Molecular Targets, Pathways, and Therapeutic Implications for Hepatocellular Carcinoma


Abstract
:1. Introduction
2. Current Molecular Targets of Approved Systemic Agents
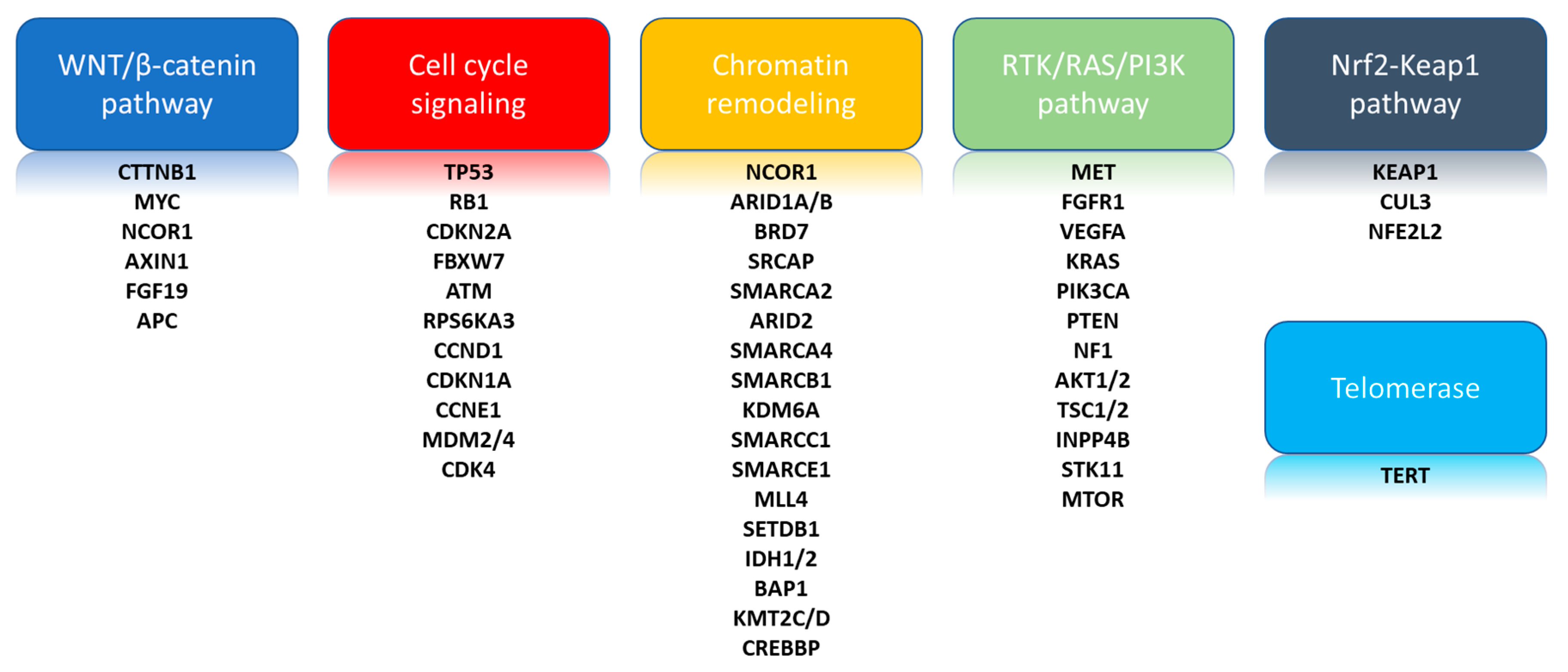
3. Landmark Comprehensive Genomic Analyses
3.1. Telomerase Reverse Transcriptase (TERT)
3.2. Cell Cycle Signaling
3.3. Chromatin Remodeling
3.4. WNT/β-Catenin Cell Signaling Pathway
3.5. Phosphatidylinositol-3-Kinase (PI3K)/AKT/Mammalian Target of Rapamycin (mTOR)
3.6. RAS/Mitogen-Activated Protein Kinase (MAPK) Pathway
3.7. Nuclear Factor-Like 2 (NFE2L2)/KEAP1 Pathway
3.8. Additional Biomarkers of Interest
4. Novel Druggable Targets
4.1. Fibroblast Growth Factors (FGFs)
4.2. c-KIT
4.3. JAK-STAT Cell Signaling Pathway
4.4. Epidermal Growth Factor Receptors (Egfrs)
4.5. c-MET
4.6. Isocitrate Dehydrogenase (IDH)
4.7. ATM
4.8. P13K/AKT/mTOR Signaling Pathway
5. Future Directions
5.1. Transcriptomics
5.2. Genomics
5.3. Integrated Transcriptomics, Genomics, and Proteomics
5.4. Molecular Heterogeneity
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HCC | Hepatocellular carcinoma |
| BCLC | Barcelona-Clínic Liver Cancer |
| VEGF | Vascular endothelial growth factor |
| EpCAM | Epithelial cell adhesion molecule |
| AFP | Alpha-fetoprotein |
| WNT | Wingless-related integration site |
| CNV | Copy number variation |
| HBV | Hepatitis B Virus |
| HCV | Hepatitis C Virus |
| NAFLD | Nonalcoholic fatty liver disease |
| OS | Median overall survival |
| RFA | Radiofrequency Ablation |
| TACE | Transarterial chemoembolization |
| MET | Metabolic equivalent of task |
| RAF | Rapidly Accelerated Fibrosarcoma |
| PDGFR | Platelet-derived growth factor |
| PD-1 | Programmed cell death protein 1 |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| RET | Rearranged during transfection |
| AST | Aspartate aminotransferase |
| ALT | Alanine transaminase |
| FMS | Fibromyalgia syndrome |
| GAS6 | Growth arrest-specific 6 |
| PFS | Progression-free survival |
| HR | Hazard ratio |
| CI | Confidence interval |
| ORR | Overall response rate |
| TCGA | The Cancer Genome Atlas |
| KMT2D | Histone-lysine N-methyltransferase 2D |
| CREBBP | cAMP response element-binding protein |
| PI3K | Phosphatidylinasitol-3-kinase |
| mTOR | Mammalian target of rapamycin |
| PTEN | Phosphatase and tensin homolog |
| NFE2L2 | Nuclear factor-like 2 |
| FGF | Fibroblast growth factors |
| TACE | Transcatheter arterial chemoembolism |
| HGF | Hepatic growth factor |
| CDKN2A | Cyclin Dependent Kinase Inhibitor 2A |
| TP53 | Tumor Protein 53 |
| MLL4 | Mixed Lineage Leukemia 4 |
| ARID1A | AT-Rich Interaction Domain 1A |
| ARID2 | AT-Rich Interaction Domain 2 |
| TERT | Telomerase reverse transcriptase |
| EGFR | Epidermal growth factor receptor |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16019. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver, European Organisation for Research and Treatment of Cancer. EASL-EORTC clinical practice guidelines: Management of hepatocellular carcinoma. Eur. J. Cancer 2012, 48, 599–641. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.M.; Hilgard, P.; Gane, E.; Blanc, J.-F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.C.C.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.; Finn, R.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.-H.; Harding, J.; Merle, P.; et al. CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2019, 30, v874–v875. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab as Second-Line Therapy in Patients with Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J. Clin. Oncol. 2019, 37, 4012. [Google Scholar] [CrossRef]
- Hung, M.H.; Wang, X.W. Molecular Alterations and Heterogeneity in Hepatocellular Carcinoma. In Hepatocellular Carcinoma: Translational Precision Medicine Approaches; Hoshida, Y., Ed.; Humana Press: Cham, Switzerland, 2019; pp. 293–316. [Google Scholar]
- West, C.A.; Black, A.P.; Mehta, A.S. Analysis of Hepatocellular Carcinoma Tissue for Biomarker Discovery. In Hepatocellular Carcinoma: Translational Precision Medicine Approaches; Hoshida, Y., Ed.; Humana Press: Cham, Switzerland, 2019; pp. 93–107. [Google Scholar]
- Whittaker, S.R.; Marais, R.; Zhu, A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010, 29, 4989–5005. [Google Scholar] [CrossRef]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; A Donehower, L.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2018, 25, 2116–2126. [Google Scholar] [CrossRef] [Green Version]
- Shampay, J.; Blackburn, E.H. Generation of telomere-length heterogeneity in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1988, 85, 534–538. [Google Scholar] [CrossRef] [Green Version]
- Amisaki, M.; Tsuchiya, H.; Sakabe, T.; Fujiwara, Y.; Shiota, G. Identification of genes involved in the regulation of TERT in hepatocellular carcinoma. Cancer Sci. 2019, 110, 550–560. [Google Scholar] [CrossRef]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.M.; Lee, D.; Ma, Y.; et al. Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Lin, H.; Zhang, R.; Wu, W.; Lei, L.; Cui, X.; Wei, C.; Tong, G.; Xing, Y. Comprehensive network analysis of the molecular mechanisms associated with sorafenib resistance in hepatocellular carcinoma. Cancer Genet. 2020, 245, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.; Wu, Y. Knockdown of miR-106a suppresses migration and invasion and enhances radiosensitivity of hepatocellular carcinoma cells by upregulating FBXW7. Int. J. Clin. Exp. Pathol. 2019, 12, 1184–1193. [Google Scholar] [PubMed]
- Ellis, L.; Atadja, P.W.; Johnstone, R.W. Epigenetics in cancer: Targeting chromatin modifications. Mol. Cancer Ther. 2009, 8, 1409–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, T.; Aburatani, H. Exploration of liver cancer genomes. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 340–349. [Google Scholar] [CrossRef]
- Jiang, T.; Chen, X.; Su, C.; Ren, S.; Zhou, C. Pan-cancer analysis of ARID1A Alterations as Biomarkers for Immunotherapy Outcomes. J. Cancer 2020, 11, 776–780. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Li, W.; Tian, F.; Jiang, K.; Liu, X.; Cen, J.; He, Q.; Qiu, Z.; Kienast, Y.; Wang, Z.; et al. Arid1a regulates response to anti-angiogenic therapy in advanced hepatocellular carcinoma. J. Hepatol. 2018, 68, 465–475. [Google Scholar] [CrossRef]
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular Function of Phosphoinositide 3-Kinases: Implications for Development, Immunity, Homeostasis, and Cancer. Ann. Rev. Cell Dev. Boil. 2001, 17, 615–675. [Google Scholar] [CrossRef]
- Carracedo, A.; Pandolfi, P.P. The PTEN–PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef] [Green Version]
- Delire, B.; Stärkel, P. The Ras/MAPK pathway and hepatocarcinoma: Pathogenesis and therapeutic implications. Eur. J. Clin. Investig. 2015, 45, 609–623. [Google Scholar] [CrossRef]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [Green Version]
- Sung, W.-K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Hoshi, T.; Miyano, S.W.; Watanabe, H.; Sonobe, R.M.K.; Seki, Y.; Ohta, E.; Nomoto, K.; Matsui, J.; Funahashi, Y. Lenvatinib induces death of human hepatocellular carcinoma cells harboring an activated FGF signaling pathway through inhibition of FGFR–MAPK cascades. Biochem. Biophys. Res. Commun. 2019, 513, 1–7. [Google Scholar] [CrossRef]
- Kudo, M.; Cheng, A.-L.; Park, J.; Park, J.H.; Liang, P.-C.; Hidaka, H.; Izumi, N.; Heo, J.; Lee, Y.J.; Sheen, I.-S.; et al. Orantinib versus placebo combined with transcatheter arterial chemoembolisation in patients with unresectable hepatocellular carcinoma (ORIENTAL): A randomised, double-blind, placebo-controlled, multicentre, phase 3 study. Lancet Gastroenterol. Hepatol. 2018, 3, 37–46. [Google Scholar] [CrossRef]
- Johnson, P.; Qin, S.; Park, J.; Poon, R.T.; Raoul, J.-L.; Philip, P.A.; Hsu, C.-H.; Hu, T.; Heo, J.; Xu, J.; et al. Brivanib Versus Sorafenib as First-Line Therapy in Patients With Unresectable, Advanced Hepatocellular Carcinoma: Results From the Randomized Phase III BRISK-FL Study. J. Clin. Oncol. 2013, 31, 3517–3524. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Decaens, T.; Raoul, J.L.; Boucher, E.; Kudo, M.; Chang, C.; Kang, Y.-K.; Assenat, E.; Lim, H.Y.; Boige, V.; et al. Brivanib in Patients with Advanced Hepatocellular Carcinoma Who Were Intolerant to Sorafenib or for Whom Sorafenib Failed: Results From the Randomized Phase III BRISK-PS Study. J. Clin. Oncol. 2013, 31, 3509–3516. [Google Scholar] [CrossRef]
- Mercade, T.M.; Moreno, V.; John, B.; Morris, J.C.; Sawyer, M.B.; Yong, W.P.; Gutierrez, M.; Karasic, T.B.; Sangro, B.; Sheng-Shun, Y.; et al. A phase I study of H3B-6527 in hepatocellular carcinoma (HCC) or intrahepatic cholangiocarcinoma (ICC) patients (pts). J. Clin. Oncol. 2019, 37, 4095. [Google Scholar] [CrossRef]
- Kim, R.D.; Sarker, D.; Meyer, T.; Yau, T.; Macarulla, T.; Park, J.-W.; Choo, S.P.; Hollebecque, A.; Sung, M.W.; Lim, H.-Y.; et al. First-in-Human Phase I Study of Fisogatinib (BLU-554) Validates Aberrant FGF19 Signaling as a Driver Event in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1696–1707. [Google Scholar] [CrossRef] [Green Version]
- Ciprotti, M.; Abraham, R.; Jansen, M.; Tokuhiro, S.; Hanai, M.; Oitate, M.; Shuster, D.E.; Martinez, A. A phase I, open label, two part, safety and tolerability study of U3-1784 in patients with advanced solid tumours. J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Becker, G.; Schmitt-Graeff, A.; Ertelt, V.; Blum, H.; Allgaier, H.-P. CD117 (c-kit) Expression in Human Hepatocellular Carcinoma. Clin. Oncol. 2007, 19, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.Y.; Fisher, G.A.; So, S.; Tang, C.; Levitt, L. Phase II Study of Imatinib in Unresectable Hepatocellular Carcinoma. Am. J. Clin. Oncol. 2008, 31, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.C.C.; Chan, P.; Curtis, C.M.; Murphy, P.S.; Suttle, A.B.; Gauvin, J.; Hodge, J.P.; Dar, M.M.; Poon, R.T.; Chen, D.-S. Phase I Dose-Finding Study of Pazopanib in Hepatocellular Carcinoma: Evaluation of Early Efficacy, Pharmacokinetics, and Pharmacodynamics. Clin. Cancer Res. 2011, 17, 6914–6923. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.-L.; Kang, Y.-K.; Lin, D.-Y.; Park, J.; Kudo, M.; Qin, S.; Chung, H.C.; Song, X.; Xu, J.; Poggi, G.; et al. Sunitinib Versus Sorafenib in Advanced Hepatocellular Cancer: Results of a Randomized Phase III Trial. J. Clin. Oncol. 2013, 31, 4067–4075. [Google Scholar] [CrossRef]
- Del Campo, S.E.M.; Levine, K.M.; Mundy-Bosse, B.L.; Grignol, V.P.; Fairchild, E.T.; Campbell, A.R.; Trikha, P.; Mace, T.A.; Paul, B.K.; Jaime-Ramirez, A.C.; et al. The Raf Kinase Inhibitor Sorafenib Inhibits JAK-STAT Signal Transduction in Human Immune Cells. J. Immunol. 2015, 195, 1995–2005. [Google Scholar] [CrossRef]
- He, L.; Deng, H.; Lei, J.; Yi, F.; Li, J.; Fan, X.; Zhang, W.; Xu, J.; Zhang, W. Efficacy of bevacizumab combined with erlotinib for advanced hepatocellular carcinoma: A single-arm meta-analysis based on prospective studies. BMC Cancer 2019, 19, 276. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.X.; Stuart, K.; Blaszkowsky, L.S.; Muzikansky, A.; Reitberg, D.P.; Clark, J.W.; Enzinger, P.C.; Bhargava, P.; Meyerhardt, J.A.; Horgan, K.; et al. Phase 2 study of cetuximab in patients with advanced hepatocellular carcinoma. Cancer 2007, 110, 581–589. [Google Scholar] [CrossRef]
- Goyal, L.; Wadlow, R.C.; Blaszkowsky, L.S.; Wolpin, B.M.; Abrams, T.A.; McCleary, N.J.; Sheehan, S.; Sundaram, E.; Karol, M.D.; Chen, J.; et al. A phase I and pharmacokinetic study of ganetespib (STA-9090) in advanced hepatocellular carcinoma. Investig. New Drugs 2014, 33, 128–137. [Google Scholar] [CrossRef]
- Hsu, C.; Yang, T.-S.; Huo, T.-I.; Hsieh, R.-K.; Yu, C.-W.; Hwang, W.-S.; Hsieh, T.-Y.; Huang, W.-T.; Chao, Y.; Meng, R.; et al. Vandetanib in patients with inoperable hepatocellular carcinoma: A phase II, randomized, double-blind, placebo-controlled study. J. Hepatol. 2012, 56, 1097–1103. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.S.; Markowitz, J.; Prescott, N.; Sadee, W.; Heerema, N.; Wei, L.; Dai, Z.; Papp, A.; Campbell, A.; Culler, K.; et al. A multi-institutional phase II study of the efficacy and tolerability of lapatinib in patients with advanced hepatocellular carcinomas. Clin. Cancer Res. 2009, 15, 5895–5901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yau, T.; Lencioni, R.; Sukeepaisarnjaroen, W.; Chao, Y.; Yen, C.-J.; Lausoontornsiri, W.; Chen, P.-J.; Sanpajit, T.; Camp, A.; Cox, D.S.; et al. A Phase I/II Multicenter Study of Single-Agent Foretinib as First-Line Therapy in Patients with Advanced Hepatocellular Carcinoma. Clin. Cancer Res. 2016, 23, 2405–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Chan, S.L.; Sukeepaisarnjaroen, W.; Han, G.; Choo, S.P.; Sriuranpong, V.; Pan, H.; Yau, T.; Guo, Y.; Chen, M.; et al. A phase II study of the efficacy and safety of the MET inhibitor capmatinib (INC280) in patients with advanced hepatocellular carcinoma. Ther. Adv. Med Oncol. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Decaens, T.; Barone, C.; Assenat, E.; Wermke, M.; Fasolo, A.; Merle, P.; Blanc, J.-F.; Grando, V.; Bruns, R.; Straub, J.; et al. Efficacy and safety of the Met inhibitor tepotinib in patients (pts) with advanced Met+ hepatocellular carcinoma (HCC) previously treated with sorafenib. Ann. Oncol. 2018, 29, ix48. [Google Scholar] [CrossRef]
- Rimassa, L.; Assenat, E.; Peck-Radosavljevic, M.; Pracht, M.; Zagonel, V.; Mathurin, P.; Caremoli, E.R.; Porta, C.; Daniele, B.; Bolondi, L.; et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): A final analysis of a phase 3, randomised, placebo-controlled study. Lancet Oncol. 2018, 19, 682–693. [Google Scholar] [CrossRef]
- O’Neil, B.H.; Bendell, J.C.; Modiano, M.R.; Machiels, J.-P.H.; Versola, M.J.; Hodge, J.P.; Sawarna, K.; Tse, N. Phase I/II study of E7050 (golvantinib) in combination with sorafenib in patients (pts) with advanced hepatocellular carcinoma (HCC): Phase I results. J. Clin. Oncol. 2013, 31, 294. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Meng, L.; Ji, B.; Yang, D. Synergistic Antitumor Effect of Sorafenib in Combination with ATM Inhibitor in Hepatocellular Carcinoma Cells. Int. J. Med. Sci. 2017, 14, 523–529. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-Y.; Hsieh, F.-S.; Wang, C.-Y.; Chen, L.-J.; Chang, S.-S.; Tsai, M.-H.; Hung, M.-H.; Kuo, C.-W.; Shih, C.-T.; Chao, T.-I.; et al. Palbociclib enhances radiosensitivity of hepatocellular carcinoma and cholangiocarcinoma via inhibiting ataxia telangiectasia–mutated kinase–mediated DNA damage response. Eur. J. Cancer 2018, 102, 10–22. [Google Scholar] [CrossRef]
- Singh, A.R.; Joshi, S.; Burgoyne, A.M.; Sicklick, J.K.; Ikeda, S.; Kono, Y.; Garlich, J.R.; Morales, G.A.; Durden, D.L. Single Agent and Synergistic Activity of the “First-in-Class” Dual PI3K/BRD4 Inhibitor SF1126 with Sorafenib in Hepatocellular Carcinoma. Mol. Cancer Ther. 2016, 15, 2553–2562. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, D.; Chiorean, E.; Harris, W.; Von Hoff, D.; Stejskal-Barnett, A.; Qi, W.; Anthony, S.; Younger, A.; Rensvold, D.; Córdova, F.; et al. Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. Eur. J. Cancer 2012, 48, 3319–3327. [Google Scholar] [CrossRef] [Green Version]
- Ewald, F.; Nörz, D.; Grottke, A.; Bach, J.; Herzberger, C.; Hofmann, B.T.; Nashan, B.; Jücker, M. Vertical Targeting of AKT and mTOR as Well as Dual Targeting of AKT and MEK Signaling Is Synergistic in Hepatocellular Carcinoma. J. Cancer 2015, 6, 1195–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.-N.; Chen, H.; Liu, T.-T.; Wu, J.; Zhu, J.; Shen, X. Targeting the mTOR regulatory network in hepatocellular carcinoma: Are we making headway? Biochim. Biophys. Acta (BBA) Rev. Cancer 2019, 1871, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Boyault, S.; Rickman, D.S.; De Reyniès, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Hérault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshida, Y.; Nijman, S.M.; Kobayashi, M.; Chan, J.A.; Brunet, J.-P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef] [Green Version]
- Hoshida, Y.; Villanueva, A.; Kobayashi, M.; Peix, J.; Chiang, D.Y.; Camargo, A.; Gupta, S.; Moore, J.; Wrobel, M.J.; Lerner, J.; et al. Gene Expression in Fixed Tissues and Outcome in Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 1995–2004. [Google Scholar] [CrossRef] [Green Version]
- Shimada, S.; Mogushi, K.; Akiyama, Y.; Furuyama, T.; Watanabe, S.; Ogura, T.; Ogawa, K.; Ono, H.; Mitsunori, Y.; Ban, D.; et al. Comprehensive molecular and immunological characterization of hepatocellular carcinoma. EBioMedicine 2019, 40, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Chiang, D.; Villanueva, A.; Hoshida, Y.; Peix, J.; Newell, P.; Minguez, B.; Leblanc, A.C.; Donovan, D.J.; Thung, S.N.; Solé, M.; et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008, 68, 6779–6788. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Forgues, M.; Wang, W.; Kim, J.W.; Ye, Q.; Jia, H.-L.; Budhu, A.; Zanetti, K.A.; Chen, Y.; Qin, L.; et al. EpCAM and -Fetoprotein Expression Defines Novel Prognostic Subtypes of Hepatocellular Carcinoma. Cancer Res. 2008, 68, 1451–1461. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Ling, S.; Zheng, S.; Xu, X. Liquid biopsy in hepatocellular carcinoma: Circulating tumor cells and circulating tumor DNA. Mol. Cancer 2019, 18, 114. [Google Scholar] [CrossRef]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Ben Maad, I.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Kenmochi, K.; Sugihara, S.; Kojiro, M. Relationship of histologic grade of hepatocellular carcinoma (HCC) to tumor size, and demonstration of tumor cells of multiple different grades in single small HCC. Liver Int. 1987, 7, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Friemel, J.; Rechsteiner, M.; Frick, L.; Böhm, F.; Struckmann, K.; Egger, M.; Moch, H.; Heikenwalder, M.; Weber, A. Intratumor Heterogeneity in Hepatocellular Carcinoma. Clin. Cancer Res. 2014, 21, 1951–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, R.; Li, R.; Guo, H.; Guo, L.; Su, Z.; Ni, X.; Qi, L.; Zhang, T.; Li, Q.; Zhang, Z.; et al. Variable Intra-Tumor Genomic Heterogeneity of Multiple Lesions in Patients With Hepatocellular Carcinoma. Gastroenterology 2016, 150, 998–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016, 26, 304–319. [Google Scholar] [CrossRef]


{kind=link}
| Study | Setting | Treatment Arms | Molecular Targets | Results | Citation |
|---|---|---|---|---|---|
| SHARP, phase III | First-line | Sorafenib (n = 299) vs. placebo (n = 303) | Sorafenib: VEGFRs, PDGFR-β, and Raf-1, and B-Raf | OS: 10.7 vs. 7.9 months (HR 0.69; 95%CI 0.55– 0.87, p < 0.001) Time to symptomatic progression: 4.1 vs. 4.9 months (p = 0.7) | [4] |
| REFLECT, Phase II | First-line | Lenvatinib (n = 478) or sorafenib (n = 476) | Lenvatinib: VEGFR1–3, FGFR 1–4, PDGFR-α, c-KIT, and RET | OS: 13.6 vs. 12.3 months HR 0.92; 95%CI 12.1–14.9) PFS: 7.3 vs. 3.6 (HR 0.64; 95%CI 0.55–0.75 p < 0.001) ORR: 41% vs. 12% | [5] |
| RESORCE, phase III | Second-line | Regorafenib (n = 374) vs. placebo (n = 193) | Regorafenib: VEGFR2 inhibitor | OS: 10.6 vs. 7.8 months (HR 0.63; 95%CI 0.50–0.79 p < 0.0001) | [6] |
| CHECKMATE-040, phase I/II | Second-line | Nivolumab (n = 262) | Nivolumab: PD-1 | Safety and tolerability with 12 patients (25%) demonstrating grade ≥3 adverse events ORR: 20% (95%CI 15–26) | [7] |
| KEYNOTE-224, phase II | Second-line | Pembrolizumab (n = 104) | Pembrolizumab: PD-1 | ORR 18/104 patients (17%) with 1 (1%) complete response and 17 (16%) partial responses | [9] |
| CELESTIAL, phase III | Second-line | Cabozantinib (n = 467) vs. placebo (n = 237) | Cabozantinib: MET, VEGFR, RET, GAS6 receptor (AXL), KIT, and FMS-like tyrosine kinase-3 (FLT3) | OS: 10.2 vs. 8.0 months (HR 0.76; 95%CI 0.63–0.92 p = 0.005) PFS: 5.2 vs. 1.9 months (HR 0.44; 95%CI 0.36–0.52 p < 0.001) ORR 4% vs <1% (p = 0.009) | [11] |
| REACH-2, phase III | Second-line | Ramucirumab (n = 197) vs. placebo (n = 95) | Ramucirumab: VEGFR2 antagonist | OS: 8.5 vs. 7.3 months (HR 0.71; 95%CI 0.531–0.949 p = 0.0199) PFS: 2.8 vs. 1.6 months (HR 0.452; 95%CI 0.339–0.603) p < 0.0001) | [12] |
| Phase I/II | Second-line | Nivolumab (N) and ipilimumab (I). Cohort A: (N 1mg/kg + I 3 mg/kg (n = 50); cohort B: N 3 mg/kg + I 1 mg/kg (n = 49); cohort C: N 3 mg/kg + I 1 mg/kg (n = 49) | Nivolumab: PD-1; Ipilimumab: CTLA-4 | Grade ≥ 3 adverse event 37% ORR: A: 16/32 (16%); B: 15/31 (15%); C: 15/31 (15%) 24-month OS: A: 61%; B: 30%; C: 42% | [14] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, J.; Chuang, J.; Cho, M.; Toomey, K.; Hendifar, A.; Li, D. Molecular Targets, Pathways, and Therapeutic Implications for Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 5232. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155232
Gong J, Chuang J, Cho M, Toomey K, Hendifar A, Li D. Molecular Targets, Pathways, and Therapeutic Implications for Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2020; 21(15):5232. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155232
Chicago/Turabian StyleGong, Jun, Jeremy Chuang, May Cho, Kyra Toomey, Andrew Hendifar, and Daneng Li. 2020. "Molecular Targets, Pathways, and Therapeutic Implications for Hepatocellular Carcinoma" International Journal of Molecular Sciences 21, no. 15: 5232. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155232