Immune Landscape in Tumor Microenvironment: Implications for Biomarker Development and Immunotherapy


 , and
, and 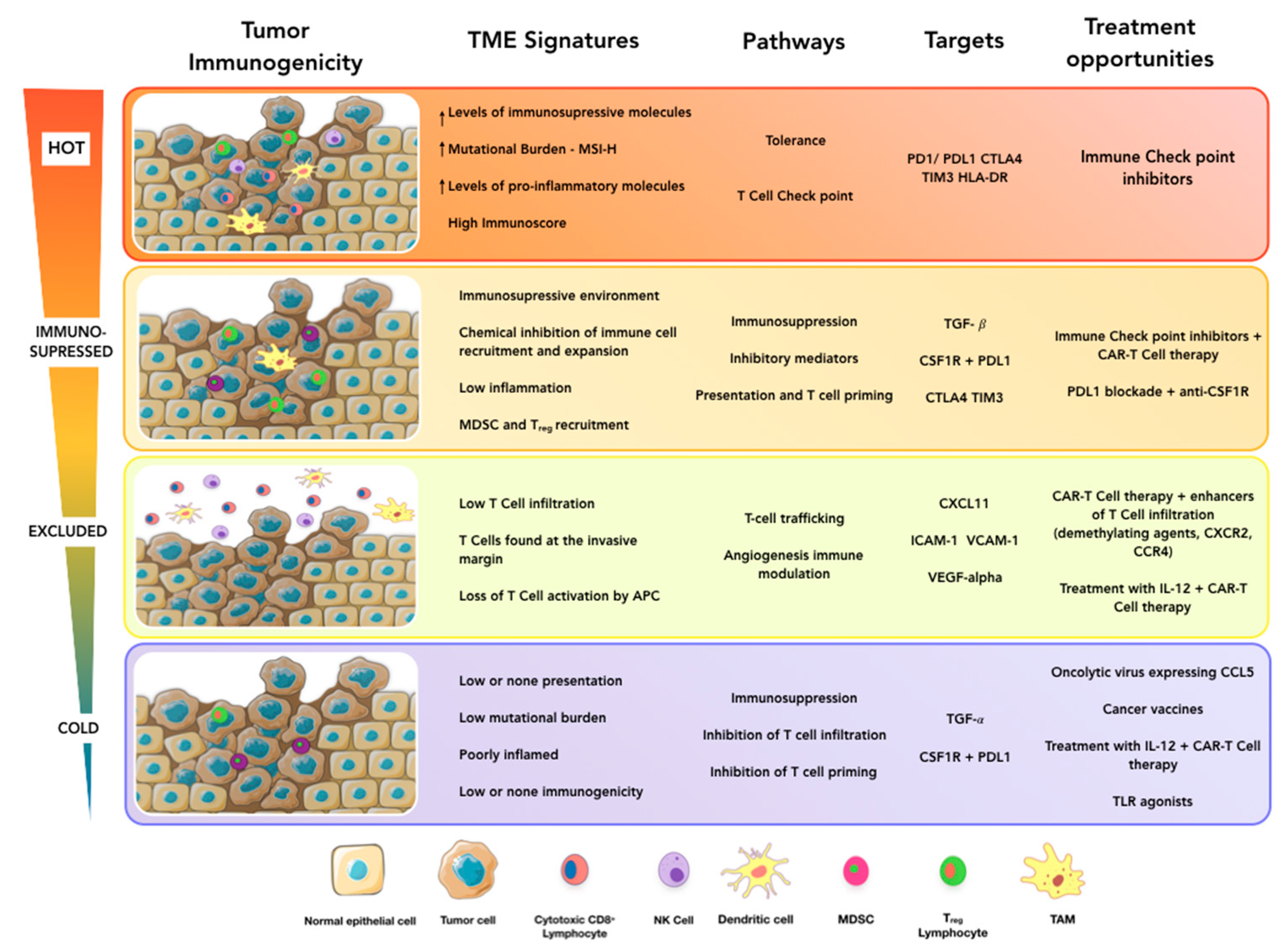{kind=link}
Abstract
:1. Introduction
2. Immunological Assessment of the TME for Cancer Prognosis
3. Evaluation of the Tumor Immune Ecosystem to Refine Current Immunotherapy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APC | antigen-presenting cells |
| CAR | chimeric antigen receptor |
| CCL | C-C motif chemokine ligand |
| CD | cluster of differentiation |
| CRC | colorectal cancer |
| CSF1R | colony-stimulating factor 1 receptor |
| CTLA4 | cytotoxic T-lymphocyte-associated protein 4 |
| CXCL | C-X-C motif chemokine ligand |
| DC | dendritic cells |
| DNA | deoxyribonucleic acid |
| EGF | epidermal growth factor |
| Erα | estrogen receptor alpha |
| FASL | FAS ligand |
| GMCSF | granulocyte macrophage colony-stimulating factor |
| HCC | hepatocellular carcinoma |
| HLA | human leukocyte antigen |
| HLADR | human leukocyte antigen DR- isotype |
| ICAM | intracellular cell adhesion molecule |
| IFNγ | interferon gamma |
| IL | interleukin |
| KLRK1 | killer cell lectin-like receptor K1 |
| LAMP | lysosome-associated membrane glycoprotein |
| MCSF | macrophage colony-stimulating factor |
| MDSC | myeloid-derived suppressor cells |
| MHC | major histocompatibility complex |
| MPE: | molecular pathological epidemiology |
| MSI | microsatellite instability |
| NK | natural killer cell |
| NKT | natural killer T cell |
| PD1 | programmed cell death protein 1 |
| PDL1 | programmed cell death ligand 1 |
| RNA | ribonucleic acid |
| TAM | tumor-associated macrophages |
| TGFβ | transforming growth factor-beta |
| Th | T helper cell |
| TIM3 | T cell immunoglobulin-mucin domain 3 |
| TLR | Toll-like receptor |
| TLS | tertiary lymphoid structures |
| TME | tumor microenvironment |
| TNFα | tumor necrosis factor-alpha |
| Treg | regulatory T cell |
| VCAM | vascular cell adhesion molecule |
| VEGF | vascular endothelial growth factor |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Three Es of Cancer Immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.L.; Galon, J.; Fridman, W.-H.; Smyth, M.J. From mice to humans: Developments in cancer immunoediting. J. Clin. Invest. 2015, 125, 3338–3346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RHOADS, C.P. Paul ehrlich and the cancer problem. Ann. N. Y. Acad. Sci. 1954, 59, 190–197. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef]
- Linette, G.P.; Becker-Hapak, M.; Skidmore, Z.L.; Baroja, M.L.; Xu, C.; Hundal, J.; Spencer, D.H.; Fu, W.; Cummins, C.; Robnett, M.; et al. Immunological ignorance is an enabling feature of the oligo-clonal T cell response to melanoma neoantigens. Proc. Natl. Acad. Sci. USA 2019, 116, 23662–23670. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef] [Green Version]
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of Tumors by the Innate Immune System and Natural Killer Cells. Adv. Immunol. 2014, 122, 91–128. [Google Scholar]
- Tiwary, S.; Berzofsky, J.A.; Terabe, M. Altered Lipid Tumor Environment and Its Potential Effects on NKT Cell Function in Tumor Immunity. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.T.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 Blocks CD8+ T Cell-Dependent Responses to Chemotherapy by Suppressing IL-12 Expression in Intratumoral Dendritic Cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Hamada, T.; Nowak, J.A.; Milner, D.A.; Song, M.; Ogino, S. Integration of microbiology, molecular pathology, and epidemiology: A new paradigm to explore the pathogenesis of microbiome-driven neoplasms. J. Pathol. 2019, 247, 615–628. [Google Scholar] [CrossRef] [Green Version]
- Koh, H.; Hamada, T.; Song, M.; Liu, L.; Cao, Y.; Nowak, J.A.; da Silva, A.; Twombly, T.; Morikawa, T.; Kim, S.A.; et al. Physical Activity and Colorectal Cancer Prognosis According to Tumor-Infiltrating T Cells. JNCI Cancer Spectr. 2018, 2, pky058. [Google Scholar] [CrossRef] [Green Version]
- Ogino, S.; Nowak, J.A.; Hamada, T.; Milner, D.A.; Nishihara, R. Insights into Pathogenic Interactions Among Environment, Host, and Tumor at the Crossroads of Molecular Pathology and Epidemiology. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 83–103. [Google Scholar] [CrossRef]
- Ogino, S.; Nowak, J.A.; Hamada, T.; Phipps, A.I.; Peters, U.; Milner, D.A.; Giovannucci, E.L.; Nishihara, R.; Giannakis, M.; Garrett, W.S.; et al. Integrative analysis of exogenous, endogenous, tumour and immune factors for precision medicine. Gut 2018, 67, 1168–1180. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Berger, A.; Camus, M.; Sanchez-Cabo, F.; Costes, A.; Molidor, R.; Mlecnik, B.; Kirilovsky, A.; Nilsson, M.; Damotte, D.; et al. Effector Memory T Cells, Early Metastasis, and Survival in Colorectal Cancer. N. Engl. J. Med. 2005, 353, 2654–2666. [Google Scholar] [CrossRef] [PubMed]
- Galon, J. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camus, M.; Tosolini, M.; Mlecnik, B.; Pages, F.; Kirilovsky, A.; Berger, A.; Costes, A.; Bindea, G.; Charoentong, P.; Bruneval, P.; et al. Coordination of Intratumoral Immune Reaction and Human Colorectal Cancer Recurrence. Cancer Res. 2009, 69, 2685–2693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Kirilovsky, A.; Mlecnik, B.; Asslaber, M.; Tosolini, M.; Bindea, G.; Lagorce, C.; Wind, P.; Marliot, F.; Bruneval, P.; et al. In Situ Cytotoxic and Memory T Cells Predict Outcome in Patients With Early-Stage Colorectal Cancer. J. Clin. Oncol. 2009, 27, 5944–5951. [Google Scholar] [CrossRef]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Mlecnik, B.; Tosolini, M.; Kirilovsky, A.; Berger, A.; Bindea, G.; Meatchi, T.; Bruneval, P.; Trajanoski, Z.; Fridman, W.-H.; Pagès, F.; et al. Histopathologic-Based Prognostic Factors of Colorectal Cancers Are Associated With the State of the Local Immune Reaction. J. Clin. Oncol. 2011, 29, 610–618. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautès–Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462. [Google Scholar] [CrossRef]
- Martinet, L.; Garrido, I.; Filleron, T.; Le Guellec, S.; Bellard, E.; Fournie, J.-J.; Rochaix, P.; Girard, J.-P. Human Solid Tumors Contain High Endothelial Venules: Association with T- and B-Lymphocyte Infiltration and Favorable Prognosis in Breast Cancer. Cancer Res. 2011, 71, 5678–5687. [Google Scholar] [CrossRef] [Green Version]
- Germain, C.; Gnjatic, S.; Tamzalit, F.; Knockaert, S.; Remark, R.; Goc, J.; Lepelley, A.; Becht, E.; Katsahian, S.; Bizouard, G.; et al. Presence of B Cells in Tertiary Lymphoid Structures Is Associated with a Protective Immunity in Patients with Lung Cancer. Am. J. Respir. Crit. Care Med. 2014, 189, 832–844. [Google Scholar] [CrossRef]
- Kroeger, D.R.; Milne, K.; Nelson, B.H. Tumor-Infiltrating Plasma Cells Are Associated with Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer. Clin. Cancer Res. 2016, 22, 3005–3015. [Google Scholar] [CrossRef] [Green Version]
- Finkin, S.; Yuan, D.; Stein, I.; Taniguchi, K.; Weber, A.; Unger, K.; Browning, J.L.; Goossens, N.; Nakagawa, S.; Gunasekaran, G.; et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat. Immunol. 2015, 16, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Medrek, C.; Pontén, F.; Jirström, K.; Leandersson, K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer 2012, 12, 306. [Google Scholar] [CrossRef]
- Jensen, T.O.; Schmidt, H.; Møller, H.J.; Høyer, M.; Maniecki, M.B.; Sjoegren, P.; Christensen, I.J.; Steiniche, T. Macrophage Markers in Serum and Tumor Have Prognostic Impact in American Joint Committee on Cancer Stage I/II Melanoma. J. Clin. Oncol. 2009, 27, 3330–3337. [Google Scholar] [CrossRef]
- Hou, Y.-C.; Chao, Y.-J.; Tung, H.-L.; Wang, H.-C.; Shan, Y.-S. Coexpression of CD44-positive/CD133-positive cancer stem cells and CD204-positive tumor-associated macrophages is a predictor of survival in pancreatic ductal adenocarcinoma. Cancer 2014, 120, 2766–2777. [Google Scholar] [CrossRef] [Green Version]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, S.; Ishii, G.; Nagai, K.; Ono, S.; Kojima, M.; Yamauchi, C.; Aokage, K.; Hishida, T.; Yoshida, J.; Suzuki, K.; et al. Prognostic Impact of CD204-Positive Macrophages in Lung Squamous Cell Carcinoma: Possible Contribution of Cd204-Positive Macrophages to the Tumor-Promoting Microenvironment. J. Thorac. Oncol. 2012, 7, 1790–1797. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, K.; Nakata, M.; Hirami, Y.; Yukawa, T.; Maeda, A.; Tanemoto, K. Tumor-Infiltrating Foxp3+ Regulatory T Cells are Correlated with Cyclooxygenase-2 Expression and are Associated with Recurrence in Resected Non-small Cell Lung Cancer. J. Thorac. Oncol. 2010, 5, 585–590. [Google Scholar] [CrossRef] [Green Version]
- Bates, G.J.; Fox, S.B.; Han, C.; Leek, R.D.; Garcia, J.F.; Harris, A.L.; Banham, A.H. Quantification of Regulatory T Cells Enables the Identification of High-Risk Breast Cancer Patients and Those at Risk of Late Relapse. J. Clin. Oncol. 2006, 24, 5373–5380. [Google Scholar] [CrossRef]
- Ling, A.; Edin, S.; Wikberg, M.L.; Öberg, Å.; Palmqvist, R. The intratumoural subsite and relation of CD8+ and FOXP3+ T lymphocytes in colorectal cancer provide important prognostic clues. Br. J. Cancer 2014, 110, 2551–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-Infiltrating FOXP3 + T Regulatory Cells Show Strong Prognostic Significance in Colorectal Cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-J.; Lee, K.S.; Cho, H.J.; Kim, Y.H.; Yang, H.K.; Kim, W.H.; Kang, G.H. Prognostic implications of tumor-infiltrating FoxP3+ regulatory T cells and CD8+ cytotoxic T cells in microsatellite-unstable gastric cancers. Hum. Pathol. 2014, 45, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Niccolai, E.; Ricci, F.; Russo, E.; Nannini, G.; Emmi, G.; Taddei, A.; Ringressi, M.N.; Melli, F.; Miloeva, M.; Cianchi, F.; et al. The Different Functional Distribution of “Not Effector” T Cells (Treg/Tnull) in Colorectal Cancer. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Badoual, C. Prognostic Value of Tumor-Infiltrating CD4+ T-Cell Subpopulations in Head and Neck Cancers. Clin. Cancer Res. 2006, 12, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Zhao, M.; Xia, M.; Li, L.; Jiang, Y. The correlation between tumor-infiltrating Foxp3+ regulatory T cells and cyclooxygenase-2 expression and their association with recurrence in resected head and neck cancers. Med. Oncol. 2012, 29, 707–713. [Google Scholar] [CrossRef]
- Kitz, A.; Dominguez-Villar, M. Molecular mechanisms underlying Th1-like Treg generation and function. Cell. Mol. Life Sci. 2017, 74, 4059–4075. [Google Scholar] [CrossRef]
- Sawant, D.V.; Vignali, D.A.A. Once a Treg, always a Treg? Immunol. Rev. 2014, 259, 173–191. [Google Scholar] [CrossRef]
- Bhela, S.; Varanasi, S.K.; Jaggi, U.; Sloan, S.S.; Rajasagi, N.K.; Rouse, B.T. The Plasticity and Stability of Regulatory T Cells during Viral-Induced Inflammatory Lesions. J. Immunol. 2017, 199, 1342–1352. [Google Scholar] [CrossRef] [Green Version]
- Voo, K.S.; Wang, Y.H.; Santori, F.R.; Boggiano, C.; Wang, Y.H.; Arima, K.; Bover, L.; Hanabuchi, S.; Khalili, J.; Marinova, E.; et al. Identification of IL-17-producing FOXP3 + regulatory T cells in humans. Proc. Natl. Acad. Sci. USA 2009, 12, 4793–4798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, L.; Mu, S.; Wang, Y.; Wang, H.; Cai, L.; Li, W.; Hu, Y. Prognostic role of myeloid-derived suppressor cells in cancers: A systematic review and meta-analysis. BMC Cancer 2018, 18, 1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e36. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e18. [Google Scholar] [CrossRef] [Green Version]
- Chevrier, S.; Levine, J.H.; Zanotelli, V.R.T.; Silina, K.; Schulz, D.; Bacac, M.; Ries, C.H.; Ailles, L.; Jewett, M.A.S.; Moch, H.; et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 2017, 169, 736–749.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niccolai, E.; Taddei, A.; Ricci, F.; Rolla, S.; D’Elios, M.M.; Benagiano, M.; Bechi, P.; Bencini, L.; Ringressi, M.N.; Pini, A.; et al. Intra-tumoral IFN-γ-producing Th22 cells correlate with TNM staging and the worst outcomes in pancreatic cancer. Clin. Sci. 2016, 130, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; He, Y.; Luo, N.; Patel, S.J.; Han, Y.; Gao, R.; Modak, M.; Carotta, S.; Haslinger, C.; Kind, D.; et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019, 179, 829–845.e20. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Friedman, C.F.; Wolchok, J.D. Combinatorial Cancer Immunotherapies. Adv. Immunol. 2016, 130, 251–277. [Google Scholar]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Invest. 2014, 124, 2246–2259. [Google Scholar] [CrossRef] [PubMed]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Magen, A.; Nie, J.; Ciucci, T.; Tamoutounour, S.; Zhao, Y.; Mehta, M.; Tran, B.; McGavern, D.B.; Hannenhalli, S.; Bosselut, R. Single-Cell Profiling Defines Transcriptomic Signatures Specific to Tumor-Reactive versus Virus-Responsive CD4+ T Cells. Cell Rep. 2019, 29, 3019–3032.e6. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Mackall, C.L. Clinical lessons learned from the first leg of the CAR T cell journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- Tarlock, K.; Alonzo, T.A.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Sung, L.; Pollard, J.A.; Aplenc, R.; Loken, M.R.; Gamis, A.S.; et al. Gemtuzumab Ozogamicin Reduces Relapse Risk in FLT3/ITD Acute Myeloid Leukemia: A Report from the Children’s Oncology Group. Clin. Cancer Res. 2016, 22, 1951–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kershaw, M.H.; Wang, G.; Westwood, J.A.; Pachynski, R.K.; Tiffany, H.L.; Marincola, F.M.; Wang, E.; Young, H.A.; Murphy, P.M.; Hwu, P. Redirecting Migration of T Cells to Chemokine Secreted from Tumors by Genetic Modification with CXCR2. Hum. Gene Ther. 2002, 13, 1971–1980. [Google Scholar] [CrossRef] [PubMed]
- Bodey, B.; Bodey, B., Jr.; Siegel, S.E.; Kaiser, H.E. Failure of cancer vaccines: The significant limitations of this approach to immunotherapy. Anticancer. Res. 2000, 20, 2665–2676. [Google Scholar] [PubMed]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e11. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, Y.; Zhang, L.; Huang, L. Nanoparticle-Delivered Transforming Growth Factor-β siRNA Enhances Vaccination against Advanced Melanoma by Modifying Tumor Microenvironment. ACS Nano 2014, 8, 3636–3645. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wrzesinski, S.H.; Stern, E.; Look, M.; Criscione, J.; Ragheb, R.; Jay, S.M.; Demento, S.L.; Agawu, A.; Licona Limon, P.; et al. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat. Mater. 2012, 11, 895–905. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.-M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Braunstein, M.J.; Kucharczyk, J.; Adams, S. Targeting Toll-Like Receptors for Cancer Therapy. Target. Oncol. 2018, 13, 583–598. [Google Scholar] [CrossRef]
- Van Herpen, C.M.L. Intratumoral Recombinant Human Interleukin-12 Administration in Head and Neck Squamous Cell Carcinoma Patients Modifies Locoregional Lymph Node Architecture and Induces Natural Killer Cell Infiltration in the Primary Tumor. Clin. Cancer Res. 2005, 11, 1899–1909. [Google Scholar] [CrossRef] [Green Version]
- Nishio, N.; Diaconu, I.; Liu, H.; Cerullo, V.; Caruana, I.; Hoyos, V.; Bouchier-Hayes, L.; Savoldo, B.; Dotti, G. Armed Oncolytic Virus Enhances Immune Functions of Chimeric Antigen Receptor-Modified T Cells in Solid Tumors. Cancer Res. 2014, 74, 5195–5205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic Agents Can Increase Lymphocyte Infiltration into Tumor and Enhance the Effectiveness of Adoptive Immunotherapy of Cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. The “cancer immunogram”. Science 2016, 352, 658–660. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Romero, K.; Rodríguez, R.M.; Amedei, A.; Barceló-Coblijn, G.; Lopez, D.H. Immune Landscape in Tumor Microenvironment: Implications for Biomarker Development and Immunotherapy. Int. J. Mol. Sci. 2020, 21, 5521. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155521
Pérez-Romero K, Rodríguez RM, Amedei A, Barceló-Coblijn G, Lopez DH. Immune Landscape in Tumor Microenvironment: Implications for Biomarker Development and Immunotherapy. International Journal of Molecular Sciences. 2020; 21(15):5521. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155521
Chicago/Turabian StylePérez-Romero, Karim, Ramón M. Rodríguez, Amedeo Amedei, Gwendolyn Barceló-Coblijn, and Daniel H. Lopez. 2020. "Immune Landscape in Tumor Microenvironment: Implications for Biomarker Development and Immunotherapy" International Journal of Molecular Sciences 21, no. 15: 5521. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155521