Modifications of Hydroxyapatite by Gallium and Silver Ions—Physicochemical Characterization, Cytotoxicity and Antibacterial Evaluation


Abstract
:1. Introduction
2. Results
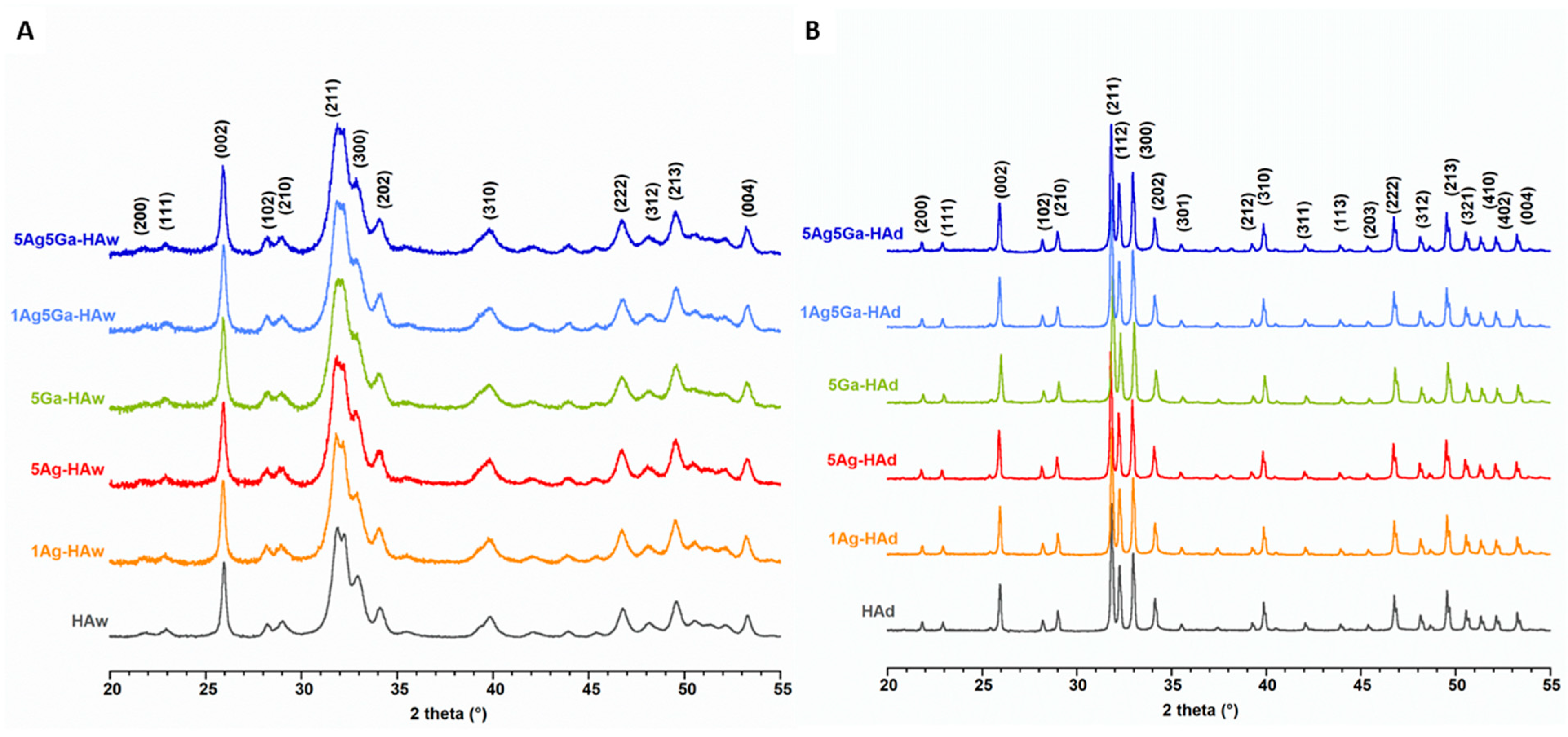
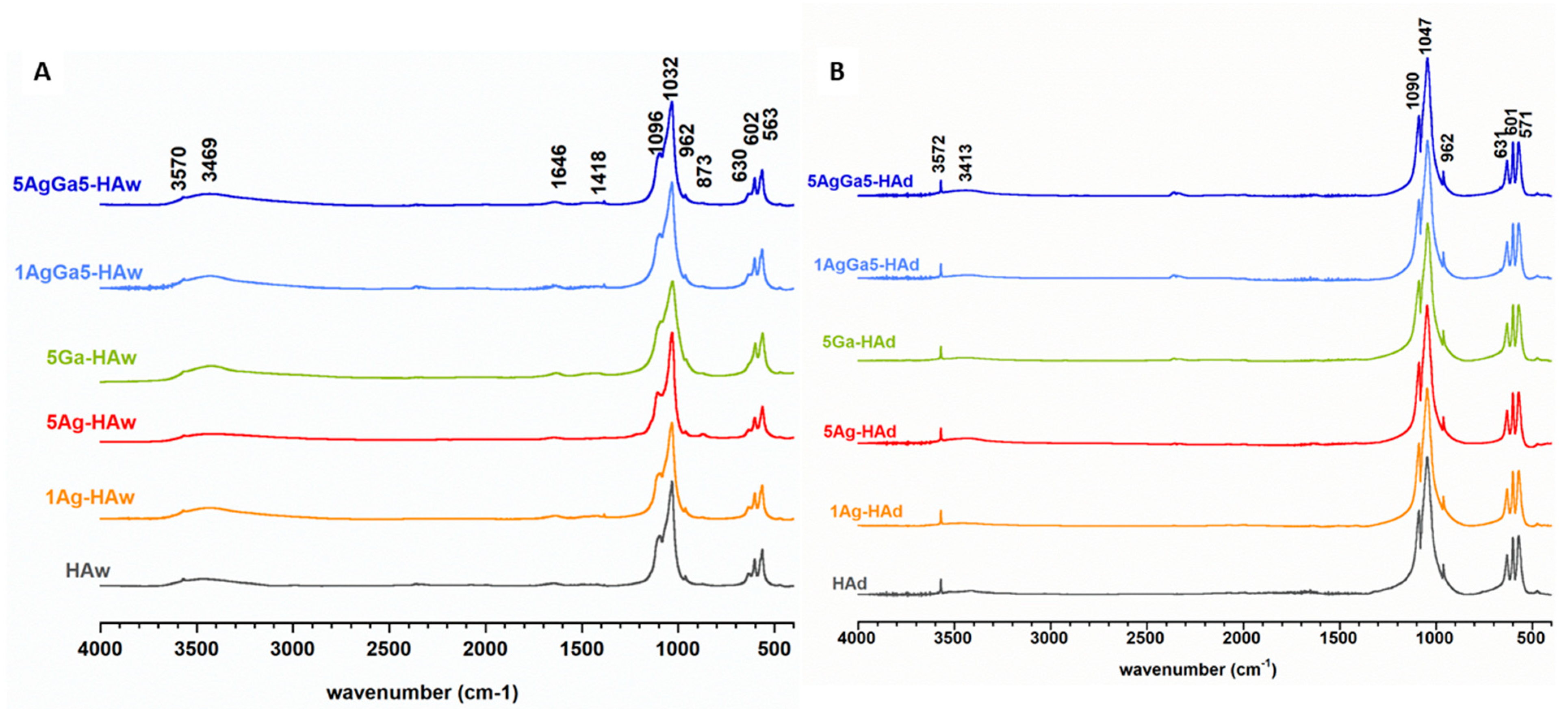
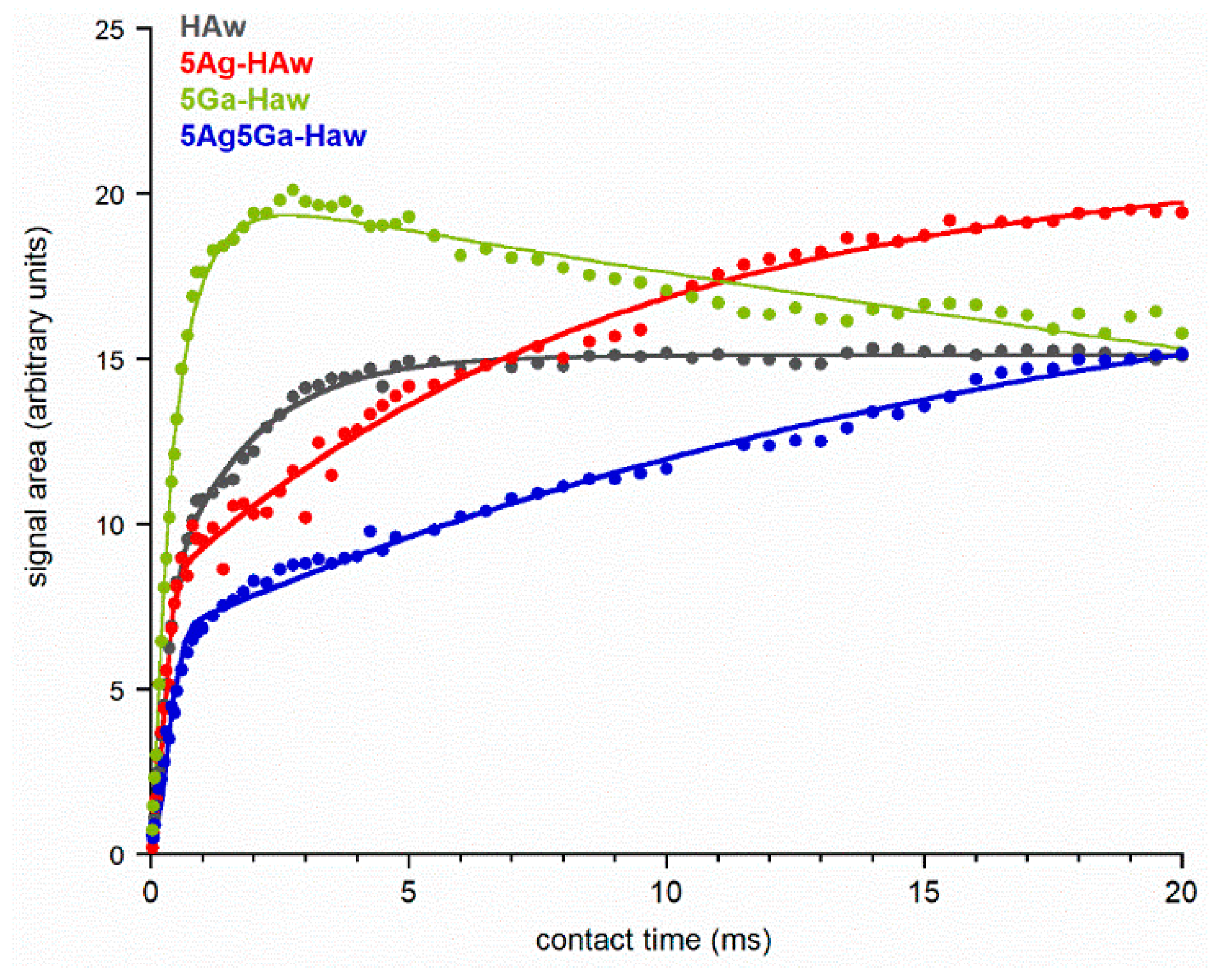
2.1. Physicochemical Properties of the Obtained Powders
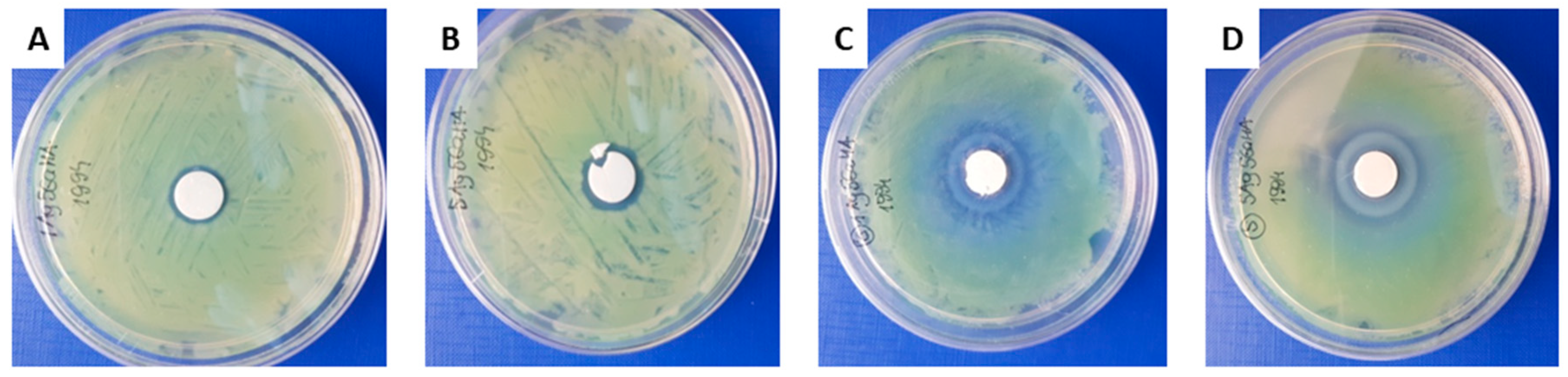
2.2. Biological Tests
3. Materials and Methods
3.1. Samples Preparation
- Pure, unsubstituted HA (HAw or HAd);
- Ag-containing HA (1Ag-HAw or 1Ag-HAd) with a nominal Ag amount of 0.11 mass% (0.01 mol);
- Ag-containing HA (5Ag-HAw or 5Ag-HAd) with a nominal Ag amount of 0.54 mass% (0.05 mol);
- Ga-containing HA (5Ga-HAw or 5Ga-HAd) with a nominal Ga amount of 0.35 mass% (0.05 mol);
- Ag and Ga containing HA (1Ag5Ga-HAw or 1Ag5Ga-HAd) with a nominal Ag and Ga amount of 0.11 mass% and 0.35 mass%, respectively (0.01 and 0.05 mol, respectively);
- Ag and Ga containing HA (5Ag5Ga-HAw or 5Ag5Ga-HAd) with a nominal Ag and Ga amount of 0.54 mass% and 0.35 mass%, respectively (0.05 mol).
3.2. Physicochemical Characterisation
3.3. Biological Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weiser, T.G.; Haynes, A.B.; Molina, G.; Lipsitz, S.R.; Esquivel, M.M.; Uribe-Leitz, T.; Fu, R.; Azad, T.; Chao, T.E.; Berry, W.R.; et al. Size and distribution of the global volume of surgery in 2012. Bull. World Health Organ. 2016, 94, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Walter, G.; Kemmerer, M.; Kappler, C.; Hoffmann, R. Treatment Algorithms for Chronic Osteomyelitis. Dtsch. Aerzteblatt Online 2012, 109, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.L.L.M.; Oliveira, P.; Carvalho, V.C.; Cimerman, S.; Savio, E. Recommendations for the treatment of osteomyelitis. Braz. J. Infect. Dis. 2014, 18, 526–534. [Google Scholar] [CrossRef] [Green Version]
- El-Husseiny, M.; Patel, S.; Macfarlane, R.J.; Haddad, F.S. Biodegradable antibiotic delivery systems. J. Bone Jt. Surgery. Br. Vol. 2011, 93, 151–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Global Guidelines for the Prevention of Surgical Site Infection; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- European Centre for Disease Prevention and Control. Healthcare-Associated Infections: Surgical Site Infections. Annual Epidemiological Report for 2017; ECDC: Stockholm, Sweden, 2019. [Google Scholar]
- Ling, M.L.; Apisarnthanarak, A.; Abbas, A.; Morikane, K.; Lee, K.Y.; Warrier, A.; Yamada, K. APSIC guidelines for the prevention of surgical site infections. Antimicrob. Resist. Infect. Control. 2019, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Goswami, K.; Stevenson, K.L.; Parvizi, J. Intraoperative and Postoperative Infection Prevention. J. Arthroplast. 2020, 35, S2–S8. [Google Scholar] [CrossRef] [Green Version]
- Tucci, G.; Romanini, E.; Zanoli, G.; Pavan, L.; Fantoni, M.; Venditti, M. Prevention of surgical site infections in orthopaedic surgery: A synthesis of current recommendations. Eur. Rev. Med Pharmacol. Sci. 2019, 23, 224–239. [Google Scholar]
- Trampuz, A.; Widmer, A.F. Infections associated with orthopedic implants. Curr. Opin. Infect. Dis. 2006, 19, 349–356. [Google Scholar] [CrossRef]
- Cortés-Penfield, N.W.; A Kulkarni, P. The History of Antibiotic Treatment of Osteomyelitis. Open Forum Infect. Dis. 2019, 6, ofz181. [Google Scholar] [CrossRef]
- Ter Boo, G.J.; Grijpma, D.; Moriarty, T.F.; Richards, R.G.; Eglin, D. Antimicrobial delivery systems for local infection prophylaxis in orthopedic- and trauma surgery. Biomaterial 2015, 52, 113–125. [Google Scholar] [CrossRef]
- Davat, M.; Wuarin, L.; Stafylakis, D.; Abbas, M.; Harbarth, S.; Hannouche, D.; Uckay, I. Should antibiotic prophylaxis before orthopedic implant surgery depend on the duration of pre-surgical hospital stay? Antimicrob. Resist. Infect. Control 2018, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Andersson, R.; Søreide, K.; Ansari, D. Surgical Infections and Antibiotic Stewardship: In Need for New Directions. Scand. J. Surg. 2019. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.G.; Mao, Z.G.; Liu, B.S.; Zhu, H.H.; Pan, H.L. Evaluating the use of antibiotic prophylaxis during open reduction and internal fixation surgery in patients at low risk of surgical site infection. Injury 2015, 46, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Verron, E.; Khairoun, I.; Guicheux, J.; Bouler, J.-M. Calcium phosphate biomaterials as bone drug delivery systems: A review. Drug Discov. Today 2010, 15, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Arcos, D.; Vallet-Regí, M. Bioceramics for drug delivery. Acta Mater. 2013, 61, 890–911. [Google Scholar] [CrossRef]
- Shirtliff, M.E.; Calhoun, J.H.; Mader, J.T. Experimental Osteomyelitis Treatment with Antibiotic-Impregnated Hydroxyapatite. Clin. Orthop. Relat. Res. 2002, 401, 239–247. [Google Scholar] [CrossRef]
- Dorozhkin, S. Bioceramics of calcium orthophosphates. Biomaterial 2010, 31, 1465–1485. [Google Scholar] [CrossRef]
- Samavedi, S.; Whittington, A.; Goldstein, A.S. Calcium phosphate ceramics in bone tissue engineering: A review of properties and their influence on cell behavior. Acta Biomater. 2013, 9, 8037–8045. [Google Scholar] [CrossRef]
- Koutsopoulos, S. Synthesis and characterization of hydroxyapatite crystals: A review study on the analytical methods. J. Biomed. Mater. Res. 2002, 62, 600–612. [Google Scholar] [CrossRef]
- Eliaz, N.; Metoki, N. Calcium Phosphate Bioceramics: A Review of Their History, Structure, Properties, Coating Technologies and Biomedical Applications. Materials 2017, 10, 334. [Google Scholar] [CrossRef] [Green Version]
- Mijnendonckx, K.; Leys, N.; Mahillon, J.; Silver, S.; Van Houdt, R. Antimicrobial silver: Uses, toxicity and potential for resistance. BioMetals 2013, 26, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Kolmas, J.; Groszyk, E.; Kwiatkowska-Różycka, D. Substituted Hydroxyapatites with Antibacterial Properties. BioMed Res. Int. 2014, 2014, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, N.A.; Oldinski, R.A.; Ma, H.; Bryers, J.D.; Williams, J.D.; Popat, K.C. Antibacterial effects of silver-doped hydroxyapatite thin films sputter deposited on titanium. Mater. Sci. Eng. C 2012, 32, 2135–2144. [Google Scholar] [CrossRef]
- Chitambar, C.R. Medical Applications and Toxicities of Gallium Compounds. Int. J. Environ. Res. Public Health 2010, 7, 2337–2361. [Google Scholar] [CrossRef] [Green Version]
- Melnikov, P.; Matos, M.D.F.C.; Malzac, A.; Teixeira, A.R.; De Albuquerque, D.M. Evaluation of in vitro toxicity of hydroxyapatite doped with gallium. Mater. Lett. 2019, 253, 343–345. [Google Scholar] [CrossRef]
- Nayak, B.; Misra, P.K. Exploration of the structural and dielectric characteristics of a potent hydroxyapatite coated gallium bioceramics for the forthcoming biomedical and orthopedic applications. Mater. Chem. Phys. 2020, 239, 121967. [Google Scholar] [CrossRef]
- Cassino, P.C.; Rosseti, L.S.; Ayala, O.I.; Martines, M.A.U.; Portugual, L.C.; De Oliveira, C.G.; Silva, I.S.; Caldas, R.D.A. Potencial of different hydroxyapatites as biomaterials in the bone remodeling. Acta Cir. Bras. 2018, 33, 816–823. [Google Scholar] [CrossRef]
- Szurkowska, K.; Drobniewska, A.; Kolmas, J. Dual Doping of Silicon and Manganese in Hydroxyapatites: Physicochemical Properties and Preliminary Biological Studies. Materials 2019, 12, 2566. [Google Scholar] [CrossRef] [Green Version]
- Stanić, V.; Janaćković, D.; Dimitrijević, S.; Tanasković, S.B.; Mitrić, M.; Pavlović, M.S.; Krstić, A.; Jovanović, D.; Raičević, S.; Dimitrijević-Branković, S.I.; et al. Synthesis of antimicrobial monophase silver-doped hydroxyapatite nanopowders for bone tissue engineering. Appl. Surf. Sci. 2011, 257, 4510–4518. [Google Scholar] [CrossRef]
- Kurtjak, M.; Vukomanovic, M.; Krajnc, A.; Kramer, L.; Suvorov, D.; Turk, B. Designing Ga( iii )-containing hydroxyapatite with antibacterial activity. RSC Adv. 2016, 6, 112839–112852. [Google Scholar] [CrossRef]
- Venkateswarlu, K.; Bose, A.C.; Rameshbabu, N. X-ray peak broadening studies of nanocrystalline hydroxyapatite by Williamson–Hall analysis. Phys. B Condens. Matter 2010, 405, 4256–4261. [Google Scholar] [CrossRef]
- Landi, E.; Tampieri, A.; Celotti, G.; Sprio, S. Densification behaviour and mechanisms of synthetic hydroxyapatites. J. Eur. Ceram. Soc. 2000, 20, 2377–2387. [Google Scholar] [CrossRef]
- Celotti, G.; Tampieri, A.; Sprio, S.; Landi, E.; Bertinetti, L.; Martra, G.; Ducati, C. Crystallinity in apatites: How can a truly disordered fraction be distinguished from nanosize crystalline domains? J. Mater. Sci. Mater. Electron. 2006, 17, 1079–1087. [Google Scholar] [CrossRef]
- Okada, M.; Furuzono, T. Hydroxylapatite nanoparticles: Fabrication methods and medical applications. Sci. Technol. Adv. Mater. 2012, 13, 064103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolmas, J.; Kalinowski, E.; Wojtowicz, A.; Kolodziejski, W. Mid-infrared reflectance microspectroscopy of human molars: Chemical comparison of the dentin–enamel junction with the adjacent tissues. J. Mol. Struct. 2010, 966, 113–121. [Google Scholar] [CrossRef]
- Geng, Z.; Wang, R.; Zhuo, X.; Li, Z.; Huang, Y.; Ma, L.; Cui, Z.; Zhu, S.; Liang, Y.; Liu, Y.; et al. Incorporation of silver and strontium in hydroxyapatite coating on titanium surface for enhanced antibacterial and biological properties. Mater. Sci. Eng. C 2017, 71, 852–861. [Google Scholar] [CrossRef]
- Gopi, D.; Shinyjoy, E.; Kavitha, L. Synthesis and spectral characterization of silver/magnesium co-substituted hydroxyapatite for biomedical applications. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 127, 286–291. [Google Scholar] [CrossRef]
- Kaflak, A.; Kolodziejski, W. Complementary information on water and hydroxyl groups in nanocrystalline carbonated hydroxyapatites from TGA, NMR and IR measurements. J. Mol. Struct. 2011, 990, 263–270. [Google Scholar] [CrossRef]
- Wopenka, B.; Pasteris, J.D. A mineralogical perspective on the apatite in bone. Mater. Sci. Eng. C 2005, 25, 131–143. [Google Scholar] [CrossRef]
- Rehman, I.; Bonfield, W. Characterization of hydroxyapatite and carbonated apatite by photo acoustic FTIR spectroscopy. J. Mater. Sci. Mater. Electron. 1997, 8, 1–4. [Google Scholar] [CrossRef]
- Pajchel, L.; Kolodziejski, W. Solid-state MAS NMR, TEM, and TGA studies of structural hydroxyl groups and water in nanocrystalline apatites prepared by dry milling. J. Nanopart. Res. 2013, 15, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellier, C.; Fayon, F.; Boukhechba, F.; Verron, E.; LeFerrec, M.; Montavon, G.; Lesoeur, J.; Schnitzler, V.; Massiot, D.; Janvier, P.; et al. Design and properties of novel gallium-doped injectable apatitic cements. Acta Biomater. 2015, 24, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Tite, T.; Popa, A.-C.; Balescu, L.M.; Bogdan, I.M.; Pasuk, I.; Ferreira, J.M.F.; Stan, G. Cationic Substitutions in Hydroxyapatite: Current Status of the Derived Biofunctional Effects and Their In Vitro Interrogation Methods. Materials 2018, 11, 2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolmas, J.; Jaklewicz, A.; Zima, A.; Bućko, M.; Paszkiewicz, Z.; Lis, J.; Ślósarczyk, A.; Kolodziejski, W. Incorporation of carbonate and magnesium ions into synthetic hydroxyapatite: The effect on physicochemical properties. J. Mol. Struct. 2011, 987, 40–50. [Google Scholar] [CrossRef]
- Mellier, C.; Fayon, F.; Schnitzler, V.; Deniard, P.; Allix, M.; Quillard, S.; Massiot, D.; Bouler, J.-M.; Bujoli, B.; Janvier, P. Characterization and Properties of Novel Gallium-Doped Calcium Phosphate Ceramics. Inorg. Chem. 2011, 50, 8252–8260. [Google Scholar] [CrossRef]
- Kolmas, J.; Kuras, M.J.; Oledzka, E.; Sobczak, M. A Solid-State NMR Study of Selenium Substitution into Nanocrystalline Hydroxyapatite. Int. J. Mol. Sci. 2015, 16, 11452–11464. [Google Scholar] [CrossRef] [Green Version]
- Kolodziejski, W. Solid-state NMR of bone. Top. Curr. Chem. 2005, 246, 235–270. [Google Scholar]
- Singh, B.; Dubey, A.K.; Kumar, S.; Saha, N.; Basu, B.; Gupta, R. In vitro biocompatibility and antimicrobial activity of wet chemically prepared Ca10− xAgx (PO4) 6 (OH) 2 (0.0 ≤ x ≤ 0.5) hydroxyapatites. Mater. Sci. Eng. C 2011, 31, 1320–1329. [Google Scholar] [CrossRef]
- Shi, C.; Gao, J.; Wang, M.; Fu, J.; Wang, D.; Zhu, Y. Ultra-trace silver-doped hydroxyapatite with non-cytotoxicity and effective antibacterial activity. Mater. Sci. Eng. C 2015, 55, 497–505. [Google Scholar] [CrossRef]
- Lim, P.N.; Chang, L.; Thian, E.S. Development of nanosized silver-substituted apatite for biomedical applications: A review. Nanomed. Nanotechnol. Boil. Med. 2015, 11, 1331–1344. [Google Scholar] [CrossRef]
- Trapet, P.; Avoscan, L.; Klinguer, A.; Pateyron, S.; Citerne, S.; Chervin, C.; Mazurier, S.; Lemanceau, P.; Wendehenne, D.; Besson-Bard, A. The Pseudomonas fluorescens Siderophore Pyoverdine Weakens Arabidopsis thaliana Defense in Favor of Growth in Iron-Deficient Conditions1. Plant Physiol. 2016, 171, 675–693. [Google Scholar] [CrossRef] [Green Version]
- Biaggini, K.; Barbey, C.; Borrel, V.; Feuilloley, M.G.J.; Tavolacci, M.-P.; Connil, N. The pathogenic potential of Pseudomonas fluorescens MFN1032 on enterocytes can be modulated by serotonin, substance P and epinephrine. Arch. Microbiol. 2015, 197, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Arciola, C.R.; An, Y.; Campoccia, D.; Donati, M.B.; Montanaro, L. Etiology of Implant Orthopedic Infections: A Survey on 1027 Clinical Isolates. Int. J. Artif. Organs 2005, 28, 1091–1100. [Google Scholar] [CrossRef]
- Kolmas, J.; Piotrowska, U.; Kuras, M.J.; Kurek, E. Effect of carbonate substitution on physicochemical and biological properties of silver containing hydroxyapatites. Mater. Sci. Eng. C 2017, 74, 124–130. [Google Scholar] [CrossRef]
- Feeney, R.E.; Petersen, I.M.; Sahinkaya, H. Liesegang-like rings of growth and inhibition of bacteria in agar caused by metal ions and chelating agents, 12. J. Bacteriol. 1957, 73, 284–290. [Google Scholar] [CrossRef] [Green Version]
- ISO 10993-5:2009 Biological Evaluation of Medical Devices—Part 5: Tests for In Vitro Cytotoxicity; International Organization for Standardization: Geneva, Switzerland, 2009.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ag Content
mol (mass%) | Efficiency (%) | Ga Content mol (mass%) | Efficiency (%) |
[Ca+(Ag)+(Ga)]/P Molar Ratio | |
|---|---|---|---|---|---|
| HAw | - | - | - | - | 1.65 ± 0.03 |
| 1Ag-HAw | 0.0056 (0.060) | 56 | - | - | 1.67 ± 0.03 |
| 5Ag-HAw | 0.0460 (0.460) | 92 | - | - | 1.63 ± 0.03 |
| 5Ga-HAw | - | - | 0.047 (0.33) | 94 | 1.59 ± 0.03 |
| 1Ag5Ga5-HAw | 0.0037 (0.04) | 37 | 0.046 (0.32) | 92 | 1.64 ± 0.04 |
| 5Ag5Ga-HAw | 0.0150 (0.16) | 30 | 0.046 (0.32) | 92 | 1.66 ± 0.02 |
| HAd | - | - | - | - | 1.70 ± 0.02 |
| 1Ag-HAd | 0.00093 (0.01) | 9.3 | - | - | 1.85 ± 0.02 |
| 5Ag-HAd | 0.0360 (0.39) | 72 | - | - | 1.83 ± 0.03 |
| 5Ga-HAd | - | - | 0.047 (0.33) | 94 | 1.88 ± 0.04 |
| 1Ag5Ga-HAd | 0.00093 (0.01) | 9.3 | 0.044 (0.31) | 88 | 1.87 ± 0.03 |
| 5Ag5Ga-HAd | 0.031 (0.33) | 62 | 0.044 (0.31) | 99 | 1.78 ± 0.03 |
| Lattice Parameters (Å) * | Crystal Size (nm) | Crystallinity Degree | |||
|---|---|---|---|---|---|
| a-axis | c-axis | (002) | (300) | ||
| HAw | 9.4113 | 6.8700 | 36 ± 2 | 14 ± 2 | 0.54 ± 0.03 |
| 1Ag-HAw | 9.4309 | 6.8784 | 34 ± 3 | 10 ± 3 | 0.44 ± 0.05 |
| 5Ag-HAw | 9.4271 | 6.8783 | 30 ± 3 | 7 ± 2 | 0.46 ± 0.04 |
| 5Ga-HAw | 9.4222 | 6.8765 | 26 ± 2 | 7 ± 2 | 0.33 ± 0.04 |
| 1Ag5Ga5-HAw | 9.4222 | 6.8765 | 29 ± 2 | 8 ± 2 | 0.40 ± 0.03 |
| 5Ag5Ga-HAw | 9.4152 | 6.8782 | 24 ± 2 | 8 ± 3 | 0.37 ± 0.02 |
| HAd | 9.4112 | 6.8754 | - | - | 0.98 ± 0.02 |
| 1Ag-HAd | 9.4113 | 6.8754 | - | - | 0.97 ± 0.03 |
| 5Ag-HAd | 9.4203 | 6.8809 | - | - | 0.95 ± 0.04 |
| 5Ga-HAd | 9.3991 | 6.8677 | - | - | 0.90 ± 0.02 |
| 1Ag5Ga-HAd | 9.4146 | 6.8777 | - | - | 0.94 ± 0.02 |
| 5Ag5Ga-HAd | 9.4152 | 6.8782 | - | - | 0.92 ± 0.03 |
| HAw | 5Ag-HAw | 5Ga-HAw | 5Ag5Ga-HAw | ||
|---|---|---|---|---|---|
| PARAMETERS | I0 | 15.1 ± 0.1 | 21.1 ± 0.4 | 20.3 ± 0.2 | 19.2 ± 0.5 |
| λ | 0.55 ± 0.02 | 0.63 ± 0.01 | 0.70 ± 0.05 | 0.67 ± 0.02 | |
| Tdf (ms) | 1.64 ± 0.04 | 8.8 ± 0.7 | 0.58 ± 0.03 | 18 ± 2 | |
| T1ρH (ms) | ∞ | ∞ | 71 ± 4 | ∞ | |
| T2 (ms) | 0.27 ± 0.01 | 0.21 ±0.01 | 0.29 ± 0.03 | 0.29 ± 0.01 |
| Wet Method | Cytotoxicity | Dry Method | Cytotoxicity |
|---|---|---|---|
| HAw | N | HAd | N |
| 1Ag-HAw | N | 1Ag-HAd | N |
| 5Ag-HAw | C 1 | 5Ag-HAd | N |
| 5Ga-HAw | N | 5Ga-HAd | C 2 |
| 1Ag5Ga5-HAw | N | 1Ag5Ga-HAd | C 3 |
| 5Ag5Ga-HAw | N | 5Ag5Ga-HAd | C 4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pajor, K.; Pajchel, Ł.; Zgadzaj, A.; Piotrowska, U.; Kolmas, J. Modifications of Hydroxyapatite by Gallium and Silver Ions—Physicochemical Characterization, Cytotoxicity and Antibacterial Evaluation. Int. J. Mol. Sci. 2020, 21, 5006. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145006
Pajor K, Pajchel Ł, Zgadzaj A, Piotrowska U, Kolmas J. Modifications of Hydroxyapatite by Gallium and Silver Ions—Physicochemical Characterization, Cytotoxicity and Antibacterial Evaluation. International Journal of Molecular Sciences. 2020; 21(14):5006. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145006
Chicago/Turabian StylePajor, Kamil, Łukasz Pajchel, Anna Zgadzaj, Urszula Piotrowska, and Joanna Kolmas. 2020. "Modifications of Hydroxyapatite by Gallium and Silver Ions—Physicochemical Characterization, Cytotoxicity and Antibacterial Evaluation" International Journal of Molecular Sciences 21, no. 14: 5006. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21145006