The Artemisinin-Derived Autofluorescent Compound BG95 Exerts Strong Anticytomegaloviral Activity Based on a Mitochondrial Targeting Mechanism

 ,
,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
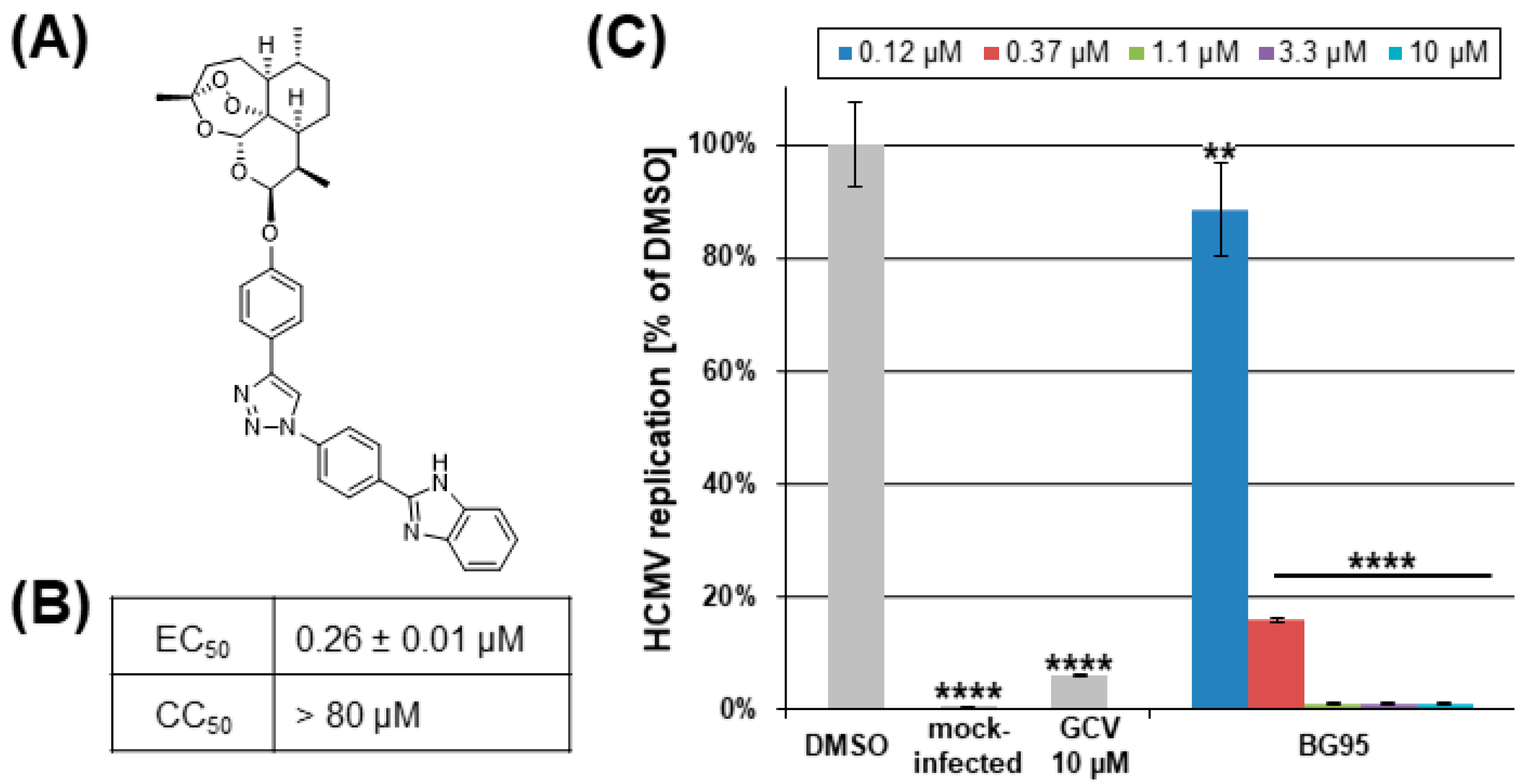
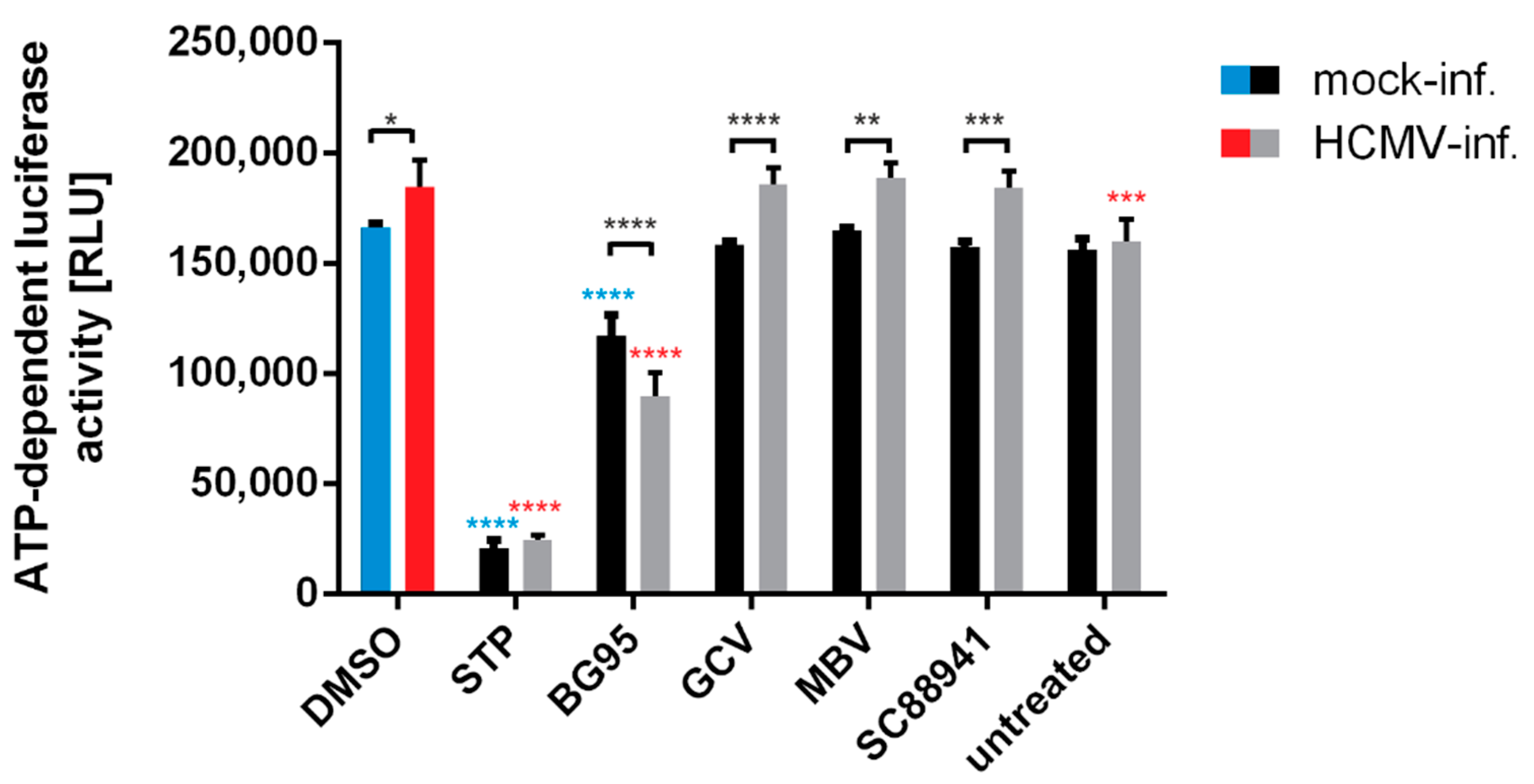
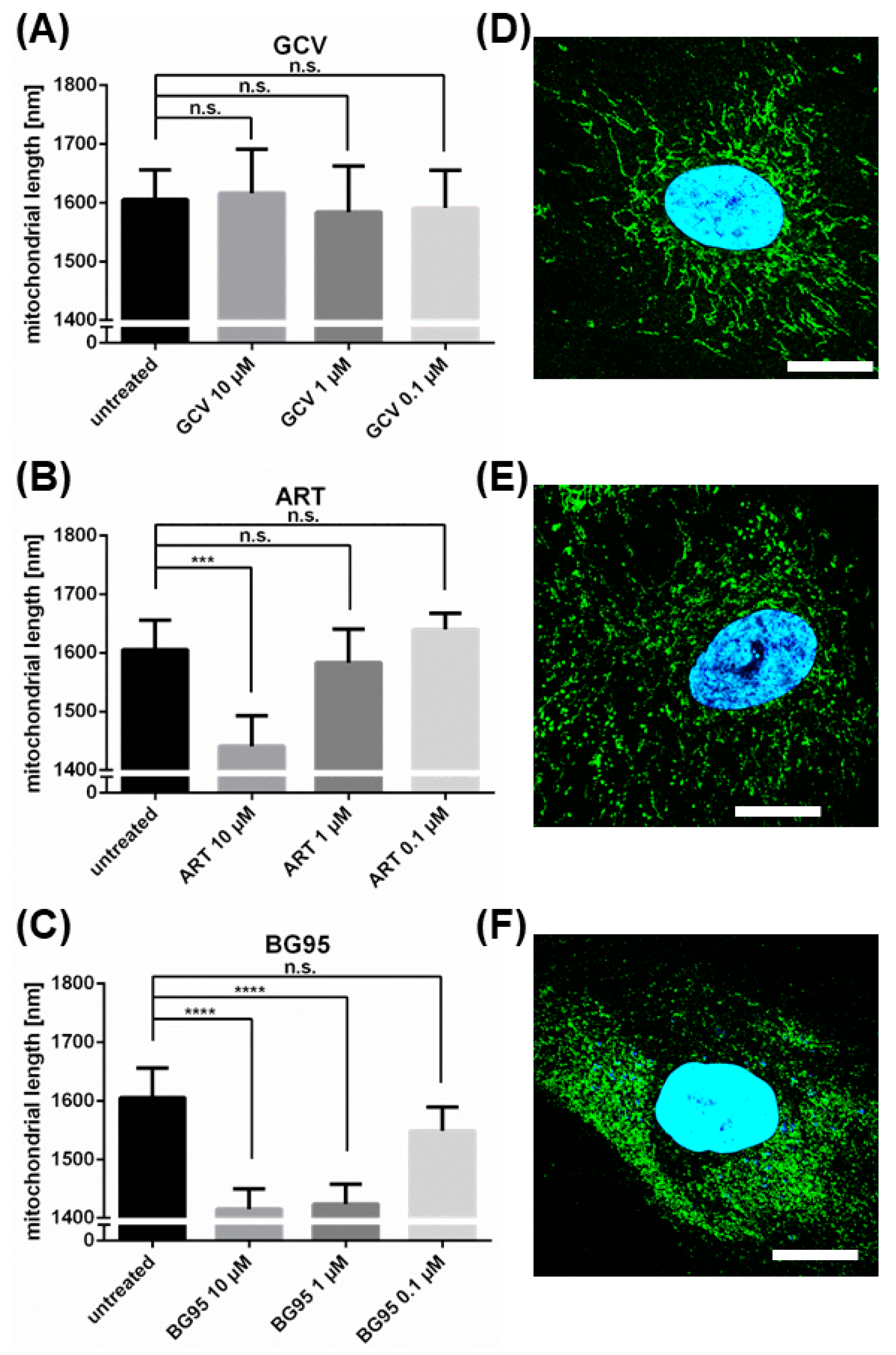
2.1. BG95 Exerts Strong Anti-HCMV Activity In Vitro in the Absence of Cytotoxicity
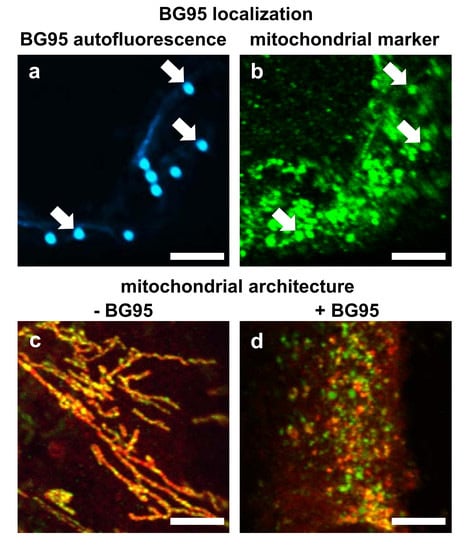
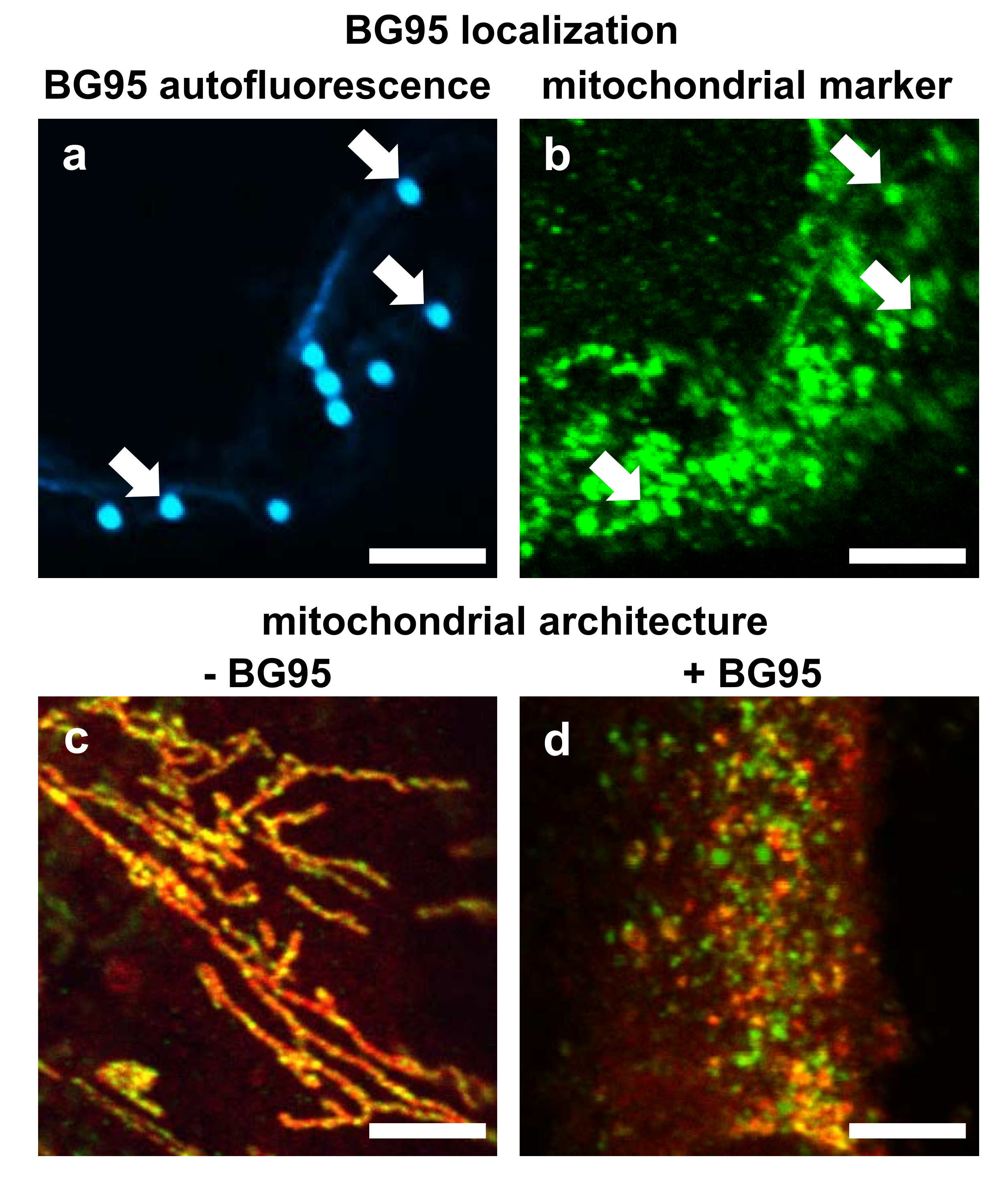
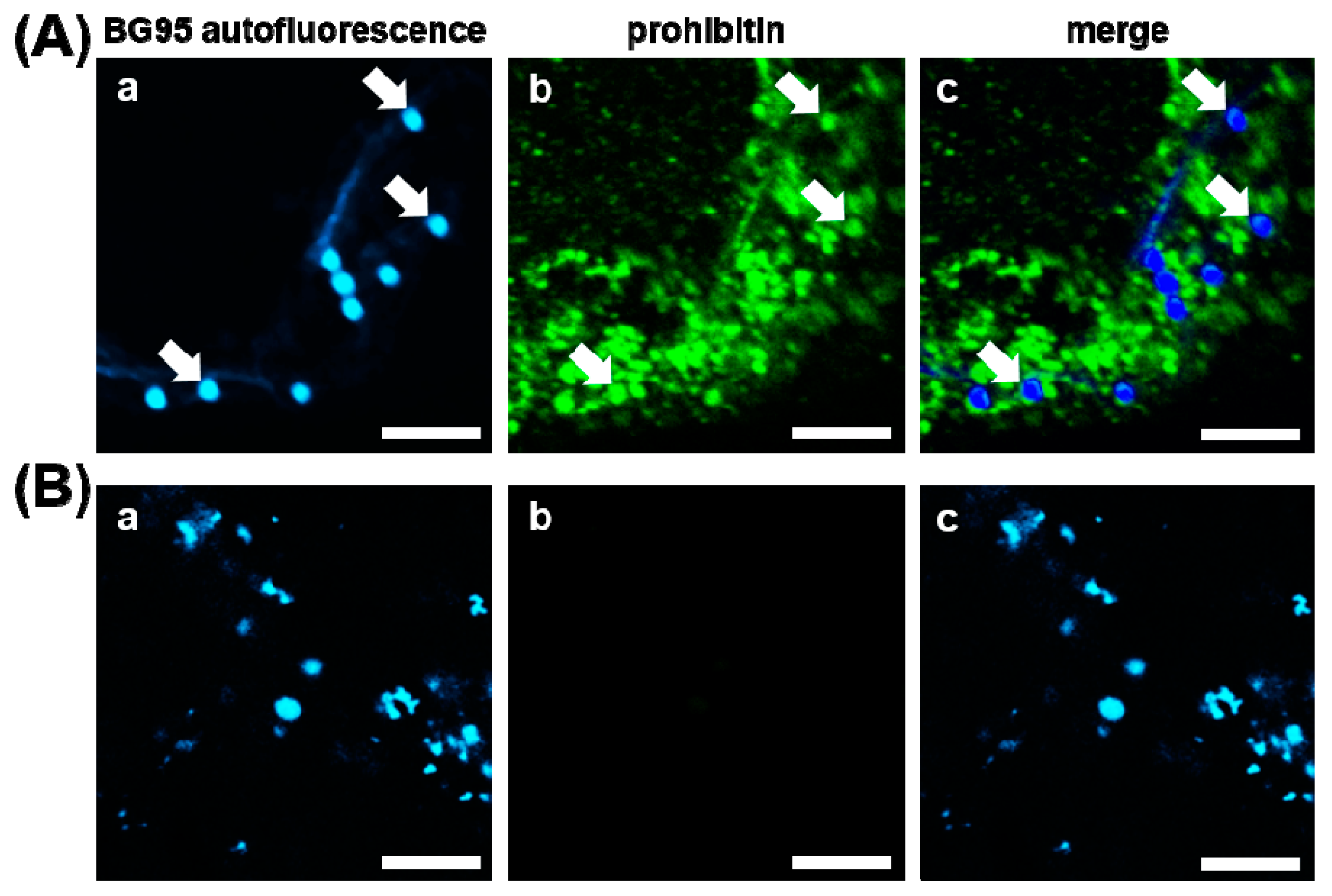
2.2. BG95 Displays Pronounced Autofluorescence and Accumulates in Mitochondria
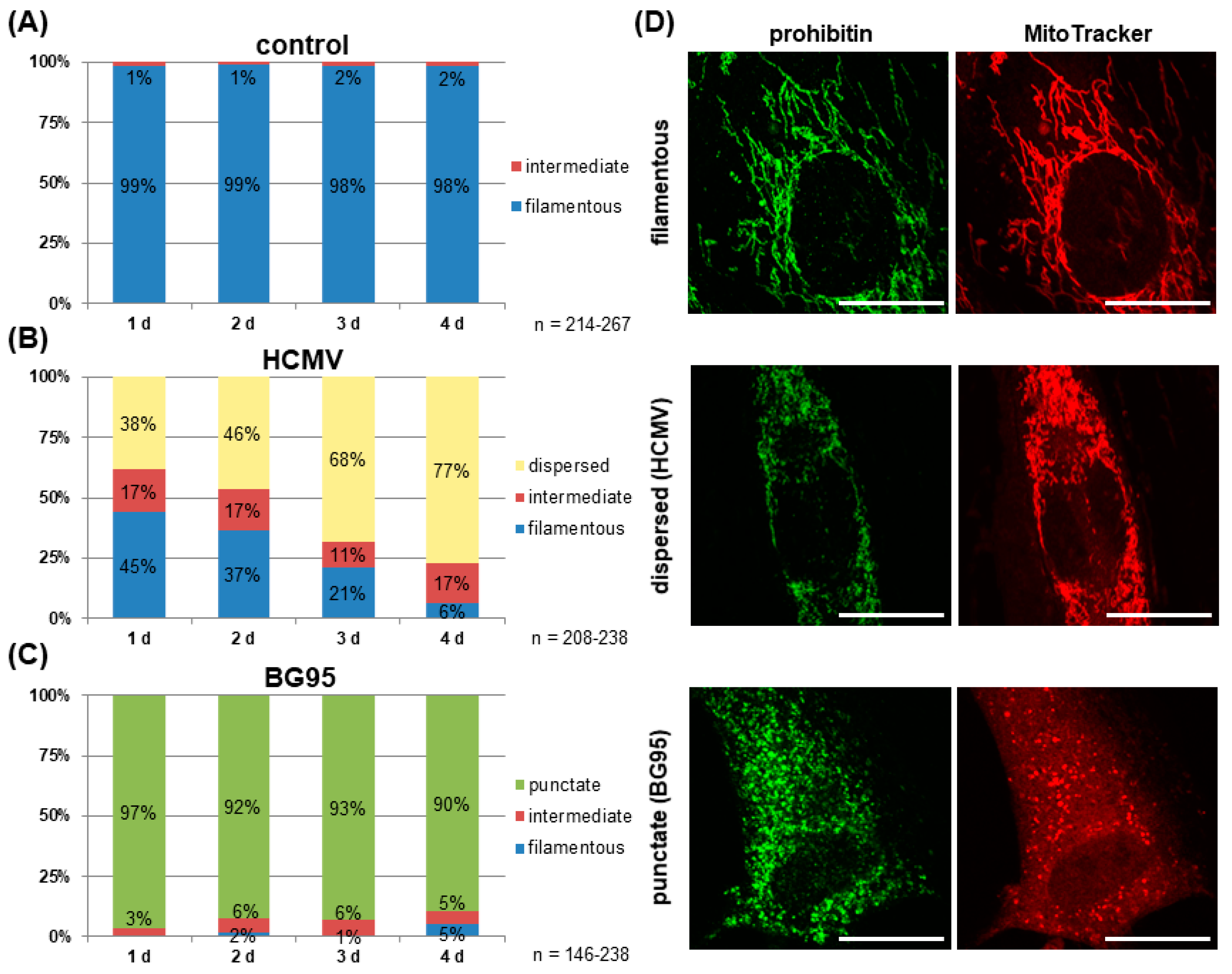
2.3. Treatment with BG95 Induces Changes in Mitochondrial Structure, Which Are Distinct from Those Induced by HCMV Infection
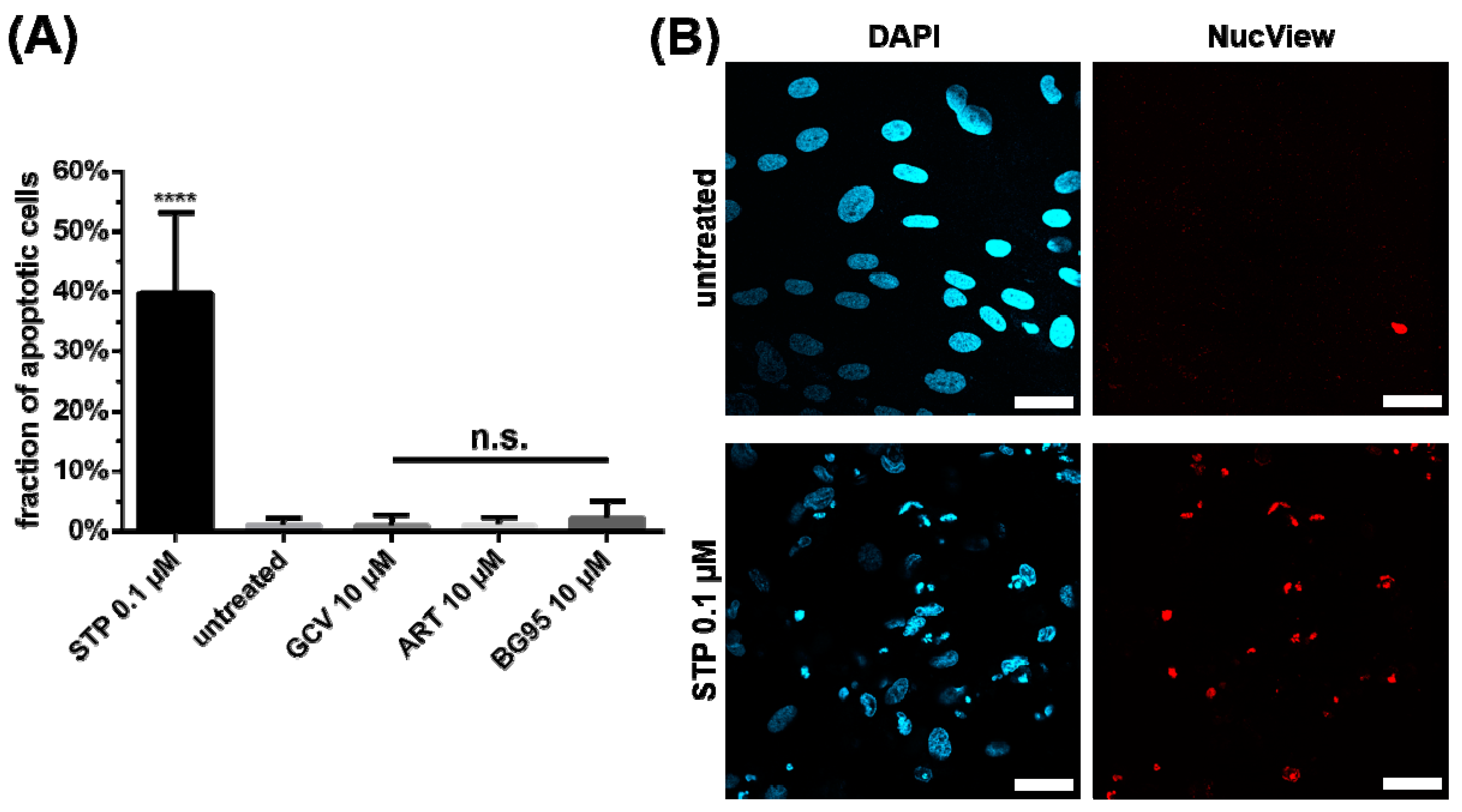
2.4. BG95 Induces Mitochondrial Changes but Does Not Induce Apoptosis
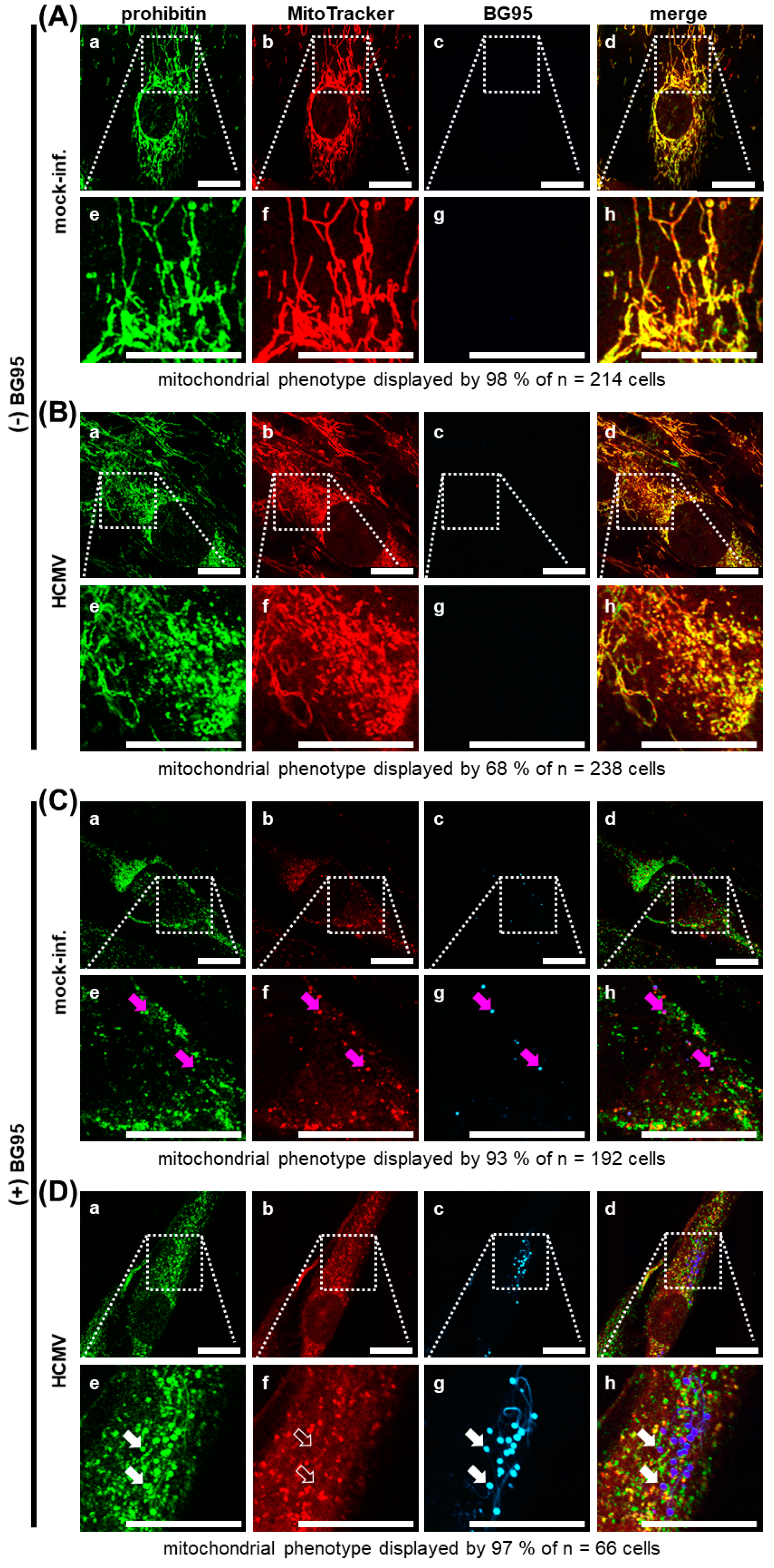
2.5. BG95 Localizes to Mitochondria and Leads to a Loss of Physiological Membrane Potential
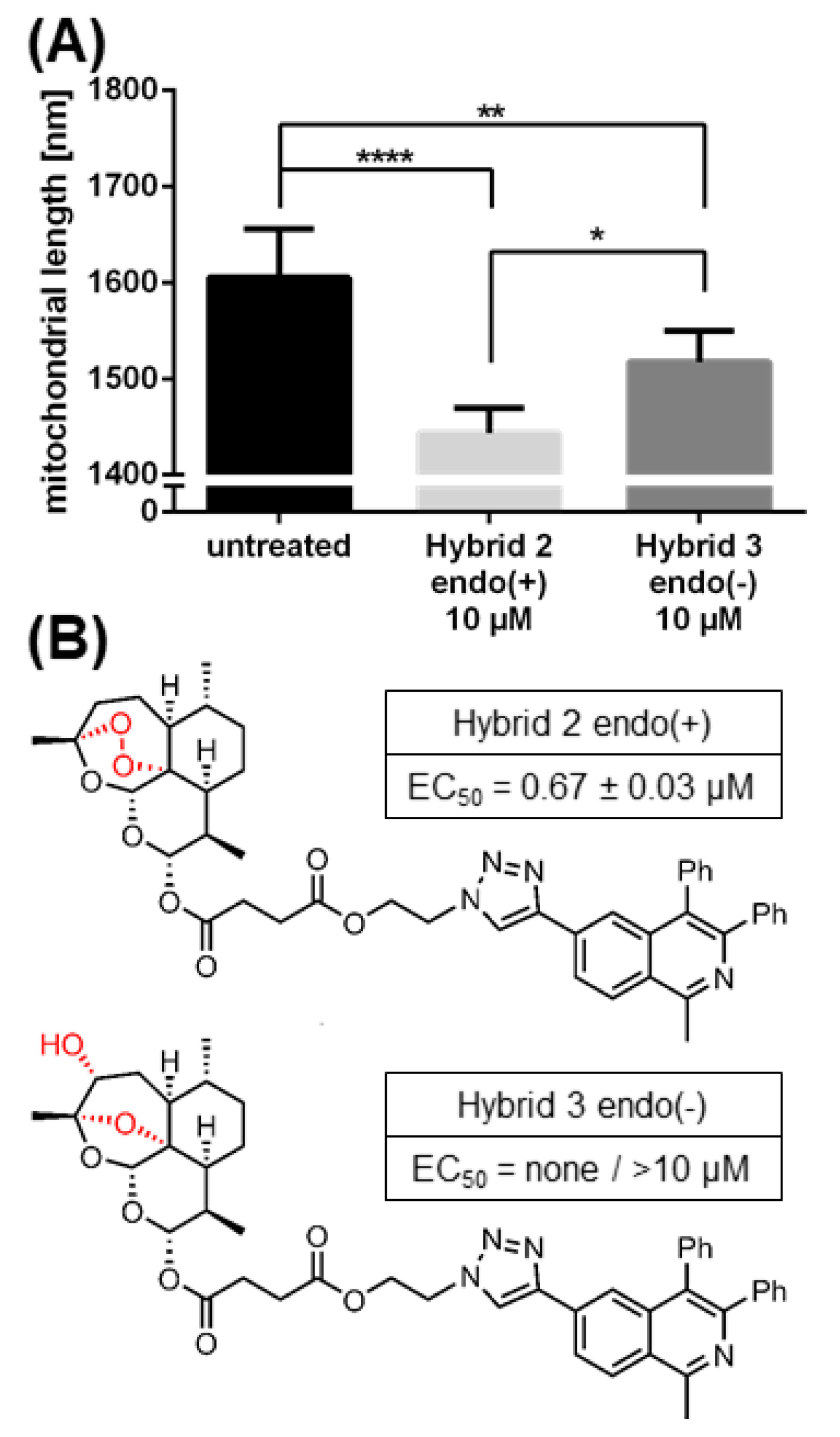
2.6. The Morphological Conversion of Mitochondria Induced by BG95 or Related Trioxanes Is Dependent on Drug Concentration and the Presence of an Intramolecular Endoperoxide Bridge
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Antiviral Compounds
4.3. Cells and Viruses
4.4. HCMV GFP-Based Replication Assay
4.5. Assessment of Cell Viability
4.6. Indirect Immunofluorescence Assay Using MitoTracker Staining and Laser Scanning Microscopy
4.7. Quantitation of Drug-Induced Morphological Changes of Mitochondria by Using the Filamentdetector Plugin of ImageJ Software
4.8. Measurement of Intracellular ATP Levels
4.9. Analysis of Mitochondrial Proteins Using Differential Centrifugation and SDS-PAGE/Western Blot
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ART | Artesunate |
| ATP | adenosine triphosphate |
| CDV | cidofovir |
| d | day(s) |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DMSO | Dimethyl sulfoxide |
| DNA | deoxyribonucleic acid |
| ETC | electron transport chain |
| FCS | fetal calf serum |
| FOS | foscarnet |
| GCV | ganciclovir |
| GFP | green fluorescent protein |
| HCMV | human cytomegalovirus |
| HFF | human foreskin fibroblast |
| IE1 | immediate-early protein 1 |
| LDH | lactate dehydrogenase |
| MBV | maribavir |
| MEF | murine embryonic fibroblast |
| MEM | Eagle’s Minimal Essential medium |
| min | minute(s) |
| MOI | multiplicity of infection |
| p.i. | post-infection |
| rpm | rotations per minute |
| SD | standard deviation |
| STP | staurosporine |
| VGCV | valganciclovir |
References
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef]
- Mocarski, E.S.; Shenk, T.; Griffiths, P.D.; Pass, R.F. Cytomegaloviruses. In Fields Virology, 6th ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1960–2014. [Google Scholar]
- Hamilton, S.; Hutterer, C.; Marschall, M. Therapeutics to prevent congenital cytomegalovirus during pregnancy: What is available now and in the future? Microbiol. Aust. 2015, 36, 156–161. [Google Scholar] [CrossRef]
- Rawlinson, W.D.; Boppana, S.B.; Fowler, K.B.; Kimberlin, D.W.; Lazzarotto, T.; Alain, S.; Daly, K.; Doutré, S.; Gibson, L.; Giles, M.L.; et al. Congenital cytomegalovirus infection in pregnancy and the neonate: Consensus recommendations for prevention, diagnosis, and therapy. Lancet Infect. Dis. 2017, 17, e177–e188. [Google Scholar] [CrossRef]
- Dropulic, L.K.; Cohen, J.I. Update on new antivirals under development for the treatment of double-stranded DNA virus infections. Clin. Pharmacol. Ther. 2010, 88, 610–619. [Google Scholar] [CrossRef] [Green Version]
- Bowman, L.J.; Melaragno, J.I.; Brennan, D.C. Letermovir for the management of cytomegalovirus infection. Expert Opin. Investig. Drugs 2017, 26, 235–241. [Google Scholar] [CrossRef]
- Chemaly, R.F.; Ullmann, A.J.; Stoelben, S.; Richard, M.P.; Bornhauser, M.; Groth, C.; Einsele, H.; Silverman, M.; Mullane, K.M.; Brown, J.; et al. Letermovir for cytomegalovirus prophylaxis in hematopoietic-cell transplantation. N. Engl. J. Med. 2014, 370, 1781–1789. [Google Scholar] [CrossRef]
- Goldner, T.; Hempel, C.; Schaeff, H.R.; Zimmermann, H.; Lischka, P. Geno- and phenotypic characterization of human cytomegalovirus mutants selected in vitro after letermovir (AIC246) exposure. Antimicrob. Agents Chemother. 2014, 58, 610–613. [Google Scholar] [CrossRef] [Green Version]
- Kropeit, D.; McCormick, D.; Erb-Zohar, K.; Moiseev, V.S.; Kobalava, Z.D.; Stobernack, H.P.; Zimmermann, H.; Schaeff, H.R. Pharmacokinetics and safety of the anti-human cytomegalovirus drug letermovir in subjects with hepatic impairment. Br. J. Clin. Pharmacol. 2017, 83, 2678–2686. [Google Scholar] [CrossRef]
- Lischka, P.; Michel, D.; Zimmermann, H. Characterization of cytomegalovirus breakthrough events in a phase 2 prophylaxis trial of letermovir (AIC246, MK 8228). J. Infect. Dis. 2016, 213, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Marschall, M.; Stamminger, T.; Urban, A.; Wildum, S.; Ruebsamen-Schaeff, H.; Zimmermann, H.; Lischka, P. In vitro evaluation of the activities of the novel anticytomegalovirus compound AIC246 (letermovir) against herpesviruses and other human pathogenic viruses. Antimicrob. Agents Chemother. 2012, 56, 1135–1137. [Google Scholar] [CrossRef] [Green Version]
- Marty, F.M.; Ljungman, P.; Chemaly, R.F.; Maertens, J.; Dadwal, S.S.; Duarte, R.F.; Haider, S.; Ullmann, A.J.; Katayama, Y.; Brown, J.; et al. Letermovir prophylaxis for cytomegalovirus in hematopoietic-cell transplantation. N. Engl. J. Med. 2017, 377, 2433–2444. [Google Scholar] [CrossRef]
- Chou, S. Rapid In vitro evolution of human cytomegalovirus UL56 mutations that confer letermovir resistance. Antimicrob. Agents Chemother. 2015, 59, 6588–6593. [Google Scholar] [CrossRef] [Green Version]
- Bock, C.M.; Parameshwarappa, G.; Bönisch, S.; Bauer, W.; Hutterer, C.; Leidenberger, M.; Friedrich, O.; Marschall, M.; Kappes, B.; Görling, A.; et al. Deeper Insight into the six-step domino reaction of aldehydes with malononitrile and evaluation of antiviral and antimalarial activities of the obtained bicyclic products. Chem. Open 2017, 6, 364–374. [Google Scholar] [CrossRef]
- Chou, S.; Marousek, G.; Auerochs, S.; Stamminger, T.; Milbradt, J.; Marschall, M. The unique antiviral activity of artesunate is broadly effective against human cytomegaloviruses including therapy-resistant mutants. Antivir. Res. 2011, 92, 364–368. [Google Scholar] [CrossRef]
- Efferth, T.; Marschall, M.; Wang, X.; Huong, S.-M.; Hauber, I.; Olbrich, A.; Kronschnabl, M.; Stamminger, T.; Huang, E.-S. Antiviral activity of artesunate towards wild-type, recombinant, and ganciclovir-resistant human cytomegaloviruses. J. Mol. Med. 2002, 80, 233–242. [Google Scholar] [CrossRef]
- Efferth, T.; Romero, M.R.; Wolf, D.G.; Stamminger, T.; Marin, J.J.G.; Marschall, M. The antiviral activities of artemisinin and artesunate. Clin. Infect. Dis. 2008, 47, 804–811. [Google Scholar] [CrossRef] [Green Version]
- Frohlich, T.; Ndreshkjana, B.; Muenzner, J.K.; Reiter, C.; Hofmeister, E.; Mederer, S.; Fatfat, M.; Baba, C.E.; Muhtasib, H.G.; Stock, R.S.; et al. Synthesis of novel hybrids of thymoquinone and artemisinin with high activity and selectivity against colon cancer. Chem. Med. Chem. 2017, 12, 226–234. [Google Scholar] [CrossRef]
- Frohlich, T.; Tsogoeva, S.B. In vivo and in vitro optimization of screening antimalarial hits toward lead molecules for preclinical development. J. Med. Chem. 2016, 59, 9668–9671. [Google Scholar] [CrossRef]
- Held, F.E.; Guryev, A.A.; Fröhlich, T.; Hampel, F.; Kahnt, A.; Hutterer, C.; Steingruber, M.; Bahsi, H.; Kninski, C.V.B.; Mattes, D.S.; et al. Facile access to potent antiviral quinazoline heterocycles with fluorescence properties via merging metal-free domino reactions. Nat. Commun. 2017, 8, 15071. [Google Scholar] [CrossRef]
- Reiter, C.; Frohlich, T.; Gruber, L.; Hutterer, C.; Marschall, M.; Voigtlander, C.; Friedrich, O.; Kappes, B.; Efferth, T.; Tsogoeva, S.B. Highly potent artemisinin-derived dimers and trimers: Synthesis and evaluation of their antimalarial, antileukemia and antiviral activities. Bioorganic Med. Chem. 2015, 23, 5452–5458. [Google Scholar] [CrossRef]
- Reiter, C.; Fröhlich, T.; Zeino, M.; Marschall, M.; Bahsi, H.; Leidenberger, M.; Friedrich, O.; Kappes, B.; Hampel, F.; Efferth, T.; et al. New efficient artemisinin derived agents against human leukemia cells, human cytomegalovirus and plasmodium falciparum: 2nd generation 1,2,4-trioxane-ferrocene hybrids. Eur. J. Med. Chem. 2015, 97, 164–172. [Google Scholar] [CrossRef]
- Sharma, B.N.; Marschall, M.; Henriksen, S.; Rinaldo, C.H. Antiviral effects of artesunate on polyomavirus BK replication in primary human kidney cells. Antimicrob. Agents Chemother. 2014, 58, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.N.; Marschall, M.; Rinaldo, C.H. Antiviral effects of artesunate on JC polyomavirus replication in COS-7 cells. Antimicrob. Agents Chemother. 2014, 58, 6724–6734. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.G.; Shimoni, A.; Resnick, I.B.; Stamminger, T.; Neumann, A.U.; Chou, S.; Efferth, T.; Caplan, O.; Rose, J.; Nagler, A.; et al. Human cytomegalovirus kinetics following institution of artesunate after hematopoietic stem cell transplantation. Antivir. Res. 2011, 90, 183–186. [Google Scholar] [CrossRef] [Green Version]
- Shapira, M.Y.; Resnick, I.B.; Chou, S.; Neumann, A.U.; Lurain, N.S.; Stamminger, T.; Caplan, O.; Saleh, N.; Efferth, T.; Marschall, M.; et al. Artesunate as a potent antiviral agent in a patient with late drug-resistant cytomegalovirus infection after hematopoietic stem cell transplantation. Clin. Infect. Dis. 2008, 46, 1455–1457. [Google Scholar] [CrossRef] [Green Version]
- Hutterer, C.; Niemann, I.; Milbradt, J.; Frohlich, T.; Reiter, C.; Kadioglu, O.; Bahsi, H.; Zeittrager, I.; Wagner, S.; Einsiedel, J.; et al. The broad-spectrum antiinfective drug artesunate interferes with the canonical nuclear factor kappa B (NF-kappaB) pathway by targeting RelA/p65. Antivir. Res. 2015, 124, 101–109. [Google Scholar] [CrossRef]
- Boger, R.A.; He, R.; Chiou, C.-J.; Liu, J.; Woodard, L.; Rosenthal, A.; Brando, L.J.; Forman, M.; Posner, G. Artemisinin-derived dimers have greatly improved anti-cytomegalovirus activity compared to artemisinin monomers. PLoS ONE 2010, 5, e10370. [Google Scholar] [CrossRef] [Green Version]
- Kamate, B.B.; Forman, M.; Sangare, C.O.; Haidara, A.S.A.; Maiga, H.; Vaidya, D.; Djimde, A.; Boger, R.A. Effect of artemether-lumefantrine (Coartem) on cytomegalovirus urine viral load during and following treatment for malaria in children. J. Clin. Virol. 2016, 77, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Kapoor, A.; He, R.; Venkatadri, R.; Forman, M.; Posner, G.H.; Boger, R.A. In vitro combination of anti-cytomegalovirus compounds acting through different targets: Role of the slope parameter and insights into mechanisms of action. Antimicrob. Agents Chemother. 2014, 58, 986–994. [Google Scholar] [CrossRef] [Green Version]
- He, R.; Park, K.; Cai, H.; Kapoor, A.; Forman, M.; Mott, B.; Posner, G.H.; Boger, R.A. Artemisinin-derived dimer diphenyl phosphate is an irreversible inhibitor of human cytomegalovirus replication. Antimicrob. Agents Chemother. 2012, 56, 3508–3515. [Google Scholar] [CrossRef] [Green Version]
- Flobinus, A.; Taudon, N.; Desbordes, M.; Labrosse, B.; Simon, F.; Mazeron, M.-C.; Schnepf, N. Stability and antiviral activity against human cytomegalovirus of artemisinin derivatives. J. Antimicrob. Chemother. 2014, 69, 34–40. [Google Scholar] [CrossRef]
- Germi, R.; Mariette, C.; Alain, S.; Lupo, J.; Thiebaut, A.; Brion, J.P.; Epaulard, O.; Saint Raymond, C.; Malvezzi, P.; Morand, P. Success and failure of artesunate treatment in five transplant recipients with disease caused by drug-resistant cytomegalovirus. Antivir. Res. 2014, 101, 57–61. [Google Scholar] [CrossRef]
- He, R.; Forman, M.; Mott, B.T.; Venkatadri, R.; Posner, G.H.; Arav-Boger, R. Unique and highly selective anticytomegalovirus activities of artemisinin-derived dimer diphenyl phosphate stem from combination of dimer unit and a diphenyl phosphate moiety. Antimicrob. Agents Chemother. 2013, 57, 4208–4214. [Google Scholar] [CrossRef] [Green Version]
- He, R.; Mott, B.T.; Rosenthal, A.S.; Genna, D.T.; Posner, G.H.; Boger, R.A. An artemisinin-derived dimer has highly potent anti-cytomegalovirus (CMV) and anti-cancer activities. PLoS ONE 2011, 6, e24334. [Google Scholar] [CrossRef] [Green Version]
- Ho, W.E.; Peh, H.Y.; Chan, T.K.; Wong, W.S.F. Artemisinins: Pharmacological actions beyond anti-malarial. Pharmacol 2014, 142, 126–139. [Google Scholar] [CrossRef]
- Roy, S.; He, R.; Kapoor, A.; Forman, M.; Mazzone, J.R.; Posner, G.H.; Boger, R.A. Inhibition of human cytomegalovirus replication by artemisinins: Effects mediated through cell cycle modulation. Antimicrob. Agents Chemother. 2015, 59, 3870–3879. [Google Scholar] [CrossRef] [Green Version]
- Stuehler, C.; Stüssi, G.; Halter, J.; Nowakowska, J.; Schibli, A.; Battegay, M.; Dirks, J.; Passweg, J.; Heim, D.; Rovo, A.; et al. Combination therapy for multidrug-resistant cytomegalovirus disease. Transpl. Infect. Dis. 2015, 17, 751–755. [Google Scholar] [CrossRef]
- Zeng, A.-H.; Ou, Y.-Y.; Guo, M.-M.; Dai, X.; Zhou, D.-Z.; Chen, R. Human embryonic lung fibroblasts treated with artesunate exhibit reduced rates of proliferation and human cytomegalovirus infection in vitro. J. Thorac. Dis. 2015, 7, 1151–1157. [Google Scholar] [CrossRef]
- Auerochs, S.; Korn, K.; Marschall, M. A reporter system for epstein-barr virus (EBV) lytic replication: Anti-EBV activity of the broad anti-herpesviral drug artesunate. J. Virol. Methods 2011, 173, 334–339. [Google Scholar] [CrossRef]
- Milbradt, J.; Auerochs, S.; Korn, K.; Marschall, M. Sensitivity of human herpesvirus 6 and other human herpesviruses to the broad-spectrum antiinfective drug artesunate. J. Clin. Virol. 2009, 46, 24–28. [Google Scholar] [CrossRef]
- Jana, S.; Iram, S.; Thomas, J.; Hayat, M.Q.; Pannecouque, C.; Dehaen, W. Application of the triazolization reaction to afford dihydroartemisinin derivatives with anti-HIV activity. Molecules 2017, 22, 303. [Google Scholar] [CrossRef]
- Morere, L.; Andouard, D.; Labrousse, F.; Saade, F.; Calliste, C.A.; Cotin, S.; Aubard, Y.; Rawlinson, W.D.; Esclaire, F.; Hantz, S.; et al. Ex vivo model of congenital cytomegalovirus infection and new combination therapies. Placenta 2015, 36, 41–47. [Google Scholar] [CrossRef]
- Paeshuyse, J.; Coelmont, L.; Vliegen, I.; Van hemel, J.; Vandenkerckhove, J.; Peys, E.; Sas, B.; De Clercq, E.; Neyts, J. Hemin potentiates the anti-hepatitis C virus activity of the antimalarial drug artemisinin. Biochem. Biophys. Res. Commun. 2006, 348, 139–144. [Google Scholar] [CrossRef]
- Qian, R.S.; Li, Z.L.; Yu, J.L.; Ma, D.J. The immunologic and antiviral effect of qinghaosu. J. Tradit. Chin. Med. 1982, 2, 271–276. [Google Scholar]
- Romero, M.R.; Efferth, T.; Serrano, M.A.; Castano, B.; Macias, R.I.; Briz, O.; Marin, J.J. Effect of artemisinin/artesunate as inhibitors of hepatitis B virus production in an “in vitro” replicative system. Antivir. Res. 2005, 68, 75–83. [Google Scholar] [CrossRef]
- Romero, M.R.; Serrano, M.A.; Vallejo, M.; Efferth, T.; Alvarez, M.; Marin, J.J.G. Antiviral effect of artemisinin from artemisia annua against a model member of the flaviviridae family, the bovine viral diarrhoea virus (BVDV). Planta Med. 2006, 72, 1169–1174. [Google Scholar] [CrossRef]
- Tsogoeva, S.B. Recent progress in the development of synthetic hybrids of natural or unnatural bioactive compounds for medicinal chemistry. Mini Rev. Med. Chem. 2010, 10, 773–793. [Google Scholar] [CrossRef]
- Hahn, F.; Fröhlich, T.; Frank, T.; Bertzbach, L.D.; Kohrt, S.; Kaufer, B.B.; Stamminger, T.; Tsogoeva, S.B.; Marschall, M. Artesunate-derived monomeric, dimeric and trimeric experimental drugs–Their unique mechanistic basis and pronounced antiherpesviral activity. Antivir. Res. 2018, 152, 104–110. [Google Scholar] [CrossRef]
- Sonntag, E.; Hahn, F.; Bertzbach, L.D.; Seyler, L.; Wangen, C.; Müller, R.; Tannig, P.; Grau, B.; Baumann, M.; Zent, E.; et al. In vivo proof-of-concept for two experimental antiviral drugs, both directed to cellular targets, using a murine cytomegalovirus model. Antivir. Res. 2019, 161, 63–69. [Google Scholar] [CrossRef]
- Wild, M.; Bertzbach, L.D.; Tannig, P.; Wangen, C.; Müller, R.; Herrmann, L.; Fröhlich, T.; Tsogoeva, S.B.; Kaufer, B.B.; Marschall, M.; et al. The trimeric artesunate derivative TF27 exerts strong anti-cytomegaloviral efficacy: Focus on prophylactic efficacy and oral treatment of immunocompetent mice. Antivir. Res. 2020, 178, 104788. [Google Scholar] [CrossRef]
- Bork, P.M.; Schmitz, M.L.; Kuhnt, M.; Escher, C.; Heinrich, M. Sesquiterpene lactone containing mexican indian medicinal plants and pure sesquiterpene lactones as potent inhibitors of transcription factor NF-kappaB. FEBS Lett. 1997, 402, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Pineres, A.J.; Castro, V.; Mora, G.; Schmidt, T.J.; Strunck, E.; Pahl, H.L.; Merfort, I. Cysteine 38 in p65/NF-kappaB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. J. Biol. Chem. 2001, 276, 39713–39720. [Google Scholar] [CrossRef] [Green Version]
- Pineres, A.J.G.; Lindenmeyer, M.T.; Merfort, I. Role of cysteine residues of p65/NF-kappaB on the inhibition by the sesquiterpene lactone parthenolide and N-ethyl maleimide, and on its transactivating potential. Life Sci. 2004, 75, 841–856. [Google Scholar] [CrossRef]
- Lyss, G.; Knorre, A.; Schmidt, T.J.; Pahl, H.L.; Merfort, I. The anti-inflammatory sesquiterpene lactone helenalin inhibits the transcription factor NF-kappaB by directly targeting p65. J. Biol. Chem. 1998, 273, 33508–33516. [Google Scholar] [CrossRef] [Green Version]
- Siedle, B.; Pineres, A.J.G.; Murillo, R.; Monting, J.S.; Castro, V.; Rungeler, P.; Klaas, C.A.; Costa, F.B.D.; Kisiel, W.; Merfort, I. Quantitative structure-activity relationship of sesquiterpene lactones as inhibitors of the transcription factor NF-kappaB. J. Med. Chem. 2004, 47, 6042–6054. [Google Scholar] [CrossRef]
- Souza, M.C.; Paixao, F.H.; Ferraris, F.K.; Ribeiro, I.; Henriques, M. Artesunate exerts a direct effect on endothelial cell activation and NF-kappaB translocation in a mechanism independent of plasmodium killing. Malar. Res. Treat. 2012, 2012, 679090. [Google Scholar] [CrossRef]
- Ismail, H.M.; Barton, V.; Phanchana, M.; Charoensutthivarakul, S.; Wong, M.H.; Hemingway, J.; Biagini, G.A.; O‘Neill, P.M.; Ward, S.A. Artemisinin activity-based probes identify multiple molecular targets within the asexual stage of the malaria parasites Plasmodium falciparum 3D7. Proc. Natl. Acad. Sci. USA 2016, 113, 2080–2085. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Casteels, T.; Frogne, T.; Ingvorsen, C.; Honore, C.; Courtney, M.; Huber, K.V.M.; Schmitner, N.; Kimmel, R.A.; Romanov, R.A.; et al. Artemisinins target GABAA receptor signaling and impair alpha cell identity. Cell 2017, 168, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, C.J.; Chia, W.N.; Loh, C.C.; Li, Z.; Lee, Y.M.; He, Y.; Yuan, L.X.; Lim, T.K.; Liu, M.; et al. Haem-activated promiscuous targeting of artemisinin in plasmodium falciparum. Nat. Commun. 2015, 6, 10111. [Google Scholar] [CrossRef]
- Hahn, F.; Niesar, A.; Wangen, C.; Wild, M.; Grau, B.; Herrmann, L.; Capci, A.; Adrait, A.; Couté, Y.; Tsogoeva, S.B.; et al. Target verification of artesunate-related antiviral drugs: Assessing the role of mitochondrial and regulatory proteins by click chemistry and fluorescence labeling. Antivir. Res. 2020. accepted. [Google Scholar]
- Nijtmans, L.G.; Jong, L.D.; Sanz, M.A.; Coates, P.J.; Berden, J.A.; Back, J.W.; Muijsers, A.O.; Spek, H.V.D.; Grivell, L.A. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 2000, 19, 2444–2451. [Google Scholar] [CrossRef] [Green Version]
- Schleicher, M.; Shepherd, B.R.; Suarez, Y.; Hernando, C.F.; Yu, J.; Pan, Y.; Acevedo, L.M.; Shadel, G.S.; Sessa, W.C. Prohibitin-1 maintains the angiogenic capacity of endothelial cells by regulating mitochondrial function and senescence. J. Cell Biol. 2008, 180, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Lea, P.J.; Temkin, R.J.; Freeman, K.B.; Mitchell, G.A.; Robinson, B.H. Variations in mitochondrial ultrastructure and dynamics observed by high resolution scanning electron microscopy (HRSEM). Microsc. Res. Tech. 1994, 27, 269–277. [Google Scholar] [CrossRef]
- Amchenkova, A.A.; Bakeeva, L.E.; Chentsov, Y.S.; Skulachev, V.P.; Zorov, D.B. Coupling membranes as energy-transmitting cables. I. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. J. Cell Biol. 1988, 107, 481–495. [Google Scholar] [CrossRef] [Green Version]
- Steingruber, M.; Marschall, M. The cytomegalovirus protein kinase pUL97:Host interactions, regulatory mechanisms and antiviral drug targeting. Microorganisms 2020, 8, 515. [Google Scholar] [CrossRef] [Green Version]
- Hahn, F.; Hutterer, C.; Henry, C.; Hamilton, S.T.; Strojan, H.; Kraut, A.; Schulte, U.; Schütz, M.; Kohrt, S.; Wangen, C.; et al. Novel cytomegalovirus-inhibitory compounds of the class pyrrolopyridines show a complex pattern of target binding that suggests an unusual mechanism of antiviral activity. Antivir. Res. 2018, 159, 84–94. [Google Scholar] [CrossRef]
- Çapcı, A.; Lorion, M.M.; Mai, C.; Hahn, F.; Hodek, J.; Wangen, C.; Weber, J.; Marschall, M.; Ackermann, L.; Tsogoeva, S.B. (Iso)Quinoline-artemisinin hybrids via click chemistry: Highly potent agents against viruses. Chem. Eur. J. 2020. [Google Scholar] [CrossRef]
- Çapcı, A.; Lorion, M.M.; Wang, H.; Simon, N.; Leidenberger, M.; Borges Silva, M.C.; Moreira, D.R.M.; Zhu, Y.; Meng, Y.; Chen, J.Y.; et al. Artemisinin-(Iso)quinoline hybrids by C-H activation and click chemistry: Combating multidrug-resistant malaria. Angew. Chem. Int. Ed. Engl. 2019, 58, 13066–13079. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Sun, X.; Wang, L.; Wong, Y.K.; Lee, Y.M.; Zhou, C.; Wu, G.; Zhao, T.; Yang, L.; Lu, L.; et al. Artesunate-induced mitophagy alters cellular redox status. Redox Biol. 2018, 19, 263–273. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, W.; Xiao, Y. Profiling of multiple targets of artemisinin activated by hemin in cancer cell proteome. ACS Chem. Biol. 2016, 11, 882–888. [Google Scholar] [CrossRef]
- Wu, G.; Cheng, B.; Qian, H.; Ma, S.; Chen, Q. Identification of HSP90 as a direct target of artemisinin for its anti-inflammatory activity via quantitative chemical proteomics. Org. Biomol. Chem. 2019, 17, 6854–6859. [Google Scholar] [CrossRef]
- Fröhlich, T.; Hahn, F.; Belmudes, L.; Leidenberger, M.; Friedrich, O.; Kappes, B.; Couté, Y.; Marschall, M.; Tsogoeva, S.B. Synthesis of artemisinin-derived dimers, trimers and dendrimers: Investigation of their antimalarial and antiviral activities including putative mechanisms of action. Chem. Eur. J. 2018, 24, 8103–8113. [Google Scholar] [CrossRef]
- Anand, S.K.; Tikoo, S.K. Viruses as modulators of mitochondrial functions. Adv. Virol. 2013, 2013, 738794. [Google Scholar] [CrossRef]
- Furukawa, T.; Sakuma, S.; Plotkin, S.A. Human cytomegalovirus infection of WI-38 cells stimulates mitochondrial DNA synthesis. Nature 1976, 262, 414–416. [Google Scholar] [CrossRef]
- Landini, M.P.; Rugolo, M. Increased accumulation of a lipophilic cation (tetraphenylphosphonium) in human embryo fibroblasts after infection with cytomegalovirus. J. Gen. Virol. 1984, 65, 2269–2272. [Google Scholar] [CrossRef]
- Kaarbo, M.; Ager-Wick, E.; Osenbroch, P.O.; Kilander, A.; Skinnes, R.; Muller, F.; Eide, L. Human cytomegalovirus infection increases mitochondrial biogenesis. Mitochondrion 2011, 11, 935–945. [Google Scholar] [CrossRef]
- Hertel, L.; Mocarski, E.S. Global analysis of host cell gene expression late during cytomegalovirus infection reveals extensive dysregulation of cell cycle gene expression and induction of pseudomitosis independent of US28 function. J. Virol. 2004, 78, 11988–12011. [Google Scholar] [CrossRef] [Green Version]
- McKinney, C.; Zavadil, J.; Bianco, C.; Shiflett, L.; Brown, S.; Mohr, I. Global reprogramming of the cellular translational landscape facilitates cytomegalovirus replication. Cell Rep. 2014, 6, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science 2007, 316, 1345–1348. [Google Scholar] [CrossRef] [Green Version]
- Munger, J.; Bajad, S.U.; Coller, H.A.; Shenk, T.; Rabinowitz, J.D. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2006, 2, e132. [Google Scholar] [CrossRef] [Green Version]
- Crowe, W.E.; Maglova, L.M.; Ponka, P.; Russell, J.M. Human cytomegalovirus-induced host cell enlargement is iron dependent. Am. J. Physiol. Cell Ph. 2004, 287, C1023–C1030. [Google Scholar] [CrossRef]
- Combs, J.A.; Norton, E.B.; Saifudeen, Z.R.; Bentrup, K.H.Z.; Katakam, P.V.; Morris, C.A.; Myers, L.; Kaur, A.; Sullivan, D.E.; Zwezdaryk, K.J. Human cytomegalovirus alters host cell mitochondrial function during acute infection. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Zhang, A.; Williamson, C.D.; Wong, D.S.; Bullough, M.D.; Brown, K.J.; Hathout, Y.; Poley, A.M.C. Quantitative proteomic analyses of human cytomegalovirus-induced restructuring of endoplasmic reticulum-mitochondrial contacts at late times of infection. Mol. Cell Proteom. 2011, 10. [Google Scholar] [CrossRef] [Green Version]
- Karniely, S.; Weekes, M.P.; Antrobus, R.; Rorbach, J.; Haute, L.V.; Umrania, Y.; Smith, D.L.; Stanton, R.J.; Minczuk, M.; Lehner, P.J.; et al. Human cytomegalovirus infection upregulates the mitochondrial transcription and translation machineries. mBio 2016, 7, e00029-16. [Google Scholar] [CrossRef] [Green Version]
- McCormick, A.L.; Smith, V.L.; Chow, D.; Mocarski, E.S. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J. Virol. 2003, 77, 631–641. [Google Scholar] [CrossRef] [Green Version]
- Colberg-Poley, A.M.; Patel, M.B.; Erezo, D.P.; Slater, J.E. Human cytomegalovirus UL37 immediate-early regulatory proteins traffic through the secretory apparatus and to mitochondria. J. Gen. Virol. 2000, 81, 1779–1789. [Google Scholar] [CrossRef]
- Norris, K.L.; Youle, R.J. Cytomegalovirus proteins vMIA and m38.5 link mitochondrial morphogenesis to Bcl-2 family proteins. J. Virol. 2008, 82, 6232–6243. [Google Scholar] [CrossRef] [Green Version]
- Kaptein, S.J.; Efferth, T.; Leis, M.; Rechter, S.; Auerochs, S.; Kalmer, M.; Bruggeman, C.A.; Vink, C.; Stamminger, T.; Marschall, M. The anti-malaria drug artesunate inhibits replication of cytomegalovirus in vitro and in vivo. Antivir. Res. 2006, 69, 60–69. [Google Scholar] [CrossRef]
- Liu, K.; Zuo, H.; Li, G.; Yu, H.; Hu, Y. Global research on artemisinin and its derivatives: Perspectives from patents. Pharmacol. Res. 2020, 159, 105048. [Google Scholar] [CrossRef]
- Hanboonkunupakarn, B.; White, N.J. Advances and roadblocks in the treatment of malaria. Br. J. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Golenser, J.; Waknine, J.H.; Krugliak, M.; Hunt, N.H.; Grau, G.E. Current perspectives on the mechanism of action of artemisinins. Int. J. Parasitol. 2006, 36, 1427–1441. [Google Scholar] [CrossRef]
- Jiang, J.B.; Jacobs, G.; Liang, D.S.; Aikawa, M. Qinghaosu-induced changes in the morphology of plasmodium inui. Am. J. Trop. Med. Hyg. 1985, 34, 424–428. [Google Scholar] [CrossRef]
- Kawai, S.; Kano, S.; Suzuki, M. Morphologic effects of artemether on plasmodium falciparum in aotus trivirgatus. Am. J. Trop. Med. Hyg. 1993, 49, 812–818. [Google Scholar] [CrossRef]
- Supale, S.; Thorel, F.; Merkwirth, C.; Gjinovci, A.; Herrera, P.L.; Scorrano, L.; Meda, P.; Langer, T.; Maechler, P. Loss of prohibitin induces mitochondrial damages altering beta-cell function and survival and is responsible for gradual diabetes development. Diabetes 2013, 62, 3488–3499. [Google Scholar] [CrossRef] [Green Version]
- Chaiyarit, S.; Thongboonkerd, V. Comparative analyses of cell disruption methods for mitochondrial isolation in high-throughput proteomics study. Anal. Biochem. 2009, 394, 249–258. [Google Scholar] [CrossRef]
- Kalpage, H.A.; Bazylianska, V.; Recanati, M.A.; Fite, A.; Liu, J.; Wan, J.; Mantena, N.; Malek, M.H.; Podgorski, I.; Heath, E.I.; et al. Tissue-specific regulation of cytochrome c by post-translational modifications: Respiration, the mitochondrial membrane potential, ROS, and apoptosis. FASEB. J. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 1540–1553. [Google Scholar] [CrossRef]
- Bertzbach, L.D.; Conradie, A.M.; Hahn, F.; Wild, M.; Marschall, M.; Kaufer, B.B. Artesunate derivative TF27 inhibits replication and pathogenesis of an oncogenic avian alphaherpesvirus. Antivir. Res. 2019, 171, 104606. [Google Scholar] [CrossRef]
- Jacquet, C.; Marschall, M.; Andouard, D.; Hamel, C.E.; Chianea, T.; Tsogoeva, S.B.; Hantz, S.; Alain, S. A highly potent trimeric derivative of artesunate shows promising treatment profiles in experimental models for congenital HCMV infection in vitro and ex vivo. Antivir. Res. 2020, 175, 104700. [Google Scholar] [CrossRef]
- Burke, C.S.; Byrne, A.; Keyes, T.E. Highly selective mitochondrial targeting by a ruthenium(II) peptide conjugate: Imaging and photoinduced damage of mitochondrial DNA. Angew. Chem. Int. Ed. Engl. 2018, 57, 12420–12424. [Google Scholar] [CrossRef]
- Antoine, T.; Fisher, N.; Amewu, R.; O‘Neill, P.M.; Ward, S.A.; Biagini, G.A. Rapid kill of malaria parasites by artemisinin and semi-synthetic endoperoxides involves ROS-dependent depolarization of the membrane potential. J. Antimicrob. Chemother. 2013, 69, 1005–1016. [Google Scholar] [CrossRef]
- Rowe, W.P.; Hartley, J.W.; Waterman, S.; Turner, H.C.; Huebner, R.J. Cytopathogenic agent resembling human salivary gland virus recovered from tissue cultures of human adenoids. Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med. 1956, 92, 418–424. [Google Scholar]
- Marschall, M.; Freitag, M.; Weiler, S.; Sorg, G.; Stamminger, T. Recombinant green fluorescent protein-expressing human cytomegalovirus as a tool for screening antiviral agents. Antimicrob. Agents Chemother. 2000, 44, 1588–1597. [Google Scholar] [CrossRef] [Green Version]
- Rechter, S.; König, T.; Auerochs, S.; Thulke, S.; Walter, H.; Dörnenburg, H.; Walter, C.; Marschall, M. Antiviral activity of arthrospira-derived spirulan-like substances. Antivir. Res. 2006, 72, 197–206. [Google Scholar] [CrossRef]
- Hutterer, C.; Eickhoff, J.; Milbradt, J.; Korn, K.; Zeittrager, I.; Bahsi, H.; Wagner, S.; Zischinsky, G.; Wolf, A.; Degenhart, C.; et al. A novel CDK7 inhibitor of the pyrazolotriazine class exerts broad-spectrum antiviral activity at nanomolar concentrations. Antimicrob. Agents Chemother. 2015, 59, 2062–2071. [Google Scholar] [CrossRef] [Green Version]
- Repetto, G.; Peso, A.D.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef]
- Hutterer, C.; Wandinger, S.K.; Wagner, S.; Müller, R.; Stamminger, T.; Zeitträger, I.; Godl, K.; Baumgartner, R.; Strobl, S.; Marschall, M. Profiling of the kinome of cytomegalovirus-infected cells reveals the functional importance of host kinases aurora A, ABL and AMPK. Antivir. Res. 2013, 99, 139–148. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wild, M.; Hahn, F.; Grau, B.; Herrmann, L.; Niesar, A.; Schütz, M.; Lorion, M.M.; Ackermann, L.; Tsogoeva, S.B.; Marschall, M. The Artemisinin-Derived Autofluorescent Compound BG95 Exerts Strong Anticytomegaloviral Activity Based on a Mitochondrial Targeting Mechanism. Int. J. Mol. Sci. 2020, 21, 5578. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155578
Wild M, Hahn F, Grau B, Herrmann L, Niesar A, Schütz M, Lorion MM, Ackermann L, Tsogoeva SB, Marschall M. The Artemisinin-Derived Autofluorescent Compound BG95 Exerts Strong Anticytomegaloviral Activity Based on a Mitochondrial Targeting Mechanism. International Journal of Molecular Sciences. 2020; 21(15):5578. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155578
Chicago/Turabian StyleWild, Markus, Friedrich Hahn, Benedikt Grau, Lars Herrmann, Aischa Niesar, Martin Schütz, Melanie M. Lorion, Lutz Ackermann, Svetlana B. Tsogoeva, and Manfred Marschall. 2020. "The Artemisinin-Derived Autofluorescent Compound BG95 Exerts Strong Anticytomegaloviral Activity Based on a Mitochondrial Targeting Mechanism" International Journal of Molecular Sciences 21, no. 15: 5578. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21155578