Modulating the Crosstalk between the Tumor and Its Microenvironment Using RNA Interference: A Treatment Strategy for Hepatocellular Carcinoma

 ,
,  , and
, and 
Abstract
:1. Hepatocellular Carcinoma
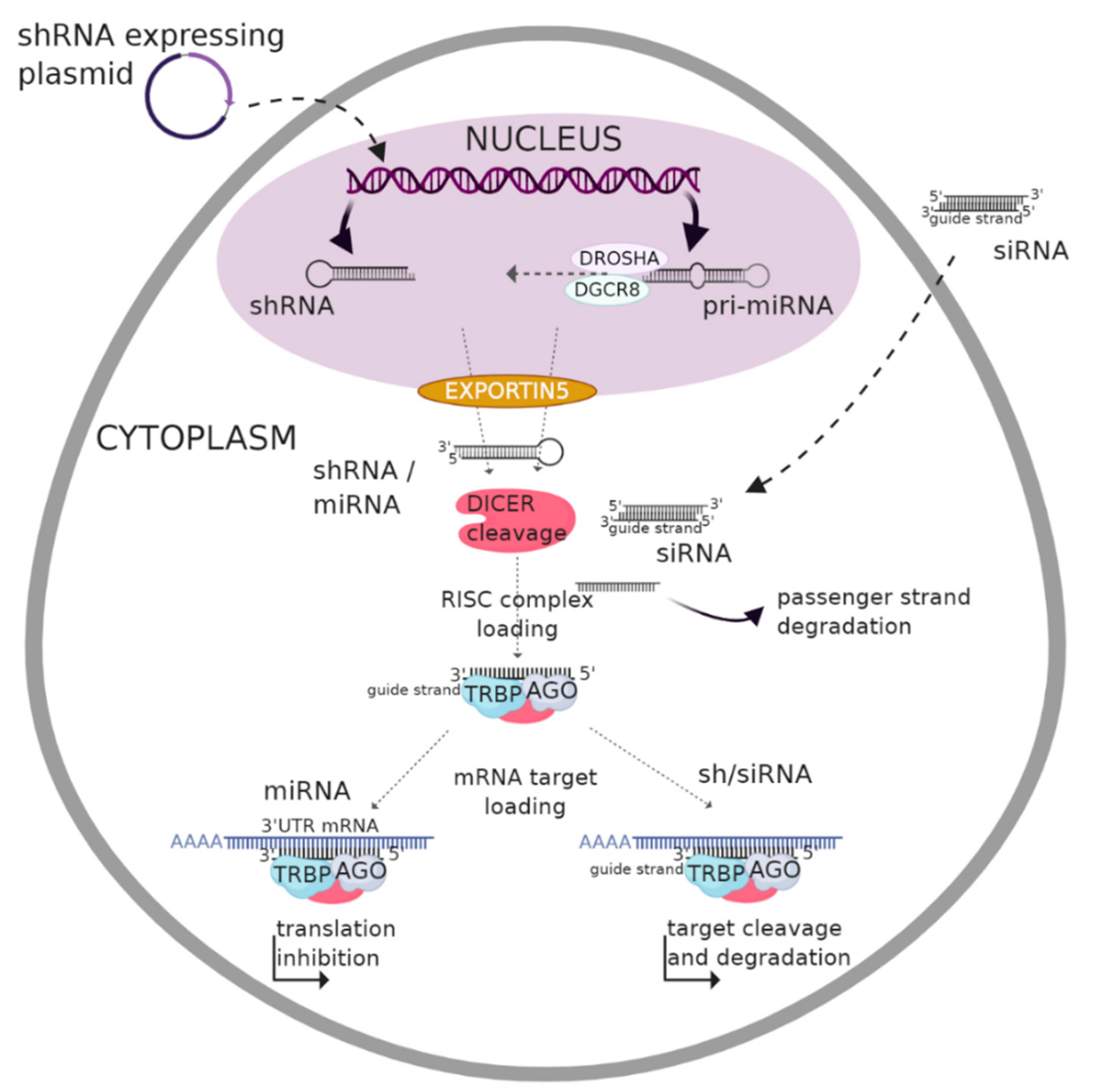
2. RNA Interference (RNAi) Overview
2.1. Biogenesis and Gene Silencing Mechanism of MicroRNA
2.2. Biogenesis and Gene Silencing Mechanism of shRNA
2.3. Biogenesis and Gene Silencing Mechanism of siRNA
3. Clinical Application of RNAi
3.1. RNAi and Cancer
3.2. RNAi-Mediated Therapeutic Intervention in the Context of HCC
4. Tackling the Crosstalk between the Tumor and Its Microenvironment Using RNA Interference
4.1. Targeting Communication with Extracellular Components
4.1.1. Extracellular Matrix (ECM)
4.1.2. Extracellular Vesicles (Exosomes)
4.2. Targeting Communication with Cellular Components
4.2.1. Cancer-Associated Fibroblasts (CAFs)
4.2.2. Endothelial Cells
4.2.3. Hepatic Stellate Cells (HSCs)
4.2.4. Immune Cells
Macrophages
Neutrophils
Dendritic Cells
Natural Killer Cells
5. Challenges and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HCC | hepatocellular carcinoma |
| RNAi | RNA interference |
| HCV | viral hepatitis C |
| HBV | viral hepatitis B |
| NASH | non-alcoholic steatohepatitis |
| PDL-1 | anti-programmed death-ligand1 |
| VEGF | anti-vascular endothelial growth factor |
| mRNA | messenger RNA |
| ncRNA | non coding RNA |
| dsRNA | double-stranded RNA |
| TLR | Toll-like receptor |
| miRNA | microRNA |
| shRNA | short hairpin RNA |
| siRNA | small interfering RNA |
| nt | nucleotide |
| pri-miRNA/shRNA/siRNA | primary–miRNA/shRNA/siRNA |
| pre-miRNA/shRNA/siRNA | precursor-miRNA/shRNA/siRNA |
| RISC | RNA induced silencing complex |
| PEI | polyethylenimine |
| EPHA2 | ephrin type-A receptor 2 |
| PLK1 | polo-like kinase 1 |
| STMN | stathmin |
| KSP | kinesin spindle protein |
| GMSF | granulocyte-macrophage colony-stimulating factor |
| TGF | transforming growth factor |
| ECM | extracellular matrix |
| CAFs | cancer associated fibroblasts |
| HSCs | hepatic stellate cells |
| TAMs | tumor associated macrophages |
| TANs | tumor-associated neutrophils |
| DCs | dendritic cells |
| T-regs | T-regulatory cells |
| HS | heparin Sulfate |
| GPC3 | glypican 3 |
| EGF | epidermal growth factor |
| HGF | hepatocyte growth factor |
| EGFR | epidermal growth factor receptor |
| NET-1 | neuroepithelial cell transforming-1 |
| EMS1 | gene encoding protein: leucine-rich repeat receptor protein kinase EMS1 |
| MIF | migration inhibition factor |
| CCN | cellular communication network factor |
| CYR61 | cysteine-rich angiogenic protein 61 |
| CTGF | connective tissue growth factor |
| NOV | nephroblastoma overexpressed |
| WISP-1 | Wnt1-Inducible Signaling pathway proteins |
| NFκB | nuclear factor kappa B |
| MSCs | mesenchymal stem cells |
| TIM-3 | T-cell immunoglobulin and mucin domain-containing molecule-3 |
| LAG3 | lymphocyte-activation gene 3 |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein-4 |
| MAPK | microtubule associated protein kinase |
| Bmi-1 | polycomb complex protein |
| CXCR | C-X-C chemokine receptor type |
| TNF | tumor necrosis factor |
| PDGF | platelet-derived growth factor |
| AATF | apoptosis-antagonizing transcription factor |
| NSG | non-obese diabetic severe combined immunodeficiency gamma mice |
| SCID | severe combined immunodeficiency |
| IL | interleukin |
| MCP-1 | monocyte chemoattractant protein-1 |
| CXCL | C-X-C motif ligand |
| FGF | fibroblast growth factor |
| CCL | chemokine C-C motif ligand |
| VEGFR | vascular endothelial growth factor receptor |
| PDGFR | platelet-derived growth factor receptor |
| MMP | matrix metalloprotease |
| GTIR | glucocorticoid-induced TNF receptor family-related |
| IGFBP-5 | insulin-like growth factor-binding protein 5 |
| REG3A | regenerating islet-derived protein 3 alpha |
| Erk | extracellular signal-regulated kinases |
| CTLs | cytotoxic T-cells |
| IFN | interferon |
| MAP4K4 | mitogen-activated protein kinase kinase kinase kinase 4 |
| SUMO | small ubiquitin-like modifier |
| SIRT6 | sirtuin 6 |
| Bcl-2 | B-cell lymphoma 2 |
| Bax | Bcl-2-associated X protein |
| ECT2 | epithelial cell transforming sequence 2 |
| RhoC | Ras homolog family member C |
| GRK2 | G protein-coupled receptor kinase 2 |
| GTIR | glucocorticoid-induced tumor necrosis factor receptor |
| STAT3 | Signal Transducer and Activators of Transcription |
| NLR | neutrophil–lymphocyte ratio |
| PD-1 | programmed cell death protein-1 |
| HIF-1 | hypoxia-inducible factor |
| APCs | antigen-presenting cells |
| IRF | interferon regulatory factor |
| ISG15 | IFN-stimulated gene 15 |
| NK | natural killer |
| AFP | α-fetoprotein |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| RGD | arginylglycylaspartic acid |
References
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; Dandona, L.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2017, 3, 524–548. [Google Scholar] [CrossRef] [PubMed]
- Ogunwobi, O.; Harricharran, T.; Huaman, J.A.; Galuza, A.; Odumuwagun, O.J.; Tan, Y.; Ma, G.X.; Nguyen, M.T. Mechanisms of hepatocellular carcinoma progression. World J. Gastroenterol. 2019, 25, 2279–2293. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Medavaram, S.; Zhang, Y. Emerging therapies in advanced hepatocellular carcinoma. Exp. Hematol. Oncol. 2018, 7, 17. [Google Scholar] [CrossRef]
- Keating, G.M. Sorafenib: A Review in Hepatocellular Carcinoma. Target. Oncol. 2017, 12, 243–253. [Google Scholar] [CrossRef]
- FDA Approves Atezolizumab Plus Bevacizumab for Unresectable Hepatocellular Carcinoma. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-atezolizumab-plus-bevacizumab-unresectable-hepatocellular-carcinoma (accessed on 15 June 2020).
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Mansoori, B.; Shotorbani, S.S.; Baradaran, B. RNA interference and its role in cancer therapy. Adv. Pharm. Bull. 2014, 4, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Setten, R.L.; Rossi, J.J.; Han, S.-P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 8, 421–446. [Google Scholar] [CrossRef]
- Wang, Q.; Carmichael, G.G. Effects of Length and Location on the Cellular Response to Double-Stranded RNA. Microbiol. Mol. Biol. Rev. 2004, 68, 432–452. [Google Scholar] [CrossRef] [Green Version]
- Chalbatani, G.M.; Dana, H.; Gharagouzloo, E.; Grijalvo, S.; Eritja, R.; Logsdon, C.D.; Memari, F.; Miri, S.R.; Rad, M.R.; Marmari, V. Small interfering RNAs (siRNAs) in cancer therapy: A nano-based approach. Int. J. Nanomed. 2019, 14, 3111–3128. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Jiang, X.; Gui, S. RNA interference-based nanosystems for inflammatory bowel disease therapy. Int. J. Nanomed. 2016, 11, 5287–5310. [Google Scholar] [CrossRef] [Green Version]
- Olena, A.F.; Patton, J.G. Genomic Organization of microRNAs. J. Cell. Physiol. 2010, 222, 540–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. (Lausanne) 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatri, N.; Rathi, M.; Baradia, D.; Trehan, S.; Misra, A. In vivo delivery aspects of miRNA, shRNA and siRNA. Crit. Rev. Ther. Drug Carrier. Syst. 2012, 29, 487–527. [Google Scholar] [CrossRef]
- Burke, J.M.; Kincaid, R.P.; Aloisio, F.; Welch, N.; Sullivan, C.S. Expression of short hairpin RNAs using the compact architecture of retroviral microRNA genes. Nucleic Acids Res. 2017, 45, 154. [Google Scholar] [CrossRef] [Green Version]
- Paddison, P.J.; Caudy, A.A.; Bernstein, E.; Hannon, G.J.; Conklin, D.S. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002, 16, 948–958. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, L.; Rossi, J.J. RNAi therapeutics: Principles, prospects and challenges. Adv. Drug Deliv. Rev. 2007, 59, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [Green Version]
- Matzke, M.A.; Birchler, J.A. RNAi-mediated pathways in the nucleus. Nat. Rev. Genet. 2005, 6, 24–35. [Google Scholar] [CrossRef]
- Sheu-Gruttadauria, J.; MacRae, I.J. Structural Foundations of RNA Silencing by Argonaute. J. Mol. Biol. 2017, 429, 2619–2639. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, P.D.A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Second RNAi Drug Approved. Nature Biotechnology, 7 April 2020. [CrossRef] [Green Version]
- Thi, E.P.; Mire, C.E.; Lee, A.C.H.; Geisbert, J.B.; Zhou, J.Z.; Agans, K.N.; Snead, N.M.; Deer, D.J.; Barnard, T.R.; Fenton, K.A.; et al. Lipid nanoparticle siRNA treatment of Ebola-virus-Makona-infected nonhuman primates. Nature 2015, 521, 362–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, M.; Katoh, M. FGFR2 and WDR11 are neighboring oncogene and tumor suppressor gene on human chromosome 10q26. Int. J. Oncol. 2003, 22, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Nieth, C.; Priebsch, A.; Stege, A.; Lage, H. Modulation of the classical multidrug resistance (MDR) phenotype by RNA interference (RNAi). FEBS Lett. 2003, 545, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Li, D.; Leng, S.; Zhu, X. RNA-based pharmacotherapy for tumors: From bench to clinic and back. Biomed. Pharm. 2020, 125, 109997. [Google Scholar] [CrossRef]
- Chiu, Y.-L.; Rana, T.M. RNAi in human cells: Basic structural and functional features of small interfering RNA. Mol. Cell 2002, 10, 549–561. [Google Scholar] [CrossRef]
- Park, J.; Park, J.; Pei, Y.; Xu, J.; Yeo, Y. Pharmacokinetics and biodistribution of recently-developed siRNA nanomedicines. Adv. Drug Deliv. Rev. 2016, 104, 93–109. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Huang, M.; Guo, W.W.; Huang, Q.; Zhang, L.Z.; Jiang, G. Nano-based delivery of RNAi in cancer therapy. Mol. Cancer 2017, 16, 134. [Google Scholar] [CrossRef] [Green Version]
- Ragelle, H.; Vandermeulen, G.; Préat, V. Chitosan-based siRNA delivery systems. J. Control. Release 2013, 172, 207–218. [Google Scholar] [CrossRef]
- Babu, A.; Muralidharan, R.; Amreddy, N.; Mehta, M.; Munshi, A.; Ramesh, R. Nanoparticles for siRNA-Based Gene Silencing in Tumor Therapy. IEEE Trans. Nanobiosci. 2016, 15, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A. Improving siRNA bio-distribution and minimizing side effects. Curr. Drug Metab. 2011, 12, 11–23. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, M.J.; Kwon, I.C.; Roberts, T.M. Delivery Strategies and Potential Targets for siRNA in Major Cancer Types. Adv. Drug Deliv. Rev. 2016, 104, 2–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Lee, S.A.; Chen, X. RNA interference as therapeutics for hepatocellular carcinoma. Recent Pat. Anticancer Drug Discov. 2011, 6, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Hajiasgharzadeh, K.; Somi, M.H.; Shanehbandi, D.; Mokhtarzadeh, A.; Baradaran, B. Small interfering RNA-mediated gene suppression as a therapeutic intervention in hepatocellular carcinoma. J. Cell. Physiol. 2019, 234, 3263–3276. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Ho, P.Y.; Tu, M.-J.; Jilek, J.L.; Chen, Q.X.; Zeng, S.; Yu, A.M. Lipidation of polyethylenimine-based polyplex increases serum stability of bioengineered RNAi agents and offers more consistent tumoral gene knockdown in vivo. Int. J. Pharm. 2018, 547, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.J.; Mitra, R.; McArthur, M.J.; Baze, W.; Barnhart, K.; Wu, S.; Rodriguez-Aguayo, C.; Zhang, X.; Coleman, R.L.; Lopez-Berestein, G. Preclinical Mammalian Safety Studies of EPHARNA (DOPC Nanoliposomal EphA2-Targeted siRNA). Mol. Cancer Ther. 2017, 16, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- BioSpace. Sirnaomics’ siRNA Therapeutic Candidate, STP705, Granted Orphan Drug Designation by US FDA for Treatment of Hepatocellular Carcinoma. Available online: https://www.biospace.com/article/sirnaomics-sirna-therapeutic-candidate-stp705-granted-orphan-drug-designation-by-us-fda-for-treatment-of-hepatocellular-carcinoma/ (accessed on 12 May 2020).
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. From bench to bed: The tumor immune microenvironment and current immunotherapeutic strategies for hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 396. [Google Scholar] [CrossRef] [Green Version]
- Filliol, A.; Schwabe, R.F. Contributions of Fibroblasts, Extracellular Matrix, Stiffness, and Mechanosensing to Hepatocarcinogenesis. Semin. Liver Dis. 2019, 39, 315–333. [Google Scholar] [CrossRef]
- Uchimura, K.; Morimoto-Tomita, M.; Bistrup, A.; Li, J.; Lyon, M.; Gallagher, J.; Werb, Z.; Rosen, S.D. HSulf-2, an extracellular endoglucosamine-6-sulfatase, selectively mobilizes heparin-bound growth factors and chemokines: Effects on VEGF, FGF-1, and SDF-1. BMC Biochem. 2006, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Montalbano, M.; Rastellini, C.; McGuire, J.T.; Prajapati, J.; Shirafkan, A.; Vento, R.; Cicalese, L. Role of Glypican-3 in the growth, migration and invasion of primary hepatocytes isolated from patients with hepatocellular carcinoma. Cell Oncol. (Dordr) 2018, 41, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Wang, Y.; Cai, M.; Lin, L.; Chen, X.; Cao, Z.; Zhu, K.; Shuai, X. Codelivery of sorafenib and GPC3 siRNA with PEI-modified liposomes for hepatoma therapy. Biomater. Sci. 2017, 5, 2468–2479. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Friedmann, Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J. Clin. Investig. 2001, 108, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Levy-Adam, F.; Feld, S.; Suss-Toby, E.; Vlodavsky, I.; Ilan, N. Heparanase facilitates cell adhesion and spreading by clustering of cell surface heparan sulfate proteoglycans. PLoS ONE 2008, 3, 2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, G.; Zhang, L.; Pu, J.; Mei, H.; Zhao, J.; Huang, K.; Zeng, F.; Tong, Q. Small RNAs targeting transcription start site induce heparanase silencing through interference with transcription initiation in human cancer cells. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ding, J.; Feng, Y.; Weng, L.; Zhao, G.; Xiang, J.; Zhang, M.; Xing, D. Targeting of growth factors in the treatment of hepatocellular carcinoma: The potentials of polysaccharides. Oncol. Lett. 2017, 13, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, B.C.; Hoshida, Y.; Fujii, T.; Wei, L.; Yamada, S.; Lauwers, G.Y.; McGinn, C.M.; DePeralta, D.K.; Chen, X.; Kuroda, T.; et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology 2014, 59, 1577–1590. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Yu, Y.; Zhang, Y.; Song, J.; Chen, H.; Li, W.; Qian, W.; Deng, L.; Kou, G.; Chen, J. EGFR-specific PEGylated immunoliposomes for active siRNA delivery in hepatocellular carcinoma. Biomaterials 2012, 33, 270–282. [Google Scholar] [CrossRef]
- Huang, Z.; Dong, L.; Chen, J.; Gao, F.; Zhang, Z.; Chen, J.; Zhang, J. Low-molecular weight chitosan/vascular endothelial growth factor short hairpin RNA for the treatment of hepatocellular carcinoma. Life Sci. 2012, 91, 1207–1215. [Google Scholar] [CrossRef]
- Li, T.; Xue, Y.; Wang, G.; Gu, T.; Li, Y.; Zhu, Y.Y.; Chen, L. Multi-target siRNA: Therapeutic Strategy for Hepatocellular Carcinoma. J. Cancer 2016, 7, 1317–1327. [Google Scholar] [CrossRef]
- Wu, Y.-Y.; Chen, L.; Wang, G.-L.; Zhang, Y.-X.; Zhou, J.-M.; He, S.; Qin, J.; Zhu, Y.-Y. Inhibition of hepatocellular carcinoma growth and angiogenesis by dual silencing of NET-1 and VEGF. J. Mol. Histol. 2013, 44, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Jian, W.; Wu, Z.; Zhao, J.; Wang, H.; Li, W.; Xia, J. Small interfering RNA (siRNA)-mediated knockdown of macrophage migration inhibitory factor (MIF) suppressed cyclin D1 expression and hepatocellular carcinoma cell proliferation. Oncotarget 2014, 5, 5570–5580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazumder, S.; DuPree, E.L.; Almasan, A. A dual role of cyclin E in cell proliferation and apoptosis may provide a target for cancer therapy. Curr. Cancer Drug Targets 2004, 4, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Lin, S.-Y.; Brunicardi, F.C.; Seu, P. Use of RNA interference to target cyclin E-overexpressing hepatocellular carcinoma. Cancer Res. 2003, 63, 3593–3597. [Google Scholar] [PubMed]
- Bai, T.; Chen, C.-C.; Lau, L.F. Matricellular protein CCN1 activates a proinflammatory genetic program in murine macrophages. J. Immunol. 2010, 184, 3223–3232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Q.; Dong, Q.; Qin, L. CCN: Core regulatory proteins in the microenvironment that affect the metastasis of hepatocellular carcinoma. Oncotarget 2016, 7, 1203–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, H.; Ogura, M.; Saito, Y.; Sekine, W.; Sano, R.; Gotou, T.; Oku, T.; Itoh, S.; Katabami, K.; Tsuji, T. Changes in adhesive and migratory characteristics of hepatocellular carcinoma (HCC) cells induced by expression of alpha3beta1 integrin. Biochim. Biophys. Acta 2008, 1780, 564–570. [Google Scholar] [CrossRef]
- Bogorad, R.L.; Yin, H.; Zeigerer, A.; Nonaka, H.; Ruda, V.M.; Zerial, M.; Anderson, D.G.; Koteliansky, V. Nanoparticle-formulated siRNA targeting integrins inhibits hepatocellular carcinoma progression in mice. Nat. Commun. 2014, 5, 3869. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Xie, F.; Wang, L.; Zhang, L.; Zhang, S.; Fang, M.; Zhou, F. The function and clinical application of extracellular vesicles in innate immune regulation. Cell. Mol. Immunol. 2020, 17, 323–334. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Fernandes, A.R.; Baptista, P.V. Exosome in tumour microenvironment: Overview of the crosstalk between normal and cancer cells. Biomed. Res. Int. 2014, 179486. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular vesicles in cancer: Cell-to-cell mediators of metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maia, J.; Caja, S.; Moraes, M.C.S.; Couto, N.; Costa-Silva, B. Exosome-Based Cell-Cell Communication in the Tumor Microenvironment. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Zhou, H.; Zhao, X.; Ding, H.; Li, W.; Qin, L.; Pan, Y. Role of exosomes and exosomal microRNAs in hepatocellular carcinoma: Potential in diagnosis and antitumour treatments (Review). Int. J. Mol. Med. 2018, 41, 1809–1816. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.; Yuan, Y.; Xie, L.; Ran, P.; Xiang, X.; Huang, Q.; Qi, G.; Guo, X.; Xiao, C.; Zheng, S. miRNA-320a inhibits tumor proliferation and invasion by targeting c-Myc in human hepatocellular carcinoma. OncoTargets Ther. 2017, 10, 885–894. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z.; Wu, J.; Wu, J.; Luo, D.; Jiang, C.; Ding, Y. Exosomes derived from HCC cells induce sorafenib resistance in hepatocellular carcinoma both in vivo and in vitro. J. Exp. Clin. Cancer Res. 2016, 35, 30. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Xu, M.; Li, X.; Su, X.; Xiao, X.; Keating, A.; Zhao, R.C. Exosomes released by hepatocarcinoma cells endow adipocytes with tumor-promoting properties. J. Hematol. Oncol. 2018, 11, 14. [Google Scholar] [CrossRef]
- Wang, X.; Shen, H.; Zhang, G.; Huang, R.; Zhang, W.; He, Q.; Jin, K.; Zhuo, H.; Zhang, Z.; Wang, J.; et al. 14-3-3ζ delivered by hepatocellular carcinoma-derived exosomes impaired anti-tumor function of tumor-infiltrating T lymphocytes. Cell Death Dis. 2018, 9, 159. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Qin, H.; Poon, T.C.W.; Sze, S.-C.; Ding, X.; Co, N.N.; Ngai, S.-M.; Chan, T.-F.; Wong, N. Hepatocellular carcinoma-derived exosomes promote motility of immortalized hepatocyte through transfer of oncogenic proteins and RNAs. Carcinogenesis 2015, 36, 1008–1018. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Jian, W.; Gao, W.; Zheng, Y.; Wang, Y.; Zhou, Z.; Zhang, H.; Wang, C. RNAi silencing of c-Myc inhibits cell migration, invasion, and proliferation in HepG2 human hepatocellular carcinoma cell line: C-Myc silencing in hepatocellular carcinoma cell. Cancer Cell Int. 2013, 13, 23. [Google Scholar] [CrossRef] [Green Version]
- Haga, Y.; Kanda, T.; Nakamura, M.; Nakamoto, S.; Sasaki, R.; Takahashi, K.; Wu, S.; Yokosuka, O. Overexpression of c-Jun contributes to sorafenib resistance in human hepatoma cell lines. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patt, Y.Z.; Charnsangavej, C.; Yoffe, B.; Smith, R.; Lawrence, D.; Chuang, V.; Carrasco, H.; Roh, M.; Chase, J.; Fischer, H.; et al. Hepatic arterial infusion of floxuridine, leucovorin, doxorubicin, and cisplatin for hepatocellular carcinoma: Effects of hepatitis B and C viral infection on drug toxicity and patient survival. J. Clin. Oncol. 1994, 12, 1204–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.E.; Bae, S.H.; Choi, J.Y.; Yoon, S.K.; You, Y.K.; Lee, M.A. Epirubicin, Cisplatin, 5-FU combination chemotherapy in sorafenib-refractory metastatic hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Kubota, T.; Abe, S.; Watanabe, Y.; Sugano, T.; Inoue, R.; Iwanaga, A.; Seki, K.; Honma, T.; Yoshida, T. Hepatic arterial infusion chemotherapy with cisplatin before radical local treatment of early hepatocellular carcinoma (JIS score 0/1) improves survival. Ann. Oncol. 2014, 25, 1379–1384. [Google Scholar] [CrossRef]
- Li, M.; Zhao, P.; Fan, T.; Chen, Y.; Zhang, X.; He, C.; Yang, T.; Lee, R.J.; Khan, M.W.; Raza, S.M.; et al. Biocompatible co-loading vehicles for delivering both nanoplatin cores and siRNA to treat hepatocellular carcinoma. Int. J. Pharm. 2019, 572, 118769. [Google Scholar] [CrossRef]
- Conigliaro, A.; Costa, V.; Lo Dico, A.; Saieva, L.; Buccheri, S.; Dieli, F.; Manno, M.; Raccosta, S.; Mancone, C.; Tripodi, M.; et al. CD90+ liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol. Cancer 2015, 14, 155. [Google Scholar] [CrossRef]
- Huang, K.-W.; Lai, Y.-T.; Chern, G.-J.; Huang, S.-F.; Tsai, C.-L.; Sung, Y.-C.; Chiang, C.-C.; Hwang, P.-B.; Ho, T.-L.; Huang, R.-L.; et al. Galactose Derivative-Modified Nanoparticles for Efficient siRNA Delivery to Hepatocellular Carcinoma. Biomacromolecules 2018, 19, 2330–2339. [Google Scholar] [CrossRef]
- Yao, Y.; Wang, T.; Liu, Y.; Zhang, N. Co-delivery of sorafenib and VEGF-siRNA via pH-sensitive liposomes for the synergistic treatment of hepatocellular carcinoma. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1374–1383. [Google Scholar] [CrossRef] [Green Version]
- De Clercq, E. AMD3100/CXCR4 Inhibitor. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef]
- Liu, J.-Y.; Chiang, T.; Liu, C.-H.; Chern, G.-G.; Lin, T.-T.; Gao, D.-Y.; Chen, Y. Delivery of siRNA Using CXCR4-targeted Nanoparticles Modulates Tumor Microenvironment and Achieves a Potent Antitumor Response in Liver Cancer. Mol. Ther. 2015, 23, 1772–1782. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderberg, C.; Pietras, K. On the origin of cancer-associated fibroblasts. Cell Cycle. 2009, 8, 1461–1462. [Google Scholar] [CrossRef] [Green Version]
- Ishii, G.; Sangai, T.; Oda, T.; Aoyagi, Y.; Hasebe, T.; Kanomata, N.; Endoh, Y.; Okumura, C.; Okuhara, Y.; Magae, J.; et al. Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem. Biophys. Res. Commun. 2003, 309, 232–240. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Elenbaas, B.; Weinberg, R.A. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp. Cell. Res. 2001, 264, 169–184. [Google Scholar] [CrossRef]
- Birgani, M.T.; Carloni, V. Tumor Microenvironment, a Paradigm in Hepatocellular Carcinoma Progression and Therapy. Int. J. Mol. Sci. 2017, 18, 405. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.D.; Nakamura, I.; Roberts, L.R. The tumor microenvironment in hepatocellular carcinoma: Current status and therapeutic targets. Semin. Cancer Biol. 2011, 21, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.P.; Santhekadur, P.K.; Seneshaw, M.; Mirshahi, F.; Uram-Tuculescu, C.; Sanyal, A.J. A Regulatory Role of Apoptosis Antagonizing Transcription Factor in the Pathogenesis of Nonalcoholic Fatty Liver Disease and Hepatocellular Carcinoma. Hepatology 2019, 69, 1520–1534. [Google Scholar] [CrossRef] [PubMed]
- Novikova, M.V.; Khromova, N.V.; Kopnin, P.B. Components of the Hepatocellular Carcinoma Microenvironment and Their Role in Tumor Progression. Biochem. Mosc. 2017, 82, 861–873. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Liu, J.; Chen, S.; Wang, W.; Ning, B.; Chen, F.; Shen, W.; Ding, J.; Chen, W.; Xie, W.; Zhang, X. Cancer-associated fibroblasts promote hepatocellular carcinoma metastasis through chemokine-activated hedgehog and TGF-β pathways. Cancer Lett. 2016, 379, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Baluk, P.; Morikawa, S.; Haskell, A.; Mancuso, M.; McDonald, D.M. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am. J. Pathol. 2003, 163, 1801–1815. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Wang, J.; Sun, Q.; Li, F.; Gao, H.; Xu, L.; Zhang, J.; Sun, X.; Tian, Y.; Zhao, Q.; et al. Interleukin-8 promotes integrin β3 upregulation and cell invasion through PI3K/Akt pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 449. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Chen, Z.; Zhang, L.; Tian, D.; Wang, D.; Fan, D.; Wu, K.; Xia, L. Interleukin-8 Induces Expression of FOXC1 to Promote Transactivation of CXCR1 and CCL2 in Hepatocellular Carcinoma Cell Lines and Formation of Metastases in Mice. Gastroenterology 2015, 149, 1053–1067. [Google Scholar] [CrossRef] [PubMed]
- Santamato, A.; Fransvea, E.; Dituri, F.; Caligiuri, A.; Quaranta, M.; Niimi, T.; Pinzani, M.; Antonaci, S.; Giannelli, G. Hepatic stellate cells stimulate HCC cell migration via laminin-5 production. Clin. Sci. (Lond.) 2011, 121, 159–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.; Park, M.J.; Kim, K.; Park, J.; Kim, J.; Kim, W.; Yoon, J.-H. Tumor-Stroma Crosstalk Enhances REG3A Expressions that Drive the Progression of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faouzi, S.; Lepreux, S.; Bedin, C.; Dubuisson, L.; Balabaud, C.; Bioulac-Sage, P.; Desmoulière, A.; Rosenbaum, J. Activation of cultured rat hepatic stellate cells by tumoral hepatocytes. Lab. Investig. 1999, 79, 485–493. [Google Scholar]
- Zhao, W.; Zhang, L.; Yin, Z.; Su, W.; Ren, G.; Zhou, C.; You, J.; Fan, J.; Wang, X. Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer Sci. 2009, 100, 646–653. [Google Scholar] [CrossRef]
- Carloni, V.; Luong, T.V.; Rombouts, K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: More complicated than ever. Liver Int. 2014, 34, 834–843. [Google Scholar] [CrossRef]
- Mehdizadeh, A.; Somi, M.H.; Darabi, M.; Farajnia, S.; Akbarzadeh, A.; Montazersaheb, S.; Yousefi, M.; Bonyadi, M. Liposome-mediated RNA interference delivery against Erk1 and Erk2 does not equally promote chemosensitivity in human hepatocellular carcinoma cell line HepG2. Artif. Cells Nanomed. Biotechnol. 2017, 45, 1612–1619. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Patel, P.; Schutzer, S.E.; Pyrsopoulos, N. Immunobiology of hepatocarcinogenesis: Ways to go or almost there. World J. Gastrointest Pathophysiol. 2016, 7, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Yeung, O.W.H.; Lo, C.-M.; Ling, C.-C.; Qi, X.; Geng, W.; Li, C.X.; Ng, K.T.P.; Forbes, S.J.; Guan, X.-Y.; Poon, R.T.P.; et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J. Hepatol. 2015, 62, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.; Shi, X.; Wang, S.; Gao, Y.; Kuang, Z.; Xie, Q.; Li, Y.; Deng, H.; Wu, Y.; Li, M.; et al. M2 macrophages mediate sorafenib resistance by secreting HGF in a feed-forward manner in hepatocellular carcinoma. Br. J. Cancer 2019, 121, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.-W.; Cai, J.; Zhao, X.-L.; Jiang, T.-H.; He, T.-F.; Fu, H.-Q.; Zhu, M.-H.; Zhang, S.-H. ShRNA-targeted MAP4K4 inhibits hepatocellular carcinoma growth. Clin. Cancer Res. 2011, 17, 710–720. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Sun, H.; Shi, X.; Wang, H.; Cui, C.; Xiao, F.; Wu, C.; Guo, X.; Wang, L. SENP1 regulates hepatocyte growth factor-induced migration and epithelial-mesenchymal transition of hepatocellular carcinoma. Tumor Biol. 2016, 37, 7741–7748. [Google Scholar] [CrossRef]
- Cho, S.-B.; Park, Y.-L.; Song, Y.-A.; Kim, K.-Y.; Lee, G.-H.; Cho, D.-H.; Myung, D.-S.; Park, K.-J.; Lee, W.-S.; Chung, I.-J.; et al. Small interfering RNA-directed targeting of RON alters invasive and oncogenic phenotypes of human hepatocellular carcinoma cells. Oncol. Rep. 2011, 26, 1581–1586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yu, Y.; Huang, Q.; Tang, K. SIRT6 regulates the proliferation and apoptosis of hepatocellular carcinoma via the ERK1/2 signaling pathway. Mol. Med. Rep. 2019, 20, 1575–1582. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Xia, H.; Zhang, X.; Karthik, S.; Pratap, S.V.; Ooi, L.L.; Hong, W.; Hui, K.M. ECT2 regulates the Rho/ERK signalling axis to promote early recurrence in human hepatocellular carcinoma. J. Hepatol. 2015, 62, 1287–1295. [Google Scholar] [CrossRef]
- Wang, W.; Wu, F.; Fang, F.; Tao, Y.; Yang, L. Inhibition of invasion and metastasis of hepatocellular carcinoma cells via targeting RhoC in vitro and in vivo. Clin. Cancer Res. 2008, 14, 6804–6812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.W.; Yan, S.X.; Wu, H.X.; Chen, J.Y.; Zhang, Y.; Li, Y.; Wei, W. The influence of TNF-α and Ang II on the proliferation, migration and invasion of HepG2 cells by regulating the expression of GRK2. Cancer Chemother. Pharmacol. 2017, 79, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Shirabe, K.; Mano, Y.; Muto, J.; Matono, R.; Motomura, T.; Toshima, T.; Takeishi, K.; Uchiyama, H.; Yoshizumi, T.; Taketomi, A.; et al. Role of tumor-associated macrophages in the progression of hepatocellular carcinoma. Surg. Today 2012, 42, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ding, T.; Pan, W.; Zhu, L.-Y.; Li, L.; Zheng, L. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. Int. J. Cancer 2009, 125, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.-M.; Peng, C.; Zhao, Q.; Wu, Y.; Chen, M.-S.; Zheng, L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma promote expansion of memory T helper 17 cells. Hepatology 2010, 51, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Li, J.; Zhang, C. STAT3 promotes the proliferation and migration of hepatocellular carcinoma cells by regulating AKT2. Oncol. Lett. 2018, 15, 3333–3338. [Google Scholar] [CrossRef]
- Nicolás-Ávila, J.Á.; Adrover, J.M.; Hidalgo, A. Neutrophils in Homeostasis, Immunity, and Cancer. Immunity 2017, 46, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Michaeli, J.; Shaul, M.E.; Mishalian, I.; Hovav, A.-H.; Levy, L.; Zolotriov, L.; Granot, Z.; Fridlender, Z.G. Tumor-associated neutrophils induce apoptosis of non-activated CD8 T-cells in a TNFα and NO-dependent mechanism, promoting a tumor-supportive environment. Oncoimmunology 2017, 6, 1356965. [Google Scholar] [CrossRef]
- Ameratunga, M.; Chénard-Poirier, M.; Candilejo, I.M.; Pedregal, M.; Lui, A.; Dolling, D.; Aversa, C.; Garces, A.I.; Ang, J.E.; Banerji, U.; et al. Neutrophil-lymphocyte ratio kinetics in patients with advanced solid tumours on phase I trials of PD-1/PD-L1 inhibitors. Eur. J. Cancer 2018, 89, 56–63. [Google Scholar] [CrossRef]
- Jeyakumar, G.; Kim, S.; Bumma, N.; Landry, C.; Silski, C.; Suisham, S.; Dickow, B.; Heath, E.; Fontana, J.; Vaishampayan, U. Neutrophil lymphocyte ratio and duration of prior anti-angiogenic therapy as biomarkers in metastatic RCC receiving immune checkpoint inhibitor therapy. J. Immunother. Cancer 2017, 5, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margetts, J.; Ogle, L.F.; Chan, S.L.; Chan, A.W.H.; Chan, K.C.A.; Jamieson, D.; Willoughby, C.E.; Mann, D.A.; Wilson, C.L.; Manas, D.M.; et al. Neutrophils: Driving progression and poor prognosis in hepatocellular carcinoma. Br. J. Cancer 2018, 118, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, K.; Fukumoto, T.; Kido, M.; Tanaka, M.; Kuramitsu, K.; Kinoshita, H.; Komatsu, S.; Tsugawa, D.; Terai, S.; Matsumoto, T.; et al. Preoperative neutrophil-to-lymphocyte ratio as a predictor of survival after reductive surgery plus percutaneous isolated hepatic perfusion for hepatocellular carcinoma: A retrospective analysis. Surg. Today 2017, 47, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Gagea, M.; Sargis, T.; Lemos, R., Jr.; Grandjean, G.; Charbono, A.; Bekiaris, V.; Sedy, J.; Kiriakova, G.; Liu, X.; et al. A new HIF-1α/RANTES-driven pathway to hepatocellular carcinoma mediated by germline haploinsufficiency of SART1/HAF in mice. Hepatology 2016, 63, 1576–1591. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.L.; Zhou, Z.J.; Hu, Z.Q.; Huang, X.W.; Wang, Z.; Chen, E.B.; Fan, J.; Cao, Y.; Dai, Z.; Zhou, J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology 2016, 150, 1646–1658. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.-L.; Dai, Z.; Zhou, Z.J.; Wang, X.Y.; Yang, G.H.; Wang, Z.; Huang, X.W.; Fan, J.; Zhou, J. Overexpression of CXCL5 mediates neutrophil infiltration and indicates poor prognosis for hepatocellular carcinoma. Hepatology 2012, 56, 2242–2254. [Google Scholar] [CrossRef] [PubMed]
- Ormandy, L.-A.; Farber, A.; Cantz, T.; Petrykowska, S.; Wedemeyer, H.; Horning, M.; Lehner, F.; Manns, M.-P.; Korangy, F.; Greten, T.-F. Direct ex vivo analysis of dendritic cells in patients with hepatocellular carcinoma. World J. Gastroenterol. 2006, 12, 3275–3282. [Google Scholar] [CrossRef]
- Streba, L.A.M.; Streba, C.T.; Săndulescu, L.; Vere, C.C.; Mitruţ, P.; Cotoi, B.V.; Popescu, L.N.; Ion, D.A. Dendritic cells and hepatocellular carcinoma. Rom. J. Morphol. Embryol. 2014, 55, 1287–1293. [Google Scholar]
- Li, P.; Du, Q.; Cao, Z.; Guo, Z.; Evankovich, J.; Yan, W.; Chang, Y.; Shao, L.; Stolz, D.B.; Tsung, A.; et al. Interferon-γ induces autophagy with growth inhibition and cell death in human hepatocellular carcinoma (HCC) cells through interferon-regulatory factor-1 (IRF-1). Cancer Lett. 2012, 314, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wang, J.; Zhang, H.; Zhu, M.; Chen, F.; Hu, Y.; Liu, H.; Zhu, H. Interferon-stimulated gene 15 (ISG15) is a trigger for tumorigenesis and metastasis of hepatocellular carcinoma. Oncotarget 2014, 5, 8429–8441. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Jiang, Y.; Wang, Z.; Wang, H. Natural killer cells involved in tumour immune escape of hepatocellular carcinoma. Int. Immunopharmacol. 2019, 73, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Hasmim, M.; Messai, Y.; Ziani, L.; Thiery, J.; Bouhris, J.-H.; Noman, M.Z.; Chouaib, S. Critical Role of Tumor Microenvironment in Shaping NK Cell Functions: Implication of Hypoxic Stress. Front. Immunol. 2015, 6, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Sun, T.; Cao, J.; Fan, S. Hypoxia-inducible factor-1α downregulation by small interfering RNA inhibits proliferation, induces apoptosis, and enhances radiosensitivity in chemical hypoxic human hepatoma SMMC-7721 cells. Cancer Biother. Radiopharm. 2011, 26, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Nishikawa, M.; Takakura, Y. Inhibition of tumor cell growth in the liver by RNA interference-mediated suppression of HIF-1alpha expression in tumor cells and hepatocytes. Gene Ther. 2008, 15, 572–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vujanovic, L.; Stahl, E.C.; Pardee, A.D.; Geller, D.A.; Tsung, A.; Watkins, S.C.; Gibson, G.A.; Storkus, W.J.; Butterfield, L.H. Tumor-Derived α-Fetoprotein Directly Drives Human Natural Killer-Cell Activation and Subsequent Cell Death. Cancer Immunol. Res. 2017, 5, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Tatsumi, T.; Miyagi, T.; Tsunematsu, H.; Aketa, H.; Hosui, A.; Kanto, T.; Hiramatsu, N.; Hayashi, N.; Takehara, T. α-Fetoprotein impairs activation of natural killer cells by inhibiting the function of dendritic cells. Clin. Exp. Immunol. 2011, 165, 211–219. [Google Scholar] [CrossRef]
- Langhans, B.; Alwan, A.W.; Krämer, B.; Glässner, A.; Lutz, P.; Strassburg, C.P.; Nattermann, J.; Spengler, U. Regulatory CD4+ T cells modulate the interaction between NK cells and hepatic stellate cells by acting on either cell type. J. Hepatol. 2015, 62, 398–404. [Google Scholar] [CrossRef]
- Li, T.; Yang, Y.; Hua, X.; Wang, G.; Liu, W.; Jia, C.; Tai, Y.; Zhang, Q.; Chen, G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012, 318, 154–161. [Google Scholar] [CrossRef]
- Venook, A.P.; Papandreou, C.; Furuse, J.; de Guevara, L.L. The incidence and epidemiology of hepatocellular carcinoma: A global and regional perspective. Oncologist 2010, 15, 5–13. [Google Scholar] [CrossRef]
- Sarker, D.; Plummer, R.; Meyer, T.; Sodergren, M.; Basu, B.; Chee, C.E.; Huang, K.-W.; Palmer, D.H.; Ma, Y.T.; Evans, T.R.J.; et al. MTL-CEBPA, a small activating RNA therapeutic up-regulating C/EBP-α, in patients with advanced liver cancer: A first-in-human, multi-centre, open-label, phase I trial. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Lin, A.; Giuliano, C.J.; Palladino, A.; John, K.M.; Abramowicz, C.; Yuan, M.L.; Sausville, E.L.; Lukow, D.A.; Liu, L.; Chait, A.R.; et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019, 11, 11. [Google Scholar] [CrossRef]
- Farra, R.; Musiani, F.; Perrone, F.; Čemažar, M.; Kamenšek, U.; Tonon, F.; Abrami, M.; Ručigaj, A.; Grassi, M.; Pozzato, G.; et al. Polymer-Mediated Delivery of siRNAs to Hepatocellular Carcinoma: Variables Affecting Specificity and Effectiveness. Molecules 2018, 23, 777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willoughby, J.L.S.; Chan, A.; Sehgal, A.; Butler, J.S.; Nair, J.K.; Racie, T.; Shulga-Morskaya, S.; Nguyen, T.; Qian, K.; Yucius, K.; et al. Evaluation of GalNAc-siRNA Conjugate Activity in Pre-clinical Animal Models with Reduced Asialoglycoprotein Receptor Expression. Mol. Ther. 2018, 26, 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thijssen, M.F.; Brüggenwirth, I.M.A.; Gillooly, A.; Khvorova, A.; Kowalik, T.F.; Martins, P.N. Gene Silencing With siRNA (RNA Interference): A New Therapeutic Option During Ex Vivo Machine Liver Perfusion Preservation. Liver Transpl. 2019, 25, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Bracho-Sanchez, E.; Xia, C.Q.; Clare-Salzler, M.J.; Keselowsky, B.G. Micro and Nano Material Carriers for Immunomodulation. Am. J. Transplant. 2016, 16, 3362–3370. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Darband, S.G.; Mirza-Aghazadeh-Attari, M.; Kaviani, M.; Mihanfar, A.; Sadighparvar, S.; Yousefi, B.; Majidinia, M. Exosomes: Natural nanoparticles as bio shuttles for RNAi delivery. J. Control. Release 2018, 289, 158–170. [Google Scholar] [CrossRef]
- Lu, M.; Xing, H.; Xun, Z.; Yang, T.; Ding, P.; Cai, C.; Wang, D.; Zhao, X. Exosome-based small RNA delivery: Progress and prospects. Asian J. Pharm. Sci. 2018, 13, 1–11. [Google Scholar] [CrossRef]
- Shahabipour, F.; Barati, N.; Johnston, T.P.; Derosa, G.; Maffioli, P.; Sahebkar, A. Exosomes: Nanoparticulate tools for RNA interference and drug delivery. J. Cell. Physiol. 2017, 232, 1660–1668. [Google Scholar] [CrossRef] [Green Version]
- Morishita, M.; Takahashi, Y.; Nishikawa, M.; Takakura, Y. Pharmacokinetics of Exosomes-An Important Factor for Elucidating the Biological Roles of Exosomes and for the Development of Exosome-Based Therapeutics. J. Pharm. Sci. 2017, 106, 2265–2269. [Google Scholar] [CrossRef] [Green Version]
- Lunavat, T.R.; Jang, S.C.; Nilsson, L.; Park, H.T.; Repiska, G.; Lässer, C.; Nilsson, J.A.; Gho, Y.S.; Lötvall, J. RNAi delivery by exosome-mimetic nanovesicles—Implications for targeting c-Myc in cancer. Biomaterials 2016, 102, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yuan, Y.; Liu, M.; Hu, X.; Quan, Y.; Chen, X. Tumor-specific delivery of KRAS siRNA with iRGD-exosomes efficiently inhibits tumor growth. ExRNA 2019, 1, 28. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Drug Name | Target | Phase | Company | NCT Reference | Status |
|---|---|---|---|---|---|
| siRNA-EphA2-DOP C | EPHA2 | Phase I | MD Anderson | NCT01591356 | ongoing |
| TKM-PLK1/TKM-080301 | PLK1 | Phase I/II | Arbutus | NCT02191878 | completed |
| pbi-shRNA STMN | STMN | Phase Ib/2 | Gradalis | NCT01505153 | completed |
| ALN-VSP02 | VEGF and KSP | Phase I | Alnylam | NCT01158079 | completed |
| Fang | Furin and GMSF | Phase I | Gradalis | NCT01061840 | completed |
| DCR-MYC | MYC | Phase I | Dicema | NCT02314052 | terminated |
| MRX34 | miR-32 mimic | Phase I | Mirna Therapeutics | NCT01829971 | terminated |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mroweh, M.; Decaens, T.; Marche, P.N.; Macek Jilkova, Z.; Clément, F. Modulating the Crosstalk between the Tumor and Its Microenvironment Using RNA Interference: A Treatment Strategy for Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 5250. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155250
Mroweh M, Decaens T, Marche PN, Macek Jilkova Z, Clément F. Modulating the Crosstalk between the Tumor and Its Microenvironment Using RNA Interference: A Treatment Strategy for Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2020; 21(15):5250. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155250
Chicago/Turabian StyleMroweh, Mariam, Thomas Decaens, Patrice N Marche, Zuzana Macek Jilkova, and Flora Clément. 2020. "Modulating the Crosstalk between the Tumor and Its Microenvironment Using RNA Interference: A Treatment Strategy for Hepatocellular Carcinoma" International Journal of Molecular Sciences 21, no. 15: 5250. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155250