31-43 endocytosis | TG2 activity seems not to be involved |
| P31-43 transcytosis | Caco-2 cells | R281 [103] | No effect on P31-43 transcytosis | TG2 activity seems not to be involved |
| Biological Process Investigated | Induced by Antibodies | Induced by P31-43 | ||||
|---|---|---|---|---|---|---|
| In Control Cells/Biopsies | In CD Cells/Biopsies | In Gliadin Sensitive Cells | In Control Cells | In CD Cells | In Gliadin Sensitive Cells | |
| Cell proliferation | No effect [94] | Increased [94] | Increased [56] | No effect [82] | Increased [82] | Increased [82] |
| ERK activation | N.d. | N.d. | Increased [56] | N.d. | N.d. | Increased [82] |
| Ca2+ ions mobilization | N.d. | N.d. | Increased [57] | N.d. | N.d. | Increased [84] |
| Protein phosphorylation | N.d. | N.d. | Several proteins modulated [106] | Increased phosphory-lation of paxillin and Fak [107]. No tyrosine phosphory-lation [46] | Increased phosphory-lation of paxillin and Fak, more than in control cells [107]. Increased tyrosine phosphory- lation [46] | Increased EGF-receptor phosphory-lation [82] |
| GRP-78 expression | N.d. | N.d. | Increased (unpublished data) | N.d. | N.d. | Increased [84] |
| Actin rearrangement | N.d. | N.d. | Increased [56] | Increased in fibroblasts, not increased in duodenal enterocytes [46] | Increased in fibroblasts [107] and in duodenal enterocytes [46] | Increased [46,82] |
| Intracellular TG2 activity | N.d. | N.d. | Increased [57] | Increased [89] | Increased (less than in control cells) [89] | Increased [84] |
| Apoptosis | No effect [94] | No effect [94] | N.d. | No effect [81,82] | Increased [81,82] | Increased [46] |
| Vesicular trafficking | N.d. | N.d. | Interference with EGF endocytosis [56] | Transient delay of EGF/EGF-receptor trafficking [87] | Prolonged delay of EGF/EGF-receptor trafficking [87] | Interference with EGF endocytosis [82] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martucciello, S.; Sposito, S.; Esposito, C.; Paolella, G.; Caputo, I. Interplay between Type 2 Transglutaminase (TG2), Gliadin Peptide 31-43 and Anti-TG2 Antibodies in Celiac Disease. Int. J. Mol. Sci. 2020, 21, 3673. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103673
Martucciello S, Sposito S, Esposito C, Paolella G, Caputo I. Interplay between Type 2 Transglutaminase (TG2), Gliadin Peptide 31-43 and Anti-TG2 Antibodies in Celiac Disease. International Journal of Molecular Sciences. 2020; 21(10):3673. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103673
Chicago/Turabian StyleMartucciello, Stefania, Silvia Sposito, Carla Esposito, Gaetana Paolella, and Ivana Caputo. 2020. "Interplay between Type 2 Transglutaminase (TG2), Gliadin Peptide 31-43 and Anti-TG2 Antibodies in Celiac Disease" International Journal of Molecular Sciences 21, no. 10: 3673. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21103673