Subcellular Localization Relevance and Cancer-Associated Mechanisms of Diacylglycerol Kinases

 , , , ,
, , , ,
Abstract
:1. Introduction
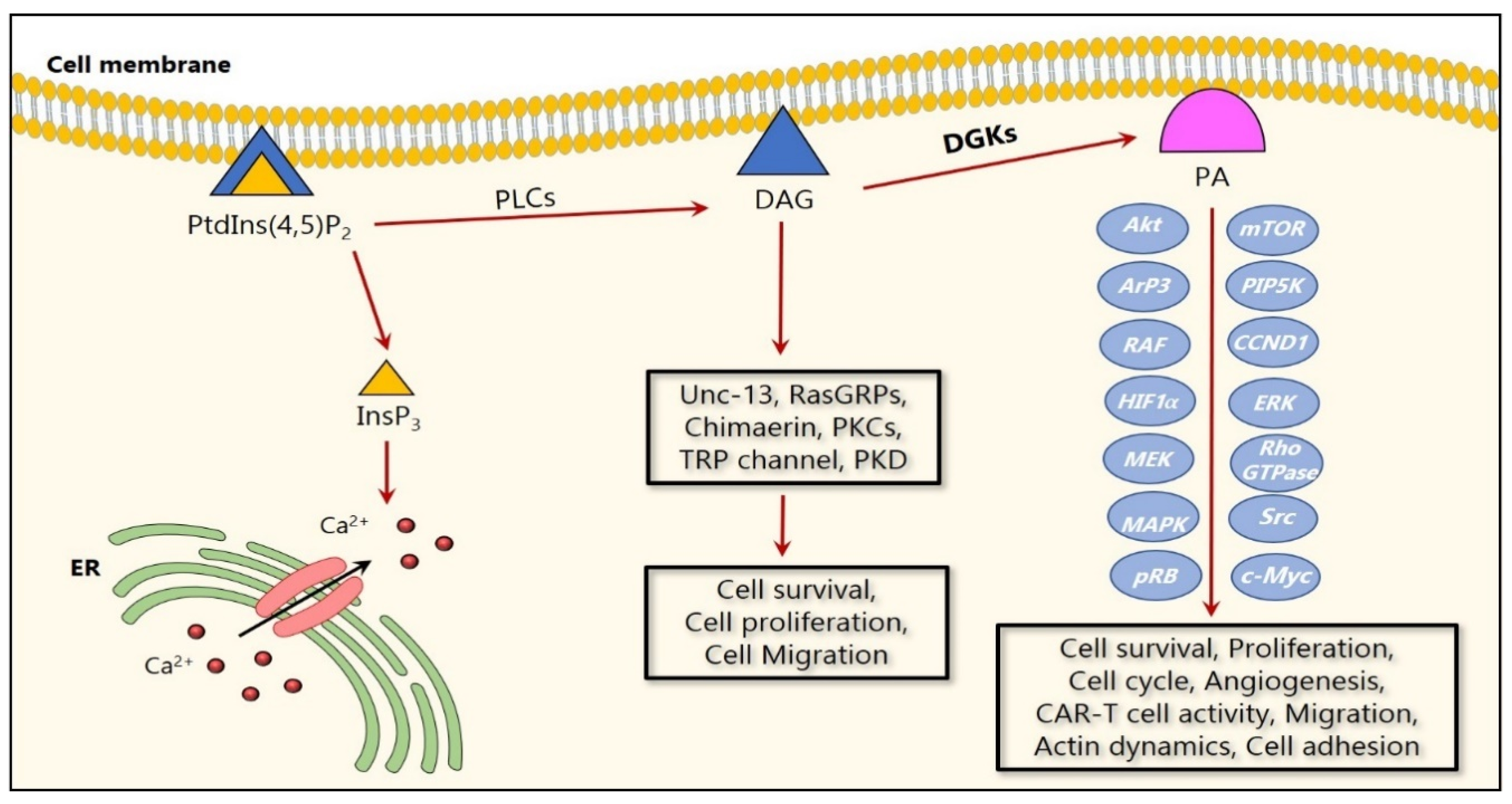
2. Activation and Regulation of DGK Isozymes
3. Cellular Localization and Distribution of DGKs
4. The Impact of DGKs in the Regulation of Cancer Cell Mechanisms
4.1. Cell Growth and Proliferation in Cancer
4.2. Cell Migration, Invasiveness, and Metastasis
5. Targeting DGKs in Cancer Therapies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3D | Three-dimensional |
| Ca2+ | Calcium |
| ACT | Adoptive cell transfer |
| AML | Acute myeloid leukemia |
| CAR | Chimeric antigen receptor |
| CCND1 | Cyclin D1 |
| CD3 | Cluster of differentiation 3 |
| CRC | Colorectal cancer |
| DAG | Diacylglycerol |
| DGKs | Diacylglycerol kinases |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial to mesenchymal transition |
| ERK | Extracellular signal-regulated kinase |
| FAK | Focal adhesion kinase |
| GAP | GTPase-activating protein |
| GLUT1 | Glucose transporter 1 |
| HCC | Hepatocellular carcinoma |
| HER2 | Human epidermal growth factor receptor-2 |
| HGF | Hepatocyte growth factor |
| HIF1α | Hypoxia-inducible factor 1-alpha |
| IL-2 | Interleukin 2 |
| InsP3R | Inositol trisphosphate receptor |
| KS | Kaposi’s sarcoma |
| MAPK | Mitogen activated protein kinase |
| MARCKS | Myristoylated alanine rich kinase substrate |
| MEK | Mitogen-activated protein kinase |
| MMP1 | Type 1 matrix metalloproteases |
| mTOR | Mammalian target of rapamycin |
| NLS | Nuclear localization sequence |
| PA | Phosphatidic acid |
| PHLPP2 | Pleckstrin homology domain leucin-rich repeat protein phosphatase 2 |
| PI | Phosphoinositide |
| PI3K | Phosphoinositide 3-kinase |
| PIP5K | Phosphatidylinositol-4-phosphate 5-kinase |
| PKC | Protein kinase C |
| PLC | Phospholipases C |
| PLD | Phospholipase D |
| pRb | Retinoblastoma protein |
| PS | Phosphatidylserine |
| PtdIns(4,5)P2 | Phosphatidylinositol 4,5 biphosphate |
| RCP Raf-1 | Rab-coupling protein Rapidly accelerated fibrosarcoma-1 |
| RasGRP | Rat sarcoma virus guanyl nucleotide-releasing protein |
| RTK | Receptor tyrosine kinase |
| SAM | Sterile α motif |
| siRNA | Short interfering RNA |
| SMSr | Sphingomyelin synthase related protein |
| STAT3 | Signal transducers and activators of transcription 3 |
| TGFβ | Transforming growth factor β |
| Unc-13 | Mammalian uncoordinated 13 |
| VEGF | Vascular endothelial growth factor |
| VEGFR | Vascular endothelial growth factor receptor |
References
- Fukumoto, M.; Ijuin, T.; Takenawa, T. PI(3,4)P2 Plays Critical Roles in the Regulation of Focal Adhesion Dynamics of MDA-MB-231 Breast Cancer Cells. Cancer Sci. 2017, 108, 941–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, A.R.; Elong Edimo, W.; Erneux, C. Phosphoinositide 5-Phosphatase Activities Control Cell Motility in Glioblastoma: Two Phosphoinositides PI(4,5)P2 and PI(3,4)P2 Are Involved. Adv. Biol. Regul. 2018, 67, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.K.; Williams, S.A.; Bindra, G.K.; Lay, F.T.; Poon, I.K.H.; Hulett, M.D. Phosphoinositides: Multipurpose Cellular Lipids with Emerging Roles in Cell Death. Cell Death Differ. 2019, 26, 781–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waugh, M.G. The Great Escape: How Phosphatidylinositol 4-Kinases and PI4P Promote Vesicle Exit from the Golgi (and Drive Cancer). Biochem. J. 2019, 476, 2321–2346. [Google Scholar] [CrossRef]
- Ulicna, L.; Kalendova, A.; Kalasova, I.; Vacik, T.; Hozák, P. PIP2 Epigenetically Represses rRNA Genes Transcription Interacting with PHF8. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 266–275. [Google Scholar] [CrossRef]
- Owusu Obeng, E.; Rusciano, I.; Marvi, M.V.; Fazio, A.; Ratti, S.; Follo, M.Y.; Xian, J.; Manzoli, L.; Billi, A.M.; Mongiorgi, S.; et al. Phosphoinositide-Dependent Signaling in Cancer: A Focus on Phospholipase C Isozymes. Int. J. Mol. Sci. 2020, 21, 2581. [Google Scholar] [CrossRef] [Green Version]
- Arranz-Nicolás, J.; Mérida, I. Biological Regulation of Diacylglycerol Kinases in Normal and Neoplastic Tissues: New Opportunities for Cancer Immunotherapy. Adv. Biol. Regul. 2020, 75, 100663. [Google Scholar] [CrossRef]
- Mérida, I.; Torres-Ayuso, P.; Ávila-Flores, A.; Arranz-Nicolás, J.; Andrada, E.; Tello-Lafoz, M.; Liébana, R.; Arcos, R. Diacylglycerol Kinases in Cancer. Adv. Biol. Regul. 2017, 63, 22–31. [Google Scholar] [CrossRef]
- Merida, I.; Arranz-Nicolás, J.; Torres-Ayuso, P.; Ávila-Flores, A. Diacylglycerol Kinase Malfunction in Human Disease and the Search for Specific Inhibitors. Handb. Exp. Pharmacol. 2020, 259, 133–162. [Google Scholar] [CrossRef]
- Ávila-Flores, A.; Santos, T.; Rincón, E.; Mérida, I. Modulation of the Mammalian Target of Rapamycin Pathway by Diacylglycerol Kinase-Produced Phosphatidic Acid. J. Biol. Chem. 2005, 280, 10091–10099. [Google Scholar] [CrossRef] [Green Version]
- Griner, E.M.; Kazanietz, M.G. Protein Kinase C and Other Diacylglycerol Effectors in Cancer. Nat. Rev. Cancer. 2007, 7, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Baldanzi, G.; Bettio, V.; Malacarne, V.; Graziani, A. Diacylglycerol Kinases: Shaping Diacylglycerol and Phosphatidic Acid Gradients to Control Cell Polarity. Front. Cell Dev. Biol. 2016, 4, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakane, F.; Mizuno, S.; Takahashi, D.; Sakai, H. Where Do Substrates of Diacylglycerol Kinases Come from? Diacylglycerol Kinases Utilize Diacylglycerol Species Supplied from Phosphatidylinositol Turnover-Independent Pathways. Adv. Biol. Regul. 2018, 67, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Cockcroft, S.; Raghu, P. Topological Organisation of the Phosphatidylinositol 4,5-Bisphosphate-Phospholipase C Resynthesis Cycle: PITPs Bridge the ER-PM Gap. Biochem. J. 2016, 473, 4289–4310. [Google Scholar] [CrossRef] [PubMed]
- Poli, A.; Ratti, S.; Finelli, C.; Mongiorgi, S.; Clissa, C.; Lonetti, A.; Cappellini, A.; Catozzi, A.; Barraco, M.; Suh, P.G.; et al. Nuclear Translocation of PKC-α Is Associated with Cell Cycle Arrest and Erythroid Differentiation in Myelodysplastic Syndromes (MDSs). FASEB J. 2018, 32, 681–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratti, S.; Mongiorgi, S.; Ramazzotti, G.; Follo, M.Y.; Mariani, G.A.; Suh, P.G.; McCubrey, J.A.; Cocco, L.; Manzoli, L. Nuclear Inositide Signaling Via Phospholipase C. J. Cell. Biochem. 2017, 118, 1969–1978. [Google Scholar] [CrossRef]
- Luo, B.; Regier, D.S.; Prescott, S.M.; Topham, M.K. Diacylglycerol Kinases. Cell. Signal. 2004, 16, 983–989. [Google Scholar] [CrossRef]
- Lo Vasco, V.R.; Galabrese, G.; Manzoli, L.; Palka, G.; Spadano, A.; Morizio, E.; Guanciali-Franchi, P.; Fantasia, D.; Cocco, L. Inositide-Specific Phospholipase c β1 Gene Deletion in the Progression of Myelodysplastic Syndrome to Acute Myeloid Leukemia. Leukemia 2004, 18, 1122–1126. [Google Scholar] [CrossRef] [Green Version]
- Manzoli, L.; Billi, A.M.; Gilmour, R.S.; Marteili, A.M.; Matteucci, A.; Rubbini, S.; Weber, G.; Cocco, L. Phosphoinositide Signaling in Nuclei of Friend Cells: Tiazofurin Down-Regulates Phospholipase C β1. Cancer Res. 1995, 55, 2978–2980. [Google Scholar]
- Irvine, R.F. Nuclear Lipid Signalling. Nat. Rev. Mol. Cell Biol. 2003, 4, 349–360. [Google Scholar] [CrossRef]
- Poli, A.; Billi, A.M.; Mongiorgi, S.; Ratti, S.; Mccubrey, J.A.; Suh, P.G.; Cocco, L.; Ramazzotti, G. Nuclear Phosphatidylinositol Signaling: Focus on Phosphatidylinositol Phosphate Kinases and Phospholipases C. J. Cell. Phys. 2016, 231, 1645–1655. [Google Scholar] [CrossRef] [PubMed]
- Sakane, F.; Imai, S.; Kai, M.; Yasuda, S.; Kanoh, H. Diacylglycerol Kinases as Emerging Potential Drug Targets for a Variety of Diseases. Curr. Drug Targets 2008, 9, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Sakane, F. Recent Progress on Type II Diacylglycerol Kinases: The Physiological Functions of Diacylglycerol Kinase Δ, η and κ and Their Involvement in Disease. J. Biochem. 2012, 152, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, H.; Samayoa, J.; Hruban, R.H.; Karchin, R. Prioritization of Driver Mutations in Pancreatic Cancer Using Cancer-Specific High-Throughput Annotation of Somatic Mutations (CHASM). Cancer Biol. Ther. 2010, 10, 582–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, I.Y.; Kim, Y.Y.; Yu, H.S.; Lee, M.; Kim, S.; Lee, J. CRISPR/Cas9-Mediated Knockout of DGK Improves Antitumor Activities of Human T Cells. Cancer Res. 2018, 78, 4692–4703. [Google Scholar] [CrossRef] [Green Version]
- Kai, M.; Yamamoto, E.; Sato, A.; Yamano, H.; Niinuma, T.; Kitajima, H.; Harada, T.; Aoki, H.; Maruyama, R.; Toyota, M.; et al. Epigenetic Silencing of Diacylglycerol Kinase Gamma in Colorectal Cancer. Mol. Carcinog. 2017, 56, 1743–1752. [Google Scholar] [CrossRef] [PubMed]
- Takeishi, K.; Taketomi, A.; Shirabe, K.; Toshima, T.; Motomura, T.; Ikegami, T.; Yoshizumi, T.; Sakane, F.; Maehara, Y. Diacylglycerol Kinase Alpha Enhances Hepatocellular Carcinoma Progression by Activation of Ras-Raf-MEK-ERK Pathway. J. Hepatol. 2012, 57, 77–83. [Google Scholar] [CrossRef]
- Torres-Ayuso, P.; Daza-Martín, M.; Martín-Pérez, J.; Ávila-Flores, A.; Mérida, I. Diacylglycerol Kinase a Promotes 3D Cancer Cell Growth and Limits Drug Sensitivity through Functional Interaction with Src. Oncotarget 2014, 5, 9710–9726. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.L.; Floyd, D.H.; Xiao, A.; Mullins, G.R.; Kefas, B.A.; Xin, W.; Yacur, M.N.; Abounader, R.; Lee, J.K.; Wilson, G.M.; et al. Diacylglycerol Kinase α Is a Critical Signaling Node and Novel Therapeutic Target in Glioblastoma and Other Cancers. Cancer Discov. 2013, 3, 782–797. [Google Scholar] [CrossRef] [Green Version]
- Cai, K.; Mulatz, K.; Ard, R.; Nguyen, T.; Gee, S.H. Increased Diacylglycerol Kinase ζ Expression in Human Metastatic Colon Cancer Cells Augments Rho GTPase Activity and Contributes to Enhanced Invasion. BMC Cancer 2014, 14, 208. [Google Scholar] [CrossRef] [Green Version]
- Torres-Ayuso, P.; Tello-Lafoz, M.; Mérida, I.; Ávila-Flores, A. Diacylglycerol Kinase-ζ Regulates mTORC1 and Lipogenic Metabolism in Cancer Cells through SREBP-1. Oncogenesis 2015, 4, e164. [Google Scholar] [CrossRef] [PubMed]
- Crotty, T.M.; Nakano, T.; Stafforini, D.M.; Topham, M.K. Diacylglycerol Kinase δ Modulates Akt Phosphorylation through Pleckstrin Homology Domain Leucine-Rich Repeat Protein Phosphatase 2 (PHLPP2). J. Biol. Chem. 2013, 288, 1439–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, S.; Kai, M.; Imai, S.I.; Takeishi, K.; Taketomi, A.; Toyota, M.; Kanoh, H.; Sakane, F. Diacylglycerol Kinase η Augments C-Raf Activity and B-Raf/C-Raf Heterodimerization. J. Biol. Chem. 2009, 284, 29559–29570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etcheverry, A.; Aubry, M.; Idbaih, A.; Vauleon, E.; Marie, Y.; Menei, P.; Boniface, R.; Figarella-Branger, D.; Karayan-Tapon, L.; Quillien, V.; et al. DGKI Methylation Status Modulates the Prognostic Value of MGMT in Glioblastoma Patients Treated with Combined Radio-Chemotherapy with Temozolomide. PLoS ONE 2014, 9, e104455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revill, K.; Wang, T.; Lachenmayer, A.; Kojima, K.; Harrington, A.; Li, J.; Hoshida, Y.; Llovet, J.M.; Powers, S. Genome-Wide Methylation Analysis and Epigenetic Unmasking Identify Tumor Suppressor Genes in Hepatocellular Carcinoma. Gastroenterology 2013, 145, 1424–1435.e1-25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mérida, I.; Ávila-Flores, A.; Merino, E. Diacylglycerol Kinases: At the Hub of Cell Signalling. Biochem. J. 2008, 409, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulga, Y.V.; Topham, M.K.; Epand, R.M. Regulation and Functions of Diacylglycerol Kinases. Chem. Rev. 2011, 111, 6186–6208. [Google Scholar] [CrossRef]
- Martinez-Moreno, M.; Garcia-Lievana, J.; Soutar, D.; Torres-Ayuso, P.; Andrada, E.; Zhong, X.-P.; Koretzky, G.A.; Merida, I.; Avila-Flores, A. FoxO-Dependent Regulation of Diacylglycerol Kinase Gene Expression. Mol. Cell. Biol. 2012, 32, 4168–4180. [Google Scholar] [CrossRef] [Green Version]
- Topham, M.K. Signaling Roles of Diacylglycerol Kinases. J. Cell. Biochem. 2006, 97, 474–484. [Google Scholar] [CrossRef]
- Sakane, F.; Imai, S.; Kai, M.; Yasuda, S.; Kanoh, H. Diacylglycerol Kinases: Why so Many of Them? Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2007, 1771, 793–806. [Google Scholar] [CrossRef]
- Topham, M.K.; Prescott, S.M. Mammalian Diacylglycerol Kinases, a Family of Lipid Kinases with Signaling Functions. J. Biol. Chem. 1999, 274, 11447–11450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramovici, H.; Hogan, A.B.; Obagi, C.; Topham, M.K.; Gee, S.H. Diacylglycerol Kinase-ζ Localization in Skeletal Muscle Is Regulated by Phosphorylation and Interaction with Syntrophins. Mol. Biol. Cell 2003, 14, 4499–4511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katagiri, Y.; Ito, T.; Saino-Saito, S.; Hozumi, Y.; Suwabe, A.; Otake, K.; Sata, M.; Kondo, H.; Sakane, F.; Kanoh, H.; et al. Expression and Localization of Diacylglycerol Kinase Isozymes and Enzymatic Features in Rat Lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L1171–L1178. [Google Scholar] [CrossRef] [PubMed]
- Topham, M.K.; Epand, R.M. Mammalian Diacylglycerol Kinases: Molecular Interactions and Biological Functions of Selected Isoforms. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 416–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, T.; Sakane, F.; Imai, S.I.; Houkin, K.; Kanoh, H. Identification and Characterization of Two Splice Variants of Human Diacylglycerol Kinase η. J. Biol. Chem. 2003, 278, 34364–34372. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Kanematsu, T.; Misumi, Y.; Sakane, F.; Konishi, H.; Kikkawa, U.; Watanabe, Y.; Katan, M.; Hirata, M. Distinct Specificity in the Binding of Inositol Phosphates by Pleckstrin Homology Domains of Pleckstrin, RAC-Protein Kinase, Diacylglycerol Kinase and a New 130 kDa Protein. Biochim. Biophys. Acta Mol. Cell Res. 1997, 1359, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Tu-Sekine, B.; Goldschmidt, H.L.; Raben, D.M. DGK-θ: Structure, Enzymology, and Physiological Roles. Front. Cell Dev. Biol. 2016, 4, 101. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.H.; Newton, A.C.; Parker, P.J.; Blumberg, P.M.; Nishizuka, Y. Taxonomy and Function of C1 Protein Kinase C Homology Domains. Protein Sci. 1997, 6, 477–480. [Google Scholar] [CrossRef] [Green Version]
- Nelson, C.D.; Perry, S.J.; Regier, D.S.; Prescott, S.M.; Topham, M.K.; Lefkowitz, R.J. Targeting of Diacylglycerol Degradation to M1 Muscarinic Receptors by β-Arrestins. Science 2007, 315, 663–666. [Google Scholar] [CrossRef]
- Mérida, I.; Arranz-Nicolás, J.; Rodríguez-Rodríguez, C.; Ávila-Flores, A. Diacylglycerol Kinase Control of Protein Kinase C. Biochem. J. 2019, 476, 1205–1219. [Google Scholar] [CrossRef]
- Ciprés, A.; Carrasco, S.; Mérida, I. Deletion of the Acidic-Rich Domain of the IL-2Rβ Chain Increases Receptor-Associated PI3K Activity. FEBS Lett. 2001, 500, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Sakane, F.; Kai, M.; Wada, I.; Imai, S.I.; Kanoh, H. The C-Terminal Part of Diacylglycerol Kinase α Lacking Zinc Fingers Serves as a Catalytic Domain. Biochem. J. 1996, 318, 583–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjuán, M.A.; Jones, D.R.; Izquierdo, M.; Mérida, I. Role of Diacylglycerol Kinase α in the Attenuation of Receptor Signaling. J. Cell Biol. 2001, 153, 207–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, I.; Casaseca, T.; Martinez-A, C.; Kanoh, H.; Merida, I. Phosphatidic Acid Generation through Interleukin 2 (IL-2)-Induced α-Diacylglycerol Kinase Activation Is an Essential Step in IL-2-Mediated Lymphocyte Proliferation. J. Biol. Chem. 1996, 271, 10334–10340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjuán, M.A.; Pradet-Balade, B.; Jones, D.R.; Martínez-A, C.; Stone, J.C.; Garcia-Sanz, J.A.; Mérida, I. T Cell Activation In Vivo Targets Diacylglycerol Kinase α to the Membrane: A Novel Mechanism for Ras Attenuation. J. Immunol. 2003, 170, 2877–2883. [Google Scholar] [CrossRef] [PubMed]
- Ciprés, A.; Carrasco, S.; Merino, E.; Díaz, E.; Krishna, U.M.; Falck, J.R.; Martínez-A, C.; Mérida, I. Regulation of Diacylglycerol Kinase α by Phosphoinositide 3-Kinase Lipid Products. J. Biol. Chem. 2003, 278, 35629–35635. [Google Scholar] [CrossRef] [Green Version]
- Thirugnanam, S.; Topham, M.K.; Epand, R.M. Physiological Implications of the Contrasting Modulation of the Activities of the ε and ζ-Isoforms of Diacylglycerol Kinase. Biochemistry 2001, 40, 10607–10613. [Google Scholar] [CrossRef]
- Topham, M.K.; Prescott, S.M. Diacylglycerol Kinase ζ Regulates Ras Activation by a Novel Mechanism. J. Cell Biol. 2001, 19, 1135–1143. [Google Scholar] [CrossRef] [Green Version]
- Regier, D.S.; Higbee, J.; Lund, K.M.; Sakane, F.; Prescott, S.M.; Topham, M.K. Diacylglycerol Kinase ι Regulates Ras Guanyl-Releasing Protein 3 and Inhibits Rap1 Signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 7595–7600. [Google Scholar] [CrossRef] [Green Version]
- Zhong, X.P.; Hainey, E.A.; Olenchock, B.A.; Zhao, H.; Topham, M.K.; Koretzky, G.A. Regulation of T Cell Receptor-Induced Activation of the Ras-ERK Pathway by Diacylglycerol Kinase ζ. J. Biol. Chem. 2002, 277, 31089–31098. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.; Prescott, S.M.; Topham, M.K. Association of Diacylglycerol Kinase ζ with Protein Kinase C α: Spatial Regulation of Diacylglycerol Signaling. J. Cell Biol. 2003, 160, 929–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topham, M.K.; Bunting, M.; Zimmerman, G.A.; McIntyre, T.M.; Blackshear, P.J.; Prescott, S.M. Protein Kinase C Regulates the Nuclear Localization of Diacylglycerol Kinase-ζ. Nature 1998, 394, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Tanaka, T.; Nakano, T.; Okada, M.; Hozumi, Y.; Topham, M.K.; Martelli, A.M. DGKζ under Stress Conditions: “To Be Nuclear or Cytoplasmic, That Is the Question.”. Adv. Biol. Regul. 2014, 54, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Kai, M.; Yasuda, S.; Imai, S.; Toyota, M.; Kanoh, H.; Sakane, F. Diacylglycerol Kinase α Enhances Protein Kinase Cζ-Dependent Phosphorylation at Ser311 of p65/RelA Subunit of Nuclear Factor-κB. FEBS Lett. 2009, 583, 3265–3268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, X.P.; Hainey, E.A.; Olenchock, B.A.; Jordan, M.S.; Maltzman, J.S.; Nichols, K.E.; Shen, H.; Koretzky, G.A. Enhanced T Cell Responses due to Diacylglycerol Kinase ζ Deficiency. Nat. Immunol. 2003, 4, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Crotty, T.; Cai, J.; Sakane, F.; Taketomi, A.; Prescott, S.M.; Topham, M.K. Diacylglycerol Kinase δ Regulates Protein Kinase C and Epidermal Growth Factor Receptor Signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 15485–15490. [Google Scholar] [CrossRef] [Green Version]
- Olenchock, B.A.; Guo, R.; Carpenter, J.H.; Jordan, M.; Topham, M.K.; Koretzky, G.A.; Zhong, X.P. Disruption of Diacylglycerol Metabolism Impairs the Induction of T Cell Anergy. Nat. Immunol. 2006, 7, 1174–1181. [Google Scholar] [CrossRef]
- Rodriguez De Turco, E.B.; Tang, W.; Topham, M.K.; Sakane, F.; Marcheselli, V.L.; Chen, C.; Taketomi, A.; Prescott, S.M.; Bazan, N.G. Diacylglycerol Kinase ε Regulates Seizure Susceptibility and Long-Term Potentiation through Arachidonoylinositol Lipid Signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 4740–4745. [Google Scholar] [CrossRef] [Green Version]
- Murakami, C.; Hoshino, F.; Sakai, H.; Hayashi, Y.; Yamashita, A.; Sakane, F. Diacylglycerol Kinase δ and Sphingomyelin Synthase-Related Protein Functionally Interact via Their Sterile α Motif Domains. J. Biol. Chem. 2020, 295, 2932–2947. [Google Scholar] [CrossRef]
- Van Der Bend, R.L.; De Widt, J.; Hilkmann, H.; Van Blitterswijk, W.J. Diacylglycerol Kinase in Receptor-Stimulated Cells Converts Its Substrate in a Topologically Restricted Manner. J. Biol. Chem. 1994, 269, 4098–4102. [Google Scholar]
- Matsubara, T.; Ikeda, M.; Kiso, Y.; Sakuma, M.; Yoshino, K.I.; Sakane, F.; Merida, I.; Saito, N.; Shirai, Y. C-Abl Tyrosine Kinase Regulates Serum-Induced Nuclear Export of Diacylglycerol Kinase α by Phosphorylation at Tyr-218. J. Biol. Chem. 2012, 287, 5507–5517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S.I.; Kai, M.; Yasuda, S.; Kanoh, H.; Sakane, F. Identification and Characterization of a Novel Human Type II Diacylglycerol Kinase, DGKκ. J. Biol. Chem. 2005, 280, 39870–39881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakane, F.; Imai, S.I.; Yamada, K.; Murakami, T.; Tsushima, S.; Kanoh, H. Alternative Splicing of the Human Diacylglycerol Kinase δ Gene Generates Two Isoforms Differing in Their Expression Patterns and in Regulatory Functions. J. Biol. Chem. 2002, 277, 43519–43526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, T.; Carrasco, S.; Jones, D.R.; Mérida, I.; Eguinoa, A. Dynamics of Diacylglycerol Kinase Zeta Translocation in Living T-Cells. Study of the Structural Domain Requirements for Translocation and Activity. J. Biol. Chem. 2002, 277, 30300–30309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Follo, M.Y.; Ratti, S.; Manzoli, L.; Ramazzotti, G.; Faenza, I.; Fiume, R.; Mongiorgi, S.; Suh, P.G.; McCubrey, J.A.; Cocco, L. Inositide-Dependent Nuclear Signalling in Health and Disease. Handb. Exp. Pharmacol. 2019, 259, 291–308. [Google Scholar] [CrossRef]
- Ratti, S.; Ramazzotti, G.; Faenza, I.; Fiume, R.; Mongiorgi, S.; Billi, A.M.; McCubrey, J.A.; Suh, P.-G.; Manzoli, L.; Follo, M.Y. Nuclear Inositide Signaling and Cell Cycle. Adv. Biol. Regul. 2018, 67, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ramazzotti, G.; Ratti, S.; Fiume, R.; Follo, M.Y.; Billi, A.M.; Rusciano, I.; Obeng, E.O.; Manzoli, L.; Cocco, L.; Faenza, I. Phosphoinositide 3 Kinase Signaling in Human Stem Cells from Reprogramming to Differentiation: A Tale in Cytoplasmic and Nuclear Compartments. Int. J. Mol. Sci. 2019, 20, 2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faenza, I.; Bavelloni, A.; Fiume, R.; Lattanzi, G.; Maraldi, N.M.; Gilmour, R.S.; Martelli, A.M.; Suh, P.G.; Billi, A.M.; Cocco, L. Up-regulation of nuclear PLCbeta1 in myogenic differentiation. J. Cell Physiol. 2003, 195, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Xian, J.; Owusu Obeng, E.; Ratti, S.; Rusciano, I.; Marvi, M.V.; Fazio, A.; De Stefano, A.; Mongiorgi, S.; Cappellini, A.; Ramazzotti, G.; et al. Nuclear Inositides and Inositide-Dependent Signaling Pathways in Myelodysplastic Syndromes. Cells 2020, 9, 697. [Google Scholar] [CrossRef] [Green Version]
- Manzoli, F.A.; Maraldi, N.M.; Cocco, L.; Capitani, S.; Facchini, A. Chromatin Phospholipids in Normal and Chronic Lymphocytic Leukemia Lymphocytes. Cancer Res. 1977, 37, 843–849. [Google Scholar]
- Ratti, S.; Follo, M.Y.; Ramazzotti, G.; Faenza, I.; Fiume, R.; Suh, P.G.; McCubrey, J.A.; Manzoli, L.; Cocco, L. Nuclear Phospholipase C Isoenzyme Imbalance Leads to Pathologies in Brain, Hematologic, Neuromuscular, and Fertility Disorders. J. Lipid Res. 2019, 60, 312–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mongiorgi, S.; Follo, M.Y.; Ryoul Yang, Y.; Ratti, S.; Manzoli, L.; A. McCubrey, J.; Maria Billi, A.; Suh, P.-G.; Cocco, L. Selective Activation of Nuclear PI-PLCbeta1 During Normal and Therapy-Related Differentiation. Curr. Pharm. Des. 2016, 22, 2345–2348. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Shirai, Y.; Miyasaka, K.; Murakami, T.; Yamaguchi, Y.; Ueyama, T.; Kai, M.; Sakane, F.; Kanoh, H.; Hashimoto, T.; et al. Nuclear Transportation of Diacylglycerol Kinase γ and Its Possible Function in the Nucleus. J. Biol. Chem. 2006, 281, 6152–6164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Blitterswijk, W.J.; Houssa, B. Properties and Functions of Diacylglycerol Kinases. Cell. Signal. 2000, 12, 595–605. [Google Scholar] [CrossRef]
- Tabellini, G.; Bortul, R.; Santi, S.; Riccio, M.; Baldini, G.; Cappellini, A.; Billi, A.M.; Berezney, R.; Ruggeri, A.; Cocco, L.; et al. Diacylglycerol Kinase-θ Is Localized in the Speckle Domains of the Nucleus. Exp. Cell Res. 2003, 287, 143–154. [Google Scholar] [CrossRef]
- Van Baal, J.; De Widt, J.; Divecha, N.; Van Blitterswijk, W.J. Translocation of Diacylglycerol Kinase θ from Cytosol to Plasma Membrane in Response to Activation of G Protein-Coupled Receptors and Protein Kinase C. J. Biol. Chem. 2005, 280, 9870–9878. [Google Scholar] [CrossRef] [Green Version]
- Nakano, T.; Matsui, H.; Tanaka, T.; Hozumi, Y.; Iseki, K.; Kawamae, K.; Goto, K. Arachidonoyl-Specific Diacylglycerol Kinase ε and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2016, 4, 132. [Google Scholar] [CrossRef] [Green Version]
- Hozumi, Y.; Akimoto, R.; Suzuki, A.; Otani, K.; Watanabe, M.; Goto, K. Expression and Localization of the Diacylglycerol Kinase Family and of Phosphoinositide Signaling Molecules in Adrenal Gland. Cell Tissue Res. 2015, 362, 295–305. [Google Scholar] [CrossRef]
- Hozumi, Y.; Fujiwara, H.; Kaneko, K.; Fujii, S.; Topham, M.K.; Watanabe, M.; Goto, K. Diacylglycerol Kinase ε Localizes to Subsurface Cisterns of Cerebellar Purkinje Cells. Cell Tissue Res. 2017, 368, 441–458. [Google Scholar] [CrossRef]
- Tolias, K.F.; Couvillon, A.D.; Cantley, L.C.; Carpenter, C.L. Characterization of a Rac1- and RhoGDI-Associated Lipid Kinase Signaling Complex. Mol. Cell. Biol. 1998, 18, 762–770. [Google Scholar] [CrossRef] [Green Version]
- Maeda, Y.; Shibata, K.; Akiyama, R.; Murakami, Y.; Takao, S.; Murakami, C.; Takahashi, D.; Sakai, H.; Sakane, F. Diacylglycerol Kinase β Induces Filopodium Formation via Its C1, Catalytic and Carboxy-Terminal Domains and Interacts with the Rac1-GTPase-Activating Protein, β2-Chimaerin. Biochem. Biophys. Res. Commun. 2018, 504, 54–60. [Google Scholar] [CrossRef]
- Houssa, B.; De Widt, J.; Kranenburg, O.; Moolenaar, W.H.; Van Blitterswijk, W.J. Diacylglycerol Kinase θ Binds to and Is Negatively Regulated by Active RhoA. J. Biol. Chem. 1999, 274, 6820–6822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramovici, H.; Mojtabaie, P.; Parks, R.J.; Zhong, X.P.; Koretzky, G.A.; Topham, M.K.; Gee, S.H. Diacylglycerol Kinase ζ Regulates Actin Cytoskeleton Reorganization Through Dissociation of rac1 from rhoGDI. Mol. Biol. Cell 2009, 20, 2049–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, N.; Hozumi, Y.; Ito, T.; Hosoya, T.; Kondo, H.; Goto, K. Differential Subcellular Targeting and Activity-Dependent Subcellular Localization of Diacylglycerol Kinase Isozymes in Transfected Cells. Eur. J. Cell Biol. 2007, 86, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Berrar, D.; Sturgeon, B.; Bradbury, I.; Downes, C.S.; Dubitzky, W. Survival Trees for Analyzing Clinical Outcome in Lung Adenocarcinomas Based on Gene Expression Profiles: Identification of Neogenin and Diacylglycerol Kinase α Expression as Critical Factors. J. Comput. Biol. 2005, 12, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained Proliferation in Cancer: Mechanisms and Novel Therapeutic Targets. Sem. Cancer Biol. 2015, 35, 25–54. [Google Scholar] [CrossRef]
- Evangelisti, C.; Tazzari, P.L.; Riccio, M.; Fiume, R.; Hozumi, Y.; Falà, F.; Goto, K.; Manzoli, L.; Cocco, L.; Martelli, A.M. Nuclear Diacylglycerol Kinase-ζ Is a Negative Regulator of Cell Cycle Progression in C2C12 Mouse Myoblasts. FASEB J. 2007, 21, 3297–3307. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dong, C.; Tian, Y.; Li, X.; Wang, B.; Zhai, D.; Bai, Y.; Chao, X. Knockdown of Diacylglycerol Kinase Zeta (DGKZ) Induces Apoptosis and G2/M Phase Arrest in Human Acute Myeloid Leukemia HL-60 Cells through MAPK/survivin/caspase Pathway. Pharmazie 2019, 74, 418–422. [Google Scholar] [CrossRef]
- Gu, X.; Wan, G.; Chen, N.; Li, J.; Chen, B.; Tang, Y.; Gu, W.; Jin, C.; Meng, J.; Zhang, P.; et al. DGKζ Plays Crucial Roles in the Proliferation and Tumorigenicity of Human Glioblastoma. Int. J. Biol. Sci. 2019, 15, 1872–1881. [Google Scholar] [CrossRef]
- Poli, A.; Fiume, R.; Baldanzi, G.; Capello, D.; Ratti, S.; Gesi, M.; Manzoli, L.; Graziani, A.; Suh, P.G.; Cocco, L.; et al. Nuclear Localization of Diacylglycerol Kinase Alpha in K562 Cells Is Involved in Cell Cycle Progression. J. Cell. Physiol. 2017, 232, 2550–2557. [Google Scholar] [CrossRef]
- Yamaki, S.; Mizuno, M.; Sato, H.; Sakai, S.; Kado, K.; Kumagai, H.; Kojima, T.; Okabe, T.; Nagano, Y.; Shirai, F.S.; et al. A Novel Diacylglycerol Kinase-Selective Inhibitor, CU-3, Induces Cancer Cell Apoptosis and Enhances Immune Response. J. Lipid Res. 2016, 57, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Jia, J.; Yao, M.; Kang, J.; Wang, Y.; Yan, X.; Zhang, L.; Lv, Q.; Chen, X.; Lu, F. Diacylglycerol Kinase γ Predicts Prognosis and Functions as a Tumor Suppressor by Negatively Regulating Glucose Transporter 1 in Hepatocellular Carcinoma. Exp. Cell Res. 2018, 373, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Iravani, A.; Kim, M.; Hozumi, Y.; Lohse, M.; Reichert, E.; Crotty, T.M.; Stafforini, D.M.; Topham, M.K. Diacylglycerol Kinase η Modulates Oncogenic Properties of Lung Cancer Cells. Clin. Transl. Oncol. 2014, 16, 29–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldanzi, G.; Pietronave, S.; Locarno, D.; Merlin, S.; Porporato, P.; Chianale, F.; Filigheddu, N.; Cantelmo, A.R.; Albini, A.; Graziani, A.; et al. Diacylglycerol Kinases Are Essential for Hepatocyte Growth Factor-Dependent Proliferation and Motility of Kaposi’s Sarcoma Cells. Cancer Sci. 2011, 102, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.G.; Weiss, S.J. Navigating ECM Barriers at the Invasive Front: The Cancer Cell–Stroma Interface. Annu. Rev. Cell Dev. Biol. 2009, 25, 567–595. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Rao, J.; Zhou, Z.; Yao, X.; Wu, F.; Yang, J.; Yang, L.; Zhang, X.; Cui, Y.; Bian, X.W.; et al. RAC1-GTP Promotes Epithelial-Mesenchymal Transition and Invasion of Colorectal Cancer by Activation of STAT3. Lab. Investig. 2018, 98, 989–998. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Buel, G.R.; Nagiec, M.J.; Han, M.-J.; Roux, P.P.; Blenis, J.; Yoon, S.-O. ERK2 Regulates Epithelial-to-Mesenchymal Plasticity through DOCK10-Dependent Rac1/FoxO1 Activation. Proc. Natl. Acad. Sci. USA 2019, 116, 2967–2976. [Google Scholar] [CrossRef] [Green Version]
- Tanimura, S.; Takeda, K. ERK Signalling as a Regulator of Cell Motility. J. Biochem. 2017, 162, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Baldanzi, G.; Mitola, S.; Cutrupi, S.; Filigheddu, N.; Van Blitterswijk, W.J.; Sinigaglia, F.; Bussolino, F.; Graziani, A. Activation of Diacylglycerol Kinase α Is Required for VEGF-Induced Angiogenic Signaling in Vitro. Oncogene 2004, 23, 4828–4838. [Google Scholar] [CrossRef] [Green Version]
- Bacchiocchi, R.; Baldanzi, G.; Carbonari, D.; Capomagi, C.; Colombo, E.; Van Blitterswijk, W.J.; Graziani, A.; Fazioli, F. Activation of α-Diacylglycerol Kinase Is Critical for the Mitogenic Properties of Anaplastic Lymphoma Kinase. Blood 2005, 106, 2175–2182. [Google Scholar] [CrossRef] [Green Version]
- Filigheddu, N.; Cutrupi, S.; Porporato, P.E.; Riboni, F.; Baldanzi, G.; Chianale, F.; Fortina, E.; Piantanida, P.; De Bortoli, M.; Vacca, G.; et al. Diacylglycerol Kinase Is Required for HGF-Induced Invasiveness and Anchorage-Independent Growth of MDA-MB-231 Breast Cancer Cells. Anticancer Res. 2007, 27, 1489–1492. [Google Scholar] [PubMed]
- Filigheddu, N.; Sampietro, S.; Chianale, F.; Porporato, P.E.; Gaggianesi, M.; Gregnanin, I.; Rainero, E.; Ferrara, M.; Perego, B.; Riboni, F.; et al. Diacylglycerol Kinase α Mediatses 17-β-Estradiol-Induced Proliferation, Motility, and Anchorage-Independent Growth of Hec-1A Endometrial Cancer Cell Line through the G Protein-Coupled Estrogen Receptor GPR30. Cell. Signal. 2011, 23, 1988–1996. [Google Scholar] [CrossRef] [PubMed]
- Rainero, E.; Caswell, P.T.; Muller, P.A.; Grindlay, J.; McCaffrey, M.W.; Zhang, Q.; Wakelam, M.J.; Vousden, K.H.; Graziani, A.; Norman, J.C. Diacylglycerol kinase α controls RCP-dependent integrin trafficking to promote invasive migration. J. Cell Biol. 2012, 196, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, Z.; Lu, M.; Yonekubo, Y.; Liang, X.; Zhang, Y.; Wu, P.; Zhou, Y.; Grinstein, S.; Hancock, J.F.; et al. Temporal Production of the Signaling Lipid Phosphatidic Acid by Phospholipase D2 Determines the Output of Extracellular Signal-Regulated Kinase Signaling in Cancer Cells. Mol. Cell. Biol. 2014, 34, 84–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, F.; He, J.; Wu, P.; Tay, L.W.R.; Cai, M.; Nian, W.; Weng, Y.; Qin, L.; Chang, J.T.; et al. Binding of PLD2-Generated Phosphatidic Acid to KIF5B Promotes MT1-MMP Surface Trafficking and Lung Metastasis of Mouse Breast Cancer Cells. Dev. Cell 2017, 43, 186–197.e7. [Google Scholar] [CrossRef] [Green Version]
- Sliva, D.; Mason, R.; Xiao, H.; English, D. Enhancement of the Migration of Metastatic Human Breast Cancer Cells by Phosphatidic Acid. Biochem. Biophys. Res. Commun. 2000, 268, 471–479. [Google Scholar] [CrossRef]
- Tanguy, E.; Wang, Q.; Moine, H.; Vitale, N. Phosphatidic Acid: From Pleiotropic Functions to Neuronal Pathology. Front. Cell. Neurosci. 2019, 13, 2. [Google Scholar] [CrossRef] [Green Version]
- Ard, R.; Mulatz, K.; Abramovici, H.; Maillet, J.C.; Fottinger, A.; Foley, T.; Byham, M.R.; Iqbal, T.A.; Yoneda, A.; Couchman, J.R.; et al. Diacylglycerol Kinase ζ Regulates RhoA Activation via a Kinase-Independent Scaffolding Mechanism. Mol. Biol. Cell 2012, 23, 4008–4019. [Google Scholar] [CrossRef]
- Lawson, C.D.; Ridley, A.J. Rho GTPase Signaling Complexes in Cell Migration and Invasion. J. Cell Biol. 2018, 217, 447–457. [Google Scholar] [CrossRef]
- Sadreddini, S.; Baradaran, B.; Aghebati-Maleki, A.; Sadreddini, S.; Shanehbandi, D.; Fotouhi, A.; Aghebati-Maleki, L. Immune Checkpoint Blockade Opens a New Way to Cancer Immunotherapy. J. Cell. Physiol. 2019, 234, 8541–8549. [Google Scholar] [CrossRef]
- Yamaki, A.; Akiyama, R.; Murakami, C.; Takao, S.; Murakami, Y.; Mizuno, S.; Takahashi, D.; Kado, S.; Taketomi, A.; Shirai, Y.; et al. Diacylglycerol Kinase A-selective Inhibitors Induce Apoptosis and Reduce Viability of Melanoma and Several Other Cancer Cell Lines. J. Cell. Biochem. 2019, 120, 10043–10056. [Google Scholar] [CrossRef] [PubMed]
- Velnati, S.; Massarotti, A.; Antona, A.; Talmon, M.; Fresu, L.G.; Galetto, A.S.; Capello, D.; Bertoni, A.; Mercalli, V.; Graziani, A.; et al. Structure Activity Relationship Studies on Amb639752: Toward the Identification of a Common Pharmacophoric Structure for DGKα Inhibitors. J. Enzyme Inhib. Med. Chem. 2020, 35, 96–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purow, B. Molecular Pathways: Targeting Diacylglycerol Kinase Alpha in Cancer. Clin. Cancer Res. 2015, 21, 5008–5012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmez, I.; Love, S.; Xiao, A.; Manigat, L.; Randolph, P.; McKenna, B.D.; Neal, B.P.; Boroda, S.; Li, M.; Brenneman, B.; et al. Targeting the Mesenchymal Subtype in Glioblastoma and Other Cancers via Inhibition of Diacylglycerol Kinase Alpha. Neuro-Oncology 2018, 20, 192–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arranz-Nicolás, J.; Ogando, J.; Soutar, D.; Arcos-Pérez, R.; Meraviglia-Crivelli, D.; Mañes, S.; Mérida, I.; Ávila-Flores, A. Diacylglycerol Kinase α Inactivation Is an Integral Component of the Costimulatory Pathway That Amplifies TCR Signals. Cancer Immunol. Immunother. 2018, 67, 965–980. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| DGK Isoforms | Subcellular Localization |
|---|---|
| DGKα | Plasma membrane, Cytosol, Nucleus |
| DGKβ | Cytoskeleton |
| DGKγ | Nucleus, Golgi, Cytosol |
| DGKδ | Plasma membrane, Endoplasmic reticulum, Endosomes |
| DGKε | Plasma membrane, Endoplasmic reticulum |
| DGKζ | Nuclear speckles, Plasma membrane, Cytosol, Cytoskeleton |
| DGKθ | Nuclear speckles, Plasma membrane, Cytosol |
| DGKι | Nucleus, Cytosol |
| DGKκ | Plasma membrane |
| DGKη | Endosomes |
| DGK Isoform | Cancer Type | Effects Caused by Downregulating DGKs |
|---|---|---|
| DGKα | Glioblastoma | Decreases Akt/mTOR, HIF1α, c-Myc activity [29] |
| HCC | Inhibits the Ras/RAF/MEK/ERK pathway [27] | |
| Acute Myeloid Leukemia | Impairs pRb signaling [100] | |
| Cervical cancer, HCC | Promotes IL-2 signaling [101] | |
| Colon, Breast cancer | Impairs Src activity [28] | |
| DGKδ | Cervical cancer, Lung adenocarcinoma | Suppresses Akt activity [32] |
| DGKε | Cervical cancer | Impairs Ras/RAF/MEK/ERK signaling [33] |
| DGKγ | HCC | Inhibition of GLUT1 expression and glycolysis [102] |
| Colorectal cancer | Inhibits Rac1 activity [26] | |
| DGKζ | Glioblastoma | Suppresses Cyclin D1 expression [99] Decreases Akt/mTOR activity [99] Increases CD3 expression to enhance T-cell functions [25] |
| Colorectal cancer | Decreases Rac1 and RhoA activity [30] | |
| Acute Myeloid Leukemia | Induces cell cycle arrest at G2M, Inhibits cell proliferation and increases apoptosis [98] | |
| DGKη | Lung cancer | Impairs MAPK signaling [103] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fazio, A.; Owusu Obeng, E.; Rusciano, I.; Marvi, M.V.; Zoli, M.; Mongiorgi, S.; Ramazzotti, G.; Follo, M.Y.; McCubrey, J.A.; Cocco, L.; et al. Subcellular Localization Relevance and Cancer-Associated Mechanisms of Diacylglycerol Kinases. Int. J. Mol. Sci. 2020, 21, 5297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155297
Fazio A, Owusu Obeng E, Rusciano I, Marvi MV, Zoli M, Mongiorgi S, Ramazzotti G, Follo MY, McCubrey JA, Cocco L, et al. Subcellular Localization Relevance and Cancer-Associated Mechanisms of Diacylglycerol Kinases. International Journal of Molecular Sciences. 2020; 21(15):5297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155297
Chicago/Turabian StyleFazio, Antonietta, Eric Owusu Obeng, Isabella Rusciano, Maria Vittoria Marvi, Matteo Zoli, Sara Mongiorgi, Giulia Ramazzotti, Matilde Yung Follo, James A. McCubrey, Lucio Cocco, and et al. 2020. "Subcellular Localization Relevance and Cancer-Associated Mechanisms of Diacylglycerol Kinases" International Journal of Molecular Sciences 21, no. 15: 5297. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155297