Phosphodiesterases in the Liver as Potential Therapeutic Targets of Cirrhotic Portal Hypertension

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Pathophysiology of Portal Hypertension
2.1. Elevated Intrahepatic Vascular Resistance
2.2. Intrahepatic Circulation
2.2.1. The Hepatic Microvascular System
2.2.2. Sinusoidal Endothelial Cell Dysfunction
2.3. Activation of Hepatic Stellate Cells
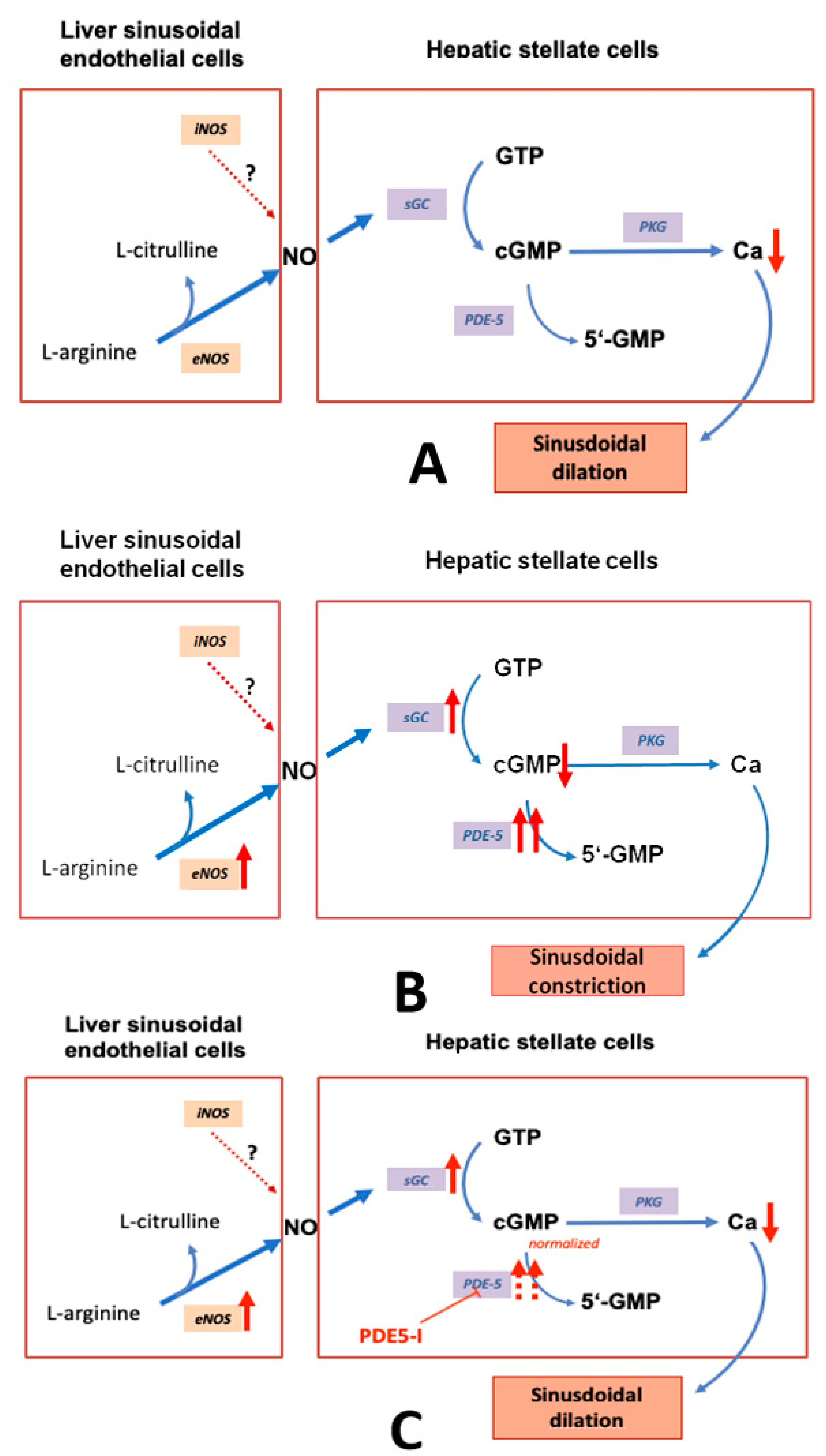
2.3.1. eNOS and iNOS
2.3.2. sGC and PDE-5
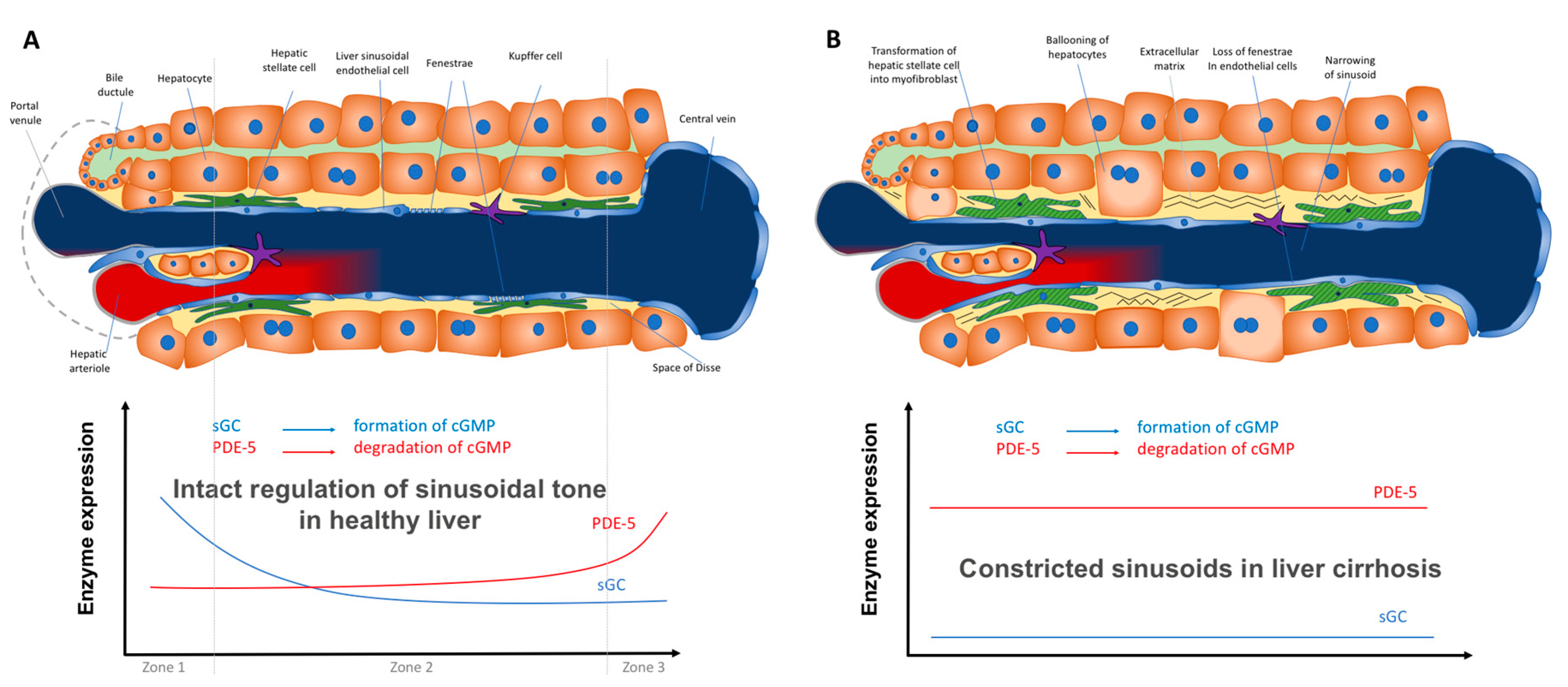
2.3.3. Zonation of the Components of the NO–cGMP Pathway: Opposing Zonation of sGC and PDE-5
2.4. Further Vascular Alterations in Portal Hypertension
2.4.1. Angiogenesis
2.4.2. Peripheral Systemic Circulation
3. Targeting Components of the NO-cGMP Pathway for Therapy of Portal Hypertension
3.1. NO
3.2. sGC
3.3. PDE-5
3.4. PDE-5 Inhibitors as Therapeutic Alternative to Treat Portal Hypertension
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NO | nitric oxide, |
| GTP | guanosine triphosphate, |
| cGMP | cyclic guanosine monophosphate, |
| PKG | protein kinase G, |
| eNOS | endothelial NO synthase, |
| iNOS | inducible NO synthase, |
| PDE-5 | phosphodiesterase 5, |
| PDE-5-I | PDE-5 inhibitor, |
| Ca | calcium |
References
- Lincoln, T.M. Cyclic GMP and phosphodiesterase 5 inhibitor therapies: What’s on the horizon? Mol. Pharmacol. 2004, 66, 11–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manallack, D.T.; Hughes, R.A.; Thompson, P.E. The next generation of phosphodiesterase inhibitors: Structural clues to ligand and substrate selectivity of phosphodiesterases. J. Med. Chem. 2005, 48, 3449–3462. [Google Scholar] [CrossRef] [PubMed]
- Corbin, J.D.; Francis, S.H. Cyclic GMP phosphodiesterase-5: Target of sildenafil. J. Biol. Chem. 1999, 274, 13729–13732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef] [PubMed]
- Yaghi, S.; Novikov, A.; Trandafirescu, T. Clinical update on pulmonary hypertension. J. Investig. Med. 2020, 68, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, Y.; Liu, J.; Qian, G. Meta-analysis of clinical efficacy of sildenafil, a phosphodiesterase type-5 inhibitor on high altitude hypoxia and its complications. High Alt. Med. Biol. 2014, 15, 46–51. [Google Scholar] [CrossRef]
- Ribaudo, G.; Pagano, M.A.; Bova, S.; Zagotto, G. New Therapeutic Applications of Phosphodiesterase 5 Inhibitors (PDE5-Is). Curr. Med. Chem. 2016, 23, 1239–1249. [Google Scholar] [CrossRef]
- Jeon, Y.H.; Heo, Y.-S.; Kim, C.M.; Hyun, Y.-L.; Lee, T.G.; Ro, S.; Cho, J.M. Phosphodiesterase: Overview of protein structures, potential therapeutic applications and recent progress in drug development. Cell. Mol. Life Sci. 2005, 62, 1198–1220. [Google Scholar] [CrossRef]
- Schwartz, B.G.; Levine, L.A.; Comstock, G.; Stecher, V.J.; Kloner, R.A. Cardiac uses of phosphodiesterase-5 inhibitors. J. Am. Coll. Cardiol. 2012, 59, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Hollas, M.A.; Ben Aissa, M.; Lee, S.H.; Gordon-Blake, J.M.; Thatcher, G.R.J. Pharmacological manipulation of cGMP and NO/cGMP in CNS drug discovery. Nitric Oxide 2019, 82, 59–74. [Google Scholar] [CrossRef]
- Groneberg, D.; König, P.; Koesling, D.; Friebe, A. Nitric Oxide–Sensitive Guanylyl Cyclase Is Dispensable for Nitrergic Signaling and Gut Motility in Mouse Intestinal Smooth Muscle. Gastroenterology 2011, 140, 1608–1617. [Google Scholar] [CrossRef]
- Solaimanzadeh, I. Acetazolamide, Nifedipine and Phosphodiesterase Inhibitors: Rationale for Their Utilization as Adjunctive Countermeasures in the Treatment of Coronavirus Disease 2019 (COVID-19). Cureus 2020, 12, e7343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, M. Molecular pathophysiology of portal hypertension. Hepatology 2015, 61, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Greuter, T.; Shah, V.H. Hepatic sinusoids in liver injury, inflammation, and fibrosis: New pathophysiological insights. J. Gastroenterol. 2016, 51, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCuskey, R.S. The hepatic microvascular system in health and disease. FASEB J. 2009, 23, S1:63.1. [Google Scholar]
- Eipel, C.; Abshagen, K.; Vollmar, B. Regulation of hepatic blood flow: The hepatic arterial buffer response revisited. World J. Gastroenterol. 2010, 16, 6046–6057. [Google Scholar] [CrossRef] [PubMed]
- Kalra, A.; Yetiskul, E.; Wehrle, C.J.; Tuma, F. Physiology, Liver; StatPearls Publishing: Treasure Island, FL, USA, 2020; Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK535438/ (accessed on 17 August 2020).
- Bosch, J.; Groszmann, R.J.; Shah, V.H. Evolution in the understanding of the pathophysiological basis of portal hypertension: How changes in paradigm are leading to successful new treatments. J. Hepatol. 2015, 62, S121–S130. [Google Scholar] [CrossRef] [Green Version]
- Iwakiri, Y.; Shah, V.; Rockey, D.C. Vascular pathobiology in chronic liver disease and cirrhosis—current status and future directions. J. Hepatol. 2014, 61, 912–924. [Google Scholar] [CrossRef] [Green Version]
- Perri, R.E.; Langer, D.A.; Chatterjee, S.; Gibbons, S.J.; Gadgil, J.; Cao, S.; Farrugia, G.; Shah, V.H. Defects in cGMP-PKG pathway contribute to impaired NO-dependent responses in hepatic stellate cells upon activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G535–G542. [Google Scholar] [CrossRef]
- Shah, V.; Haddad, F.G.; Garcia-Cardena, G.; Frangos, J.A.; Mennone, A.; Groszmann, R.J.; Sessa, W.C. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J. Clin. Investig. 1997, 100, 2923–2930. [Google Scholar] [CrossRef] [Green Version]
- De Franchis, R.; Baveno VI Faculty. Expanding consensus in portal hypertension: Report of the Baveno VI Consensus Workshop: Stratifying risk and individualizing care for portal hypertension. J. Hepatol. 2015, 63, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakab, S.S.; Garcia-Tsao, G. Evaluation and Management of Esophageal and Gastric Varices in Patients with Cirrhosis. Clin. Liver Dis. 2020, 24, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Turco, L.; Garcia-Tsao, G. Portal Hypertension: Pathogenesis and Diagnosis. Clin. Liver Dis. 2019, 23, 573–587. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Maeso-Díaz, R.; Bosch, J. Pathophysiology and a Rational Basis of Therapy. Dig. Dis. 2015, 33, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, S.M.; Waldman, B. The Circulatory System in Liver Disease. Crit. Care Clin. 2016, 32, 331–342. [Google Scholar] [CrossRef]
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- Tripathi, D. Drugs used in therapy of portal hypertension. Clin. Liver Dis. 2012, 1, 136–138. [Google Scholar] [CrossRef]
- Tetangco, E.P.; Silva, R.G.; Lerma, E.V. Portal hypertension: Etiology, evaluation, and management. Dis. Mon. 2016, 62, 411–426. [Google Scholar] [CrossRef]
- Møller, S.; Bendtsen, F. Complications of cirrhosis. A 50 years flashback. Scand. J. Gastroenterol. 2015, 50, 763–780. [Google Scholar] [CrossRef]
- La Mura, V.; Abraldes, J.G.; Raffa, S.; Retto, O.; Berzigotti, A.; García-Pagán, J.C.; Bosch, J. Prognostic value of acute hemodynamic response to i.v. propranolol in patients with cirrhosis and portal hypertension. J. Hepatol. 2009, 51, 279–287. [Google Scholar] [CrossRef]
- Rockey, D.C. Hepatic Blood Flow Regulation by Stellate Cells in Normal and Injured Liver. Semin. Liver Dis. 2001, 21, 337–350. [Google Scholar] [CrossRef] [PubMed]
- García-Pagán, J.-C.; Gracia-Sancho, J.; Bosch, J. Functional aspects on the pathophysiology of portal hypertension in cirrhosis. J. Hepatol. 2012, 57, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.; Di Pascoli, M.; Verardo, A.; Gatta, A. Splanchnic vasodilation and hyperdynamic circulatory syndrome in cirrhosis. World J. Gastroenterol. 2014, 20, 2555–2563. [Google Scholar] [CrossRef]
- Münzel, T.; Feil, R.; Mülsch, A.; Lohmann, S.M.; Hofmann, F.; Walter, U. Physiology and Pathophysiology of Vascular Signaling Controlled by Cyclic Guanosine 3′,5′-Cyclic Monophosphate–Dependent Protein Kinase. Circulation 2003, 108, 2172–2183. [Google Scholar] [CrossRef] [Green Version]
- Münzel, T. Endotheliale Dysfunktion: Pathophysiologie, Diagnostik und prognostische Bedeutung. DMW Dtsch. Med. Wochenschr. 2008, 133, 2465–2470. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Palmer, R.M.; Higgs, E.A. Biosynthesis of nitric oxide from L-arginine. A pathway for the regulation of cell function and communication. Biochem. Pharmacol. 1989, 38, 1709–1715. [Google Scholar] [CrossRef]
- Vallance, P.; Moncada, S. Hyperdynamic circulation in cirrhosis: A role for nitric oxide? Lancet 1991, 337, 776–778. [Google Scholar] [CrossRef]
- Hanafy, K.A.; Krumenacker, J.S.; Murad, F. NO, nitrotyrosine, and cyclic GMP in signal transduction. Med. Sci. Monit. 2001, 7, 801–819. [Google Scholar]
- Vallance, P.; Leiper, J. Blocking NO synthesis: How, where and why? Nat. Rev. Drug Discov. 2002, 1, 939–950. [Google Scholar] [CrossRef]
- Wobst, J.; Kessler, T.; Dang, T.A.; Erdmann, J.; Schunkert, H. Role of sGC-dependent NO signalling and myocardial infarction risk. J. Mol. Med. 2015, 93, 383–394. [Google Scholar] [CrossRef]
- Wobst, J.; von Ameln, S.; Wolf, B.; Wierer, M.; Dang, T.A.; Sager, H.B.; Tennstedt, S.; Hengstenberg, C.; Koesling, D.; Friebe, A.; et al. Stimulators of the soluble guanylyl cyclase: Promising functional insights from rare coding atherosclerosis-related GUCY1A3 variants. Basic Res. Cardiol. 2016, 111. [Google Scholar] [CrossRef] [PubMed]
- Theilig, F.; Bostanjoglo, M.; Pavenstädt, H.; Grupp, C.; Holland, G.; Slosarek, I.; Gressner, A.M.; Russwurm, M.; Koesling, D.; Bachmann, S. Cellular distribution and function of soluble guanylyl cyclase in rat kidney and liver. J. Am. Soc. Nephrol. 2001, 12, 2209–2220. [Google Scholar] [PubMed]
- Ni, Y.; Li, J.-M.; Liu, M.-K.; Zhang, T.-T.; Wang, D.-P.; Zhou, W.-H.; Hu, L.-Z.; Lv, W.-L. Pathological process of liver sinusoidal endothelial cells in liver diseases. World J. Gastroenterol. 2017, 23, 7666–7677. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, Y.; Tsai, M.-H.; McCabe, T.J.; Gratton, J.-P.; Fulton, D.; Groszmann, R.J.; Sessa, W.C. Phosphorylation of eNOS initiates excessive NO production in early phases of portal hypertension. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H2084–H2090. [Google Scholar] [CrossRef] [PubMed]
- Tsai, E.J.; Kass, D.A. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol. Ther. 2009, 122, 216–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifer, A.; Kilić, A.; Hoffmann, L.S. Regulation of metabolism by cGMP. Pharmacol. Ther. 2013, 140, 81–91. [Google Scholar] [CrossRef]
- Yang, J.; Clark, J.W.; Bryan, R.M.; Robertson, C.S. Mathematical modeling of the nitric oxide/cGMP pathway in the vascular smooth muscle cell. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H886–H897. [Google Scholar] [CrossRef]
- Kim, N.N. Phosphodiesterase type 5 inhibitors: A biochemical and clinical correlation survey. Int. J. Impot. Res. 2003, 15 (Suppl. 5), S13–S19. [Google Scholar] [CrossRef] [Green Version]
- Wall, M.E.; Francis, S.H.; Corbin, J.D.; Grimes, K.; Richie-Jannetta, R.; Kotera, J.; Macdonald, B.A.; Gibson, R.R.; Trewhella, J. Mechanisms associated with cGMP binding and activation of cGMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 2003, 100, 2380–2385. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.Y.; Jeong, W.K.; Baik, S.K. Invasive and non-invasive diagnosis of cirrhosis and portal hypertension. World J. Gastroenterol. 2014, 20, 4300–4315. [Google Scholar] [CrossRef]
- Krawutschke, C.; Koesling, D.; Russwurm, M. Cyclic GMP in Vascular Relaxation: Export Is of Similar Importance as Degradation. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2011–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybalkin, S.D.; Yan, C.; Bornfeldt, K.E.; Beavo, J.A. Cyclic GMP Phosphodiesterases and Regulation of Smooth Muscle Function. Circ. Res. 2003, 93, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A.; et al. Sildenafil Use in Pulmonary Arterial Hypertension (SUPER) Study Group. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakiri, Y. Pathophysiology of Portal Hypertension. Clin. Liver Dis. 2014, 18, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, Y.S.; Shah, V.H. Pathophysiology of portal hypertension and its clinical links. J. Clin. Exp. Hepatol. 2011, 1, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Shah, V.; Lyford, G.; Gores, G.; Farrugia, G. Nitric oxide in gastrointestinal health and disease. Gastroenterology 2004, 126, 903–913. [Google Scholar] [CrossRef]
- Schaffner, D. Investigations of Hepatic Hemodynamics and Alterations in the NO-cGMP Pathway in an Animal Model of Liver Fibrosis/Cirrhosis Suggest PDE5 Inhibitors as Promising Adjunct in Portal Hypertension Therapy. 2018. Available online: https://freidok.uni-freiburg.de/data/16021 (accessed on 22 August 2020). [CrossRef]
- Müsch, A. The unique polarity phenotype of hepatocytes. Exp. Cell Res. 2014, 328, 276–283. [Google Scholar] [CrossRef] [Green Version]
- Rockey, D.C. The Molecular Basis of Portal Hypertension. Trans. Am. Clin. Climatol. Assoc. 2017, 128, 330–345. [Google Scholar]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Fleming, I. Molecular mechanisms underlying the activation of eNOS. Pflügers Arch. 2010, 459, 793–806. [Google Scholar] [CrossRef]
- Iwakiri, Y.; Kim, M.Y. Nitric oxide in liver diseases. Trends Pharmacol. Sci. 2015, 36, 524–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, X.; Keller, T.C.S.; Begandt, D.; Butcher, J.T.; Biwer, L.; Keller, A.S.; Columbus, L.; Isakson, B.E. Endothelial nitric oxide synthase in the microcirculation. Cell Mol. Life Sci. 2015, 72, 4561–4575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, V.; Toruner, M.; Haddad, F.; Cadelina, G.; Papapetropoulos, A.; Choo, K.; Sessa, W.C.; Groszmann, R.J. Impaired endothelial nitric oxide synthase activity associated with enhanced caveolin binding in experimental cirrhosis in the rat. Gastroenterology 1999, 117, 1222–1228. [Google Scholar] [CrossRef]
- Steib, C.J.; Gerbes, A.L.; Bystron, M.; Op den Winkel, M.; Härtl, J.; Roggel, F.; Prüfer, T.; Göke, B.; Bilzer, M. Kupffer cell activation in normal and fibrotic livers increases portal pressure via thromboxane A(2). J. Hepatol. 2007, 47, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Xu, H.; Kresge, N.; Keller, S.; Sarmadi, A.H.; Baveja, R.; Clemens, M.G.; Zhang, J.X. Role of thromboxane A2 in early BDL-induced portal hypertension. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G453–G460. [Google Scholar] [CrossRef] [Green Version]
- Iglarz, M.; Steiner, P.; Wanner, D.; Rey, M.; Hess, P.; Clozel, M. Vascular Effects of Endothelin Receptor Antagonists Depends on Their Selectivity for ETA Versus ETB Receptors and on the Functionality of Endothelial ETB Receptors. J. Cardiovasc. Pharmacol. 2015, 66, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Rockey, D.C.; Weisiger, R.A. Endothelin induced contractility of stellate cells from normal and cirrhotic rat liver: Implications for regulation of portal pressure and resistance. Hepatology 1996, 24, 233–240. [Google Scholar] [CrossRef]
- Theodorakis, N.; Maluccio, M.; Skill, N. Murine study of portal hypertension associated endothelin-1 hypo-response. World J. Gastroenterol. 2015, 21, 4817–4828. [Google Scholar] [CrossRef] [Green Version]
- Yokomori, H.; Oda, M.; Yasogawa, Y.; Nishi, Y.; Ogi, M.; Takahashi, M.; Ishii, H. Enhanced expression of endothelin B receptor at protein and gene levels in human cirrhotic liver. Am. J. Pathol. 2001, 159, 1353–1362. [Google Scholar] [CrossRef]
- Watanabe, N.; Takashimizu, S.; Nishizaki, Y.; Kawazoe, K.; Ilzuka, T.; Kojima, S.; Kamochi, J.; Shiraishi, H.; Matsuzaki, S. Effects of endothelin receptor antagonists on hepatic hemodynamics and sinusoidal endothelial fenestrae in cirrhotic rats. Gastroenterology 2000, 118, A968. [Google Scholar] [CrossRef]
- Hellerbrand, C. Hepatic stellate cells—The pericytes in the liver. Pflügers Arch. 2013, 465, 775–778. [Google Scholar] [CrossRef] [PubMed]
- Gressner, A.M. Transdifferentiation of hepatic stellate cells (Ito cells) to myofibroblasts: A key event in hepatic fibrogenesis. Kidney Int. Suppl. 1996, 54, S39–S45. [Google Scholar] [PubMed]
- Brusilovskaya, K.; Königshofer, P.; Schwabl, P.; Reiberger, T. Vascular Targets for the Treatment of Portal Hypertension. Sem. Liver Dis. 2019, 39, 483–501. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Zhou, Q.; Hennenberg, M.; Trebicka, J.; Jochem, K.; Leifeld, L.; Biecker, E.; Sauerbruch, T.; Heller, J. Intrahepatic upregulation of RhoA and Rho-kinase signalling contributes to increased hepatic vascular resistance in rats with secondary biliary cirrhosis. Gut 2006, 55, 1296–1305. [Google Scholar] [CrossRef]
- Klein, S.; Rick, J.; Lehmann, J.; Schierwagen, R.; Schierwagen, I.G.; Verbeke, L.; Hittatiya, K.; Uschner, F.E.; Manekeller, S.; Strassburg, C.P.; et al. Janus-kinase-2 relates directly to portal hypertension and to complications in rodent and human cirrhosis. Gut 2017, 66, 145–155. [Google Scholar] [CrossRef]
- Klein, S.; Van Beuge, M.M.; Granzow, M.; Beljaars, L.; Schierwagen, R.; Kilic, S.; Heidari, I.; Huss, S.; Sauerbruch, T.; Poelstra, K.; et al. HSC-specific inhibition of Rho-kinase reduces portal pressure in cirrhotic rats without major systemic effects. J. Hepatol. 2012, 57, 1220–1227. [Google Scholar] [CrossRef]
- Trebicka, J.; Hennenberg, M.; Odenthal, M.; Shir, K.; Klein, S.; Granzow, M.; Vogt, A.; Dienes, H.-P.; Lammert, F.; Reichen, J.; et al. Atorvastatin attenuates hepatic fibrosis in rats after bile duct ligation via decreased turnover of hepatic stellate cells. J. Hepatol. 2010, 53, 702–712. [Google Scholar] [CrossRef]
- Hu, L.S.; George, J.; Wang, J.H. Current concepts on the role of nitric oxide in portal hypertension. World J. Gastroenterol. 2013, 19, 1707–1717. [Google Scholar] [CrossRef]
- Huang, H.-C.; Wang, S.-S.; Chan, C.-Y.; Chen, Y.-C.; Lee, F.-Y.; Chang, F.-Y.; Chu, C.-J.; Lin, H.-C.; Lu, R.-H.; Lee, S.-D. Role of Hepatic Nitric Oxide Synthases in Rats with Thioacetamide-induced Acute Liver Failure and Encephalopathy. J. Chin. Med. Assoc. 2007, 70, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Leung, T.-M.; Tipoe, G.L.; Liong, E.C.; Lau, T.Y.H.; Fung, M.-L.; Nanji, A.A. Endothelial nitric oxide synthase is a critical factor in experimental liver fibrosis. Int. J. Exp. Pathol. 2008, 89, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Trebicka, J.; Hennenberg, M.; Laleman, W.; Shelest, N.; Biecker, E.; Schepke, M.; Nevens, F.; Sauerbruch, T.; Heller, J. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology 2007, 46, 242–253. [Google Scholar] [CrossRef] [PubMed]
- McNaughton, L.; Puttagunta, L.; Martinez-Cuesta, M.A.; Kneteman, N.; Mayers, I.; Moqbel, R.; Hamid, Q.; Radomski, M.W. Distribution of nitric oxide synthase in normal and cirrhotic human liver. Proc. Natl. Acad. Sci. USA 2002, 99, 17161–17166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, C. Inducible nitric oxide synthase: What difference does it make? J. Clin. Investig. 1997, 100, 2417–2423. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J. Nitric oxide is not just blowing in the wind. Br. J. Pharmacol. 2019, 176, 131–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaffner, D.; Lazaro, A.; Deibert, P.; Hasselblatt, P.; Stoll, P.; Fauth, L.; Baumstark, M.W.; Merfort, I.; Schmitt-Graeff, A.; Kreisel, W. Analysis of the nitric oxide-cyclic guanosine monophosphate pathway in experimental liver cirrhosis suggests phosphodiesterase-5 as potential target to treat portal hypertension. World J. Gastroenterol. 2018, 24, 4356–4368. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, J.W.; Dover, A.R.; Chia, S.; Cruden, N.L.M.; Hayes, P.C.; Newby, D.E. Inducible nitric oxide synthase activity contributes to the regulation of peripheral vascular tone in patients with cirrhosis and ascites. Gut 2006, 55, 542–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albillos, A.; de la Hera, A.; González, M.; Moya, J.-L.; Calleja, J.-L.; Monserrat, J.; Ruiz-del-Arbol, L.; Alvarez-Mon, M. Increased lipopolysaccharide binding protein in cirrhotic patients with marked immune and hemodynamic derangement. Hepatology 2003, 37, 208–217. [Google Scholar] [CrossRef]
- Bauer, T.M.; Schwacha, H.; Steinbrückner, B.; Brinkmann, F.E.; Ditzen, A.K.; Aponte, J.J.; Pelz, K.; Berger, D.; Kist, M.; Blum, H.E. Small intestinal bacterial overgrowth in human cirrhosis is associated with systemic endotoxemia. Am. J. Gastroenterol. 2002, 97, 2364–2370. [Google Scholar] [CrossRef]
- Guarner, C.; Soriano, G.; Tomas, A.; Bulbena, O.; Novella, M.T.; Balanzo, J.; Vilardell, F.; Mourelle, M.; Moncada, S. Increased serum nitrite and nitrate levels in patients with cirrhosis: Relationship to endotoxemia. Hepatology 1993, 18, 1139–1143. [Google Scholar] [CrossRef]
- Murad, F. Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N. Engl. J. Med. 2006, 355, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Matei, V.; Rodríguez-Vilarrupla, A.; Deulofeu, R.; Colomer, D.; Fernández, M.; Bosch, J.; Garcia-Pagán, J.-C. The eNOS cofactor tetrahydrobiopterin improves endothelial dysfunction in livers of rats with CCl4 cirrhosis. Hepatology 2006, 44, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Cui, J.; Cun, Y.; Shi, A. Tetrahydrobiopterin ameliorates hepatic ischemia-reperfusion Injury by coupling with eNOS in mice. J. Surg. Res. 2012, 176, e65–e71. [Google Scholar] [CrossRef] [PubMed]
- Schwabl, P.; Brusilovskaya, K.; Supper, P.; Bauer, D.; Königshofer, P.; Riedl, F.; Hayden, H.; Fuchs, C.D.; Stift, J.; Oberhuber, G.; et al. The soluble guanylate cyclase stimulator riociguat reduces fibrogenesis and portal pressure in cirrhotic rats. Sci. Rep. 2018, 8. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pmc/articles/PMC6008436/ (accessed on 5 November 2018). [CrossRef] [PubMed]
- Uschner, F.E.; Glückert, K.; Paternostro, R.; Gnad, T.; Schierwagen, R.; Mandorfer, M.; Magdaleno, F.; Ortiz, C.; Schwarzkopf, K.; Kamath, P.; et al. Combination of phosphodiesterase-5-inhibitors and beta blockers improves experimental portal hypertension and erectile dysfunction. Liver Int. 2020. [Google Scholar] [CrossRef]
- Davies, N.A.; Hodges, S.J.; Pitsillides, A.A.; Mookerjee, R.P.; Jalan, R.; Mehdizadeh, S. Hepatic guanylate cyclase activity is decreased in a model of cirrhosis: A quantitative cytochemistry study. FEBS Lett. 2006, 580, 2123–2128. [Google Scholar] [CrossRef] [Green Version]
- Loureiro-Silva, M.R.; Iwakiri, Y.; Abraldes, J.G.; Haq, O.; Groszmann, R.J. Increased phosphodiesterase-5 expression is involved in the decreased vasodilator response to nitric oxide in cirrhotic rat livers. J. Hepatol. 2006, 44, 886–893. [Google Scholar] [CrossRef]
- Lee, K.-C.; Yang, Y.-Y.; Huang, Y.-T.; Lee, F.-Y.; Hou, M.-C.; Lin, H.-C.; Lee, S.-D. Administration of a low dose of sildenafil for 1 week decreases intrahepatic resistance in rats with biliary cirrhosis: The role of NO bioavailability. Clin. Sci. 2010, 119, 45–55. [Google Scholar] [CrossRef]
- Hall, K.C.; Bernier, S.G.; Jacobson, S.; Liu, G.; Zhang, P.Y.; Sarno, R.; Catanzano, V.; Currie, M.G.; Masferrer, J.L. sGC stimulator praliciguat suppresses stellate cell fibrotic transformation and inhibits fibrosis and inflammation in models of NASH. Proc. Natl. Acad. Sci. USA 2019, 116, 11057–11062. [Google Scholar] [CrossRef] [Green Version]
- Jungermann, K. Metabolic zonation of liver parenchyma. Semin Liver Dis 1988, 8, 329–341. [Google Scholar] [CrossRef]
- Jungermann, K.; Katz, N. Functional specialization of different hepatocyte populations. Physiol. Rev. 1989, 69, 708–764. [Google Scholar] [CrossRef] [PubMed]
- Jungermann, K.; Thurman, R.G. Hepatocyte heterogeneity in the metabolism of carbohydrates. Enzyme 1992, 46, 33–58. [Google Scholar] [CrossRef]
- Gebhardt, R.; Matz-Soja, M. Liver zonation: Novel aspects of its regulation and its impact on homeostasis. World J. Gastroenterol. 2014, 20, 8491–8504. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W.; Dorrell, C.; Grompe, M. Stem cells and liver regeneration. Gastroenterology 2009, 137, 466–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kietzmann, T. Metabolic zonation of the liver: The oxygen gradient revisited. Redox Biol. 2017, 11, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T. Liver Zonation in Health and Disease: Hypoxia and Hypoxia-Inducible Transcription Factors as Concert Masters. Int. J. Mol. Sci. 2019, 20, 2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.; Martínez-Ramírez, A.S.; Borders, T.L.; Gao, F.; Sosa-Pineda, B. Metabolic and non-metabolic liver zonation is established non-synchronously and requires sinusoidal Wnts. Elife 2020, 9. [Google Scholar] [CrossRef]
- Su, T.; Yang, Y.; Lai, S.; Jeong, J.; Jung, Y.; McConnell, M.; Utsumi, T.; Iwakiri, Y. Single-cell transcriptomics reveals zone-specific alterations of liver sinusoidal endothelial cells in cirrhosis. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Halpern, K.B.; Shenhav, R.; Massalha, H.; Toth, B.; Egozi, A.; Massasa, E.E.; Medgalia, C.; David, E.; Giladi, A.; Moor, A.E.; et al. Paired-cell sequencing enables spatial gene expression mapping of liver endothelial cells. Nat. Biotechnol. 2018, 36, 962–970. [Google Scholar] [CrossRef]
- Ben-Moshe, S.; Shapira, Y.; Moor, A.E.; Manco, R.; Veg, T.; Bahar Halpern, K.; Itzkovitz, S. Spatial sorting enables comprehensive characterization of liver zonation. Nat. Metab. 2019, 1, 899–911. [Google Scholar] [CrossRef]
- Ben-Moshe, S.; Itzkovitz, S. Spatial heterogeneity in the mammalian liver. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 395–410. [Google Scholar] [CrossRef] [PubMed]
- Dobie, R.; Wilson-Kanamori, J.R.; Henderson, B.E.P.; Smith, J.R.; Matchett, K.P.; Portman, J.R.; Wallenborg, K.; Picelli, S.; Zagorska, A.; Pendem, S.V.; et al. Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis. Cell Rep. 2019, 29, 1832–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aizarani, N.; Saviano, A.; Sagar null Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grün, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Ghallab, A.; Myllys, M.; Holland, C.H.; Zaza, A.; Murad, W.; Hassan, R.; Ahmed, Y.A.; Abbas, T.; Abdelrahim, E.A.; Schneider, K.M.; et al. Influence of Liver Fibrosis on Lobular Zonation. Cells 2019, 8, 1556. [Google Scholar] [CrossRef] [Green Version]
- Meier, R.P.H.; Meyer, J.; Montanari, E.; Lacotte, S.; Balaphas, A.; Muller, Y.D.; Clément, S.; Negro, F.; Toso, C.; Morel, P.; et al. Interleukin-1 Receptor Antagonist Modulates Liver Inflammation and Fibrosis in Mice in a Model-Dependent Manner. Int. J. Mol. Sci. 2019, 20, 1295. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pmc/articles/PMC6471711/ (accessed on 18 August 2020). [CrossRef] [Green Version]
- Coulon, S.; Heindryckx, F.; Geerts, A.; Van Steenkiste, C.; Colle, I.; Van Vlierberghe, H. Angiogenesis in chronic liver disease and its complications. Liver Int. 2011, 31, 146–162. [Google Scholar] [CrossRef]
- Thabut, D.; Shah, V. Intrahepatic angiogenesis and sinusoidal remodeling in chronic liver disease: New targets for the treatment of portal hypertension? J. Hepatol. 2010, 53, 976–980. [Google Scholar] [CrossRef] [Green Version]
- Gana, J.C.; Serrano, C.A.; Ling, S.C. Angiogenesis and portal-systemic collaterals in portal hypertension. Ann. Hepatol. 2016, 15, 303–313. [Google Scholar] [CrossRef]
- Taura, K.; De Minicis, S.; Seki, E.; Hatano, E.; Iwaisako, K.; Osterreicher, C.H.; Kodama, Y.; Miura, K.; Ikai, I.; Uemoto, S.; et al. Hepatic stellate cells secrete angiopoietin 1 that induces angiogenesis in liver fibrosis. Gastroenterology 2008, 135, 1729–1738. [Google Scholar] [CrossRef]
- Møller, S.; Bendtsen, F. The pathophysiology of arterial vasodilatation and hyperdynamic circulation in cirrhosis. Liver Int. 2018, 38, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Krag, A.; Bendtsen, F.; Henriksen, J.H.; Møller, S. Low cardiac output predicts development of hepatorenal syndrome and survival in patients with cirrhosis and ascites. Gut 2010, 59, 105–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennenberg, M.; Trebicka, J.; Sauerbruch, T.; Heller, J. Mechanisms of extrahepatic vasodilation in portal hypertension. Gut 2008, 57, 1300–1314. [Google Scholar] [CrossRef] [PubMed]
- Wiest, R.; Groszmann, R.J. The paradox of nitric oxide in cirrhosis and portal hypertension: Too much, not enough. Hepatology 2002, 35, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.; Wiest, R.; Garcia-Cardena, G.; Cadelina, G.; Groszmann, R.J.; Sessa, W.C. Hsp90 regulation of endothelial nitric oxide synthase contributes to vascular control in portal hypertension. Am. J. Physiol. 1999, 277, G463–G468. [Google Scholar] [CrossRef]
- Hori, N.; Wiest, R.; Groszmann, R.J. Enhanced release of nitric oxide in response to changes in flow and shear stress in the superior mesenteric arteries of portal hypertensive rats. Hepatology 1998, 28, 1467–1473. [Google Scholar] [CrossRef]
- Abraldes, J.G.; Iwakiri, Y.; Loureiro-Silva, M.; Haq, O.; Sessa, W.C.; Groszmann, R.J. Mild increases in portal pressure upregulate vascular endothelial growth factor and endothelial nitric oxide synthase in the intestinal microcirculatory bed, leading to a hyperdynamic state. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G980–G987. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-C.; Haq, O.; Utsumi, T.; Sethasine, S.; Abraldes, J.G.; Groszmann, R.J.; Iwakiri, Y. Intestinal and plasma VEGF levels in cirrhosis: The role of portal pressure. J. Cell Mol. Med. 2012, 16, 1125–1133. [Google Scholar] [CrossRef]
- Francque, S.; Wamutu, S.; Chatterjee, S.; Van Marck, E.; Herman, A.; Ramon, A.; Jung, A.; Vermeulen, W.; De Winter, B.; Pelckmans, P.; et al. Non-alcoholic steatohepatitis induces non-fibrosis-related portal hypertension associated with splanchnic vasodilation and signs of a hyperdynamic circulation in vitro and in vivo in a rat model. Liver Int. 2010, 30, 365–375. [Google Scholar] [CrossRef]
- Wiest, R.; Cadelina, G.; Milstien, S.; McCuskey, R.S.; Garcia-Tsao, G.; Groszmann, R.J. Bacterial translocation up-regulates GTP-cyclohydrolase I in mesenteric vasculature of cirrhotic rats. Hepatology 2003, 38, 1508–1515. [Google Scholar] [CrossRef]
- Grace, J.A.; Klein, S.; Herath, C.B.; Granzow, M.; Schierwagen, R.; Masing, N.; Walther, T.; Sauerbruch, T.; Burrell, L.M.; Angus, P.W.; et al. Activation of the MAS receptor by angiotensin-(1-7) in the renin-angiotensin system mediates mesenteric vasodilatation in cirrhosis. Gastroenterology 2013, 145, 874–884. [Google Scholar] [CrossRef]
- Gunarathne, L.S.; Angus, P.W.; Herath, C.B. Blockade of Mas Receptor or Mas-Related G-Protein Coupled Receptor Type D Reduces Portal Pressure in Cirrhotic but Not in Non-cirrhotic Portal Hypertensive Rats. Front. Physiol. 2019, 10. Available online: https://www.frontiersin.org/article/10.3389/fphys.2019.01169/full (accessed on 16 April 2020). [CrossRef] [PubMed] [Green Version]
- Niederberger, M.; Ginès, P.; Tsai, P.; Martin, P.Y.; Morris, K.; Weigert, A.; McMurtry, I.; Schrier, R.W. Increased aortic cyclic guanosine monophosphate concentration in experimental cirrhosis in rats: Evidence for a role of nitric oxide in the pathogenesis of arterial vasodilation in cirrhosis. Hepatology 1995, 21, 1625–1631. [Google Scholar] [PubMed]
- Martin, P.Y.; Ohara, M.; Gines, P.; Xu, D.L.; St John, J.; Niederberger, M.; Schrier, R.W. Nitric oxide synthase (NOS) inhibition for one week improves renal sodium and water excretion in cirrhotic rats with ascites. J. Clin. Investig. 1998, 101, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Montoliu, C.; Rodrigo, R.; Monfort, P.; Llansola, M.; Cauli, O.; Boix, J.; Elmlili, N.; Agusti, A.; Felipo, V. Cyclic GMP pathways in hepatic encephalopathy. Neurological and therapeutic implications. Metab. Brain Dis. 2010, 25, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Corbalán, R.; Montoliu, C.; Miñana, M.D.; Del Olmo, J.A.; Serra, M.A.; Aparisi, L.; Rodrigo, J.M.; Felipo, V. Altered modulation of soluble guanylate cyclase by nitric oxide in patients with liver disease. Metab. Brain Dis. 2002, 17, 295–301. [Google Scholar] [CrossRef]
- Felipo, V.; Urios, A.; Giménez-Garzó, C.; Cauli, O.; Andrés-Costa, M.-J.; González, O.; Serra, M.A.; Sánchez-González, J.; Aliaga, R.; Giner-Durán, R.; et al. Non invasive blood flow measurement in cerebellum detects minimal hepatic encephalopathy earlier than psychometric tests. World J. Gastroenterol. 2014, 20, 11815–11825. [Google Scholar] [CrossRef] [Green Version]
- Kirstetter, P.; Moreau, R.; Vachiery, F.; Gadano, A.; Soupison, T.; Pilette, C.; Pussard, E.; Cailmail, S.; Takahashi, H.; Lebrec, D. Plasma concentrations of cyclic 3′,5′-guanosine monophosphate in patients with cirrhosis: Relationship with atrial natriuretic peptide and haemodynamics. J. Gastroenterol. Hepatol. 1997, 12, 233–236. [Google Scholar] [CrossRef]
- Pasmanter, N.; Iheanacho, F.; Hashmi, M.F. Biochemistry, Cyclic GMP; StatPearls Publishing: Treasure Island, FL, USA, 2020; Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK542234/ (accessed on 20 May 2020).
- Catalano, S.; Campana, A.; Giordano, C.; Győrffy, B.; Tarallo, R.; Rinaldi, A.; Bruno, G.; Ferraro, A.; Romeo, F.; Lanzino, M.; et al. Expression and Function of Phosphodiesterase Type 5 in Human Breast Cancer Cell Lines and Tissues: Implications for Targeted Therapy. Clin. Cancer Res. 2016, 22, 2271–2282. [Google Scholar] [CrossRef] [Green Version]
- Pokreisz, P.; Vandenwijngaert, S.; Bito, V.; Van den Bergh, A.; Lenaerts, I.; Busch, C.; Marsboom, G.; Gheysens, O.; Vermeersch, P.; Biesmans, L.; et al. Ventricular phosphodiesterase 5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation 2009, 119, 408–416. [Google Scholar] [CrossRef] [Green Version]
- Lawless, M.; Caldwell, J.L.; Radcliffe, E.J.; Smith, C.E.R.; Madders, G.W.P.; Hutchings, D.C.; Woods, L.S.; Church, S.J.; Unwin, R.D.; Kirkwood, G.J.; et al. Phosphodiesterase 5 inhibition improves contractile function and restores transverse tubule loss and catecholamine responsiveness in heart failure. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Elhwuegi, A. The Wonders of Phosphodiesterase-5 Inhibitors: A Majestic History. Ann. Med. Health Sci. Res. 2016, 6, 139–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buys, E.S.; Zimmer, D.P.; Chickering, J.; Graul, R.; Chien, Y.T.; Profy, A.; Hadcock, J.R.; Masferrer, J.L.; Milne, G.T. Discovery and development of next generation sGC stimulators with diverse multidimensional pharmacology and broad therapeutic potential. Nitric Oxide 2018, 78, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Sandner, P.; Zimmer, D.P.; Milne, G.T.; Follmann, M.; Hobbs, A.; Stasch, J.-P. Soluble Guanylate Cyclase Stimulators and Activators. Handb. Exp. Pharmacol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Sandner, P.; Stasch, J.P. Anti-fibrotic effects of soluble guanylate cyclase stimulators and activators: A review of the preclinical evidence. Respir. Med. 2017, 122 (Suppl. 1), S1–S9. [Google Scholar] [CrossRef] [Green Version]
- Reinke, Y.; Gross, S.; Eckerle, L.G.; Hertrich, I.; Busch, M.; Busch, R.; Riad, A.; Rauch, B.H.; Stasch, J.-P.; Dörr, M.; et al. The soluble guanylate cyclase stimulator riociguat and the soluble guanylate cyclase activator cinaciguat exert no direct effects on contractility and relaxation of cardiac myocytes from normal rats. Eur. J. Pharmacol. 2015, 767, 1–9. [Google Scholar] [CrossRef]
- Montfort, W.R.; Wales, J.A.; Weichsel, A. Structure and Activation of Soluble Guanylyl Cyclase, the Nitric Oxide Sensor. Antioxid. Redox Signal. 2017, 26, 107–121. [Google Scholar] [CrossRef]
- Flores-Costa, R.; Alcaraz-Quiles, J.; Titos, E.; López-Vicario, C.; Casulleras, M.; Duran-Güell, M.; Rius, B.; Diaz, A.; Hall, K.; Shea, C.; et al. The soluble guanylate cyclase stimulator IW-1973 prevents inflammation and fibrosis in experimental non-alcoholic steatohepatitis. Br. J. Pharmacol. 2018, 175, 953–967. [Google Scholar] [CrossRef]
- Frey, R.; Becker, C.; Saleh, S.; Unger, S.; van der Mey, D.; Mück, W. Clinical Pharmacokinetic and Pharmacodynamic Profile of Riociguat. Clin. Pharmacokinet. 2018, 57, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Knorr, A.; Hirth-Dietrich, C.; Alonso-Alija, C.; Härter, M.; Hahn, M.; Keim, Y.; Wunder, F.; Stasch, J.-P. Nitric Oxide-independent Activation of Soluble Guanylate Cyclase by BAY 60-2770 in Experimental Liver Fibrosis. Arzneimittelforschung 2011, 58, 71–80. [Google Scholar] [CrossRef]
- Xie, G.; Wang, X.; Wang, L.; Wang, L.; Atkinson, R.D.; Kanel, G.C.; Gaarde, W.A.; DeLeve, L.D. Role of Differentiation of Liver Sinusoidal Endothelial Cells in Progression and Regression of Hepatic Fibrosis in Rats. Gastroenterology 2012, 142, 918–927. [Google Scholar] [CrossRef] [Green Version]
- Colle, I.; De Vriese, A.S.; Van Vlierberghe, H.; Lameire, N.H.; DeVos, M. Systemic and splanchnic haemodynamic effects of sildenafil in an in vivo animal model of cirrhosis support for a risk in cirrhotic patients. Liver Int. 2004, 24, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Halverscheid, L.; Deibert, P.; Schmidt, R.; Blum, H.E.; Dunkern, T.; Pannen, B.H.J.; Kreisel, W. Phosphodiesterase-5 inhibitors have distinct effects on the hemodynamics of the liver. BMC Gastroenterol. 2009, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Uschner, F.E.; Glückert, K.; Klein, S.; Magdaleno, F.; Schierwagen, R.; Trebicka, J. FRI-247—Udenafil decreases portal pressure and improves erectile dysfunction in liver cirrhosis. J. Hepatol. 2018, 68, S471. [Google Scholar] [CrossRef]
- Choi, S.-M.; Shin, J.-H.; Kim, J.-M.; Lee, C.-H.; Kang, K.-K.; Ahn, B.-O.; Yoo, M. Effect of udenafil on portal venous pressure and hepatic fibrosis in rats. A novel therapeutic option for portal hypertension. Arzneimittelforschung 2009, 59, 641–646. [Google Scholar] [CrossRef]
- Ali, F.E.M.; Azouz, A.A.; Bakr, A.G.; Abo-Youssef, A.M.; Hemeida, R.A.M. Hepatoprotective effects of diosmin and/or sildenafil against cholestatic liver cirrhosis: The role of Keap-1/Nrf-2 and P38-MAPK/NF-κB/iNOS signaling pathway. Food Chem. Toxicol. 2018, 120, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Deibert, P.; Schumacher, Y.-O.; Ruecker, G.; Opitz, O.G.; Blum, H.E.; Rössle, M.; Kreisel, W. Effect of vardenafil, an inhibitor of phosphodiesterase-5, on portal haemodynamics in normal and cirrhotic liver—Results of a pilot study. Aliment. Pharmacol. Ther. 2006, 23, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Bremer, H.C.; Kreisel, W.; Roecker, K.; Dreher, M.; Koenig, D.; Kurz-Schmieg, A.K.; Blum, H.E.; Roessle, M.; Deibert, P. Phosphodiesterase 5 inhibitors lower both portal and pulmonary pressure in portopulmonary hypertension: A case report. J. Med. Case Rep. 2007, 1, 46. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-C.; Yang, Y.-Y.; Wang, Y.-W.; Hou, M.-C.; Lee, F.-Y.; Lin, H.-C.; Lee, S.-D. Acute administration of sildenafil enhances hepatic cyclic guanosine monophosphate production and reduces hepatic sinusoid resistance in cirrhotic patients. Hepatol. Res. 2008, 38, 1186–1193. [Google Scholar] [CrossRef]
- Clemmesen, J.-O.; Giraldi, A.; Ott, P.; Dalhoff, K.; Hansen, B.-A.; Larsen, F.-S. Sildenafil does not influence hepatic venous pressure gradient in patients with cirrhosis. World J. Gastroenterol. 2008, 14, 6208–6212. [Google Scholar] [CrossRef]
- Tandon, P.; Inayat, I.; Tal, M.; Spector, M.; Shea, M.; Groszmann, R.J.; Garcia-Tsao, G. Sildenafil has no effect on portal pressure but lowers arterial pressure in patients with compensated cirrhosis. Clin. Gastroenterol. Hepatol. 2010, 8, 546–549. [Google Scholar] [CrossRef] [Green Version]
- Kreisel, W.; Deibert, P.; Kupcinskas, L.; Sumskiene, J.; Appenrodt, B.; Roth, S.; Neagu, M.; Rössle, M.; Zipprich, A.; Caca, K.; et al. The phosphodiesterase-5-inhibitor udenafil lowers portal pressure in compensated preascitic liver cirrhosis. A dose-finding phase-II-study. Dig. Liver Dis. 2015, 47, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Deibert, P.; Lazaro, A.; Stankovic, Z.; Schaffner, D.; Rössle, M.; Kreisel, W. Beneficial long term effect of a phosphodiesterase-5-inhibitor in cirrhotic portal hypertension: A case report with 8 years follow-up. World J. Gastroenterol. 2018, 24, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Sauerbruch, T.; Trebicka, J. Future therapy of portal hypertension in liver cirrhosis—A guess. F1000 Prime Rep. 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauerbruch, T.; Schierwagen, R.; Trebicka, J. Managing portal hypertension in patients with liver cirrhosis. F1000 Res. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Berzigotti, A.; Bosch, J. Pharmacologic management of portal hypertension. Clin. Liver Dis. 2014, 18, 303–317. [Google Scholar] [CrossRef]
- Abraldes, J.G.; Trebicka, J.; Chalasani, N.; D’Amico, G.; Rockey, D.C.; Shah, V.H.; Bosch, J.; Garcia-Tsao, G. Prioritization of Therapeutic Targets and Trial Design in Cirrhotic Portal Hypertension. Hepatology 2019, 69, 1287–1299. [Google Scholar] [CrossRef] [Green Version]
- Pascal, J.P.; Cales, P. Propranolol in the prevention of first upper gastrointestinal tract hemorrhage in patients with cirrhosis of the liver and esophageal varices. N. Engl. J. Med. 1987, 317, 856–861. [Google Scholar] [CrossRef]
- Abraldes, J.G.; Garcia-Tsao, G. The Design of Clinical Trials in Portal Hypertension. Semin. Liver Dis. 2017, 37, 73–84. [Google Scholar] [CrossRef]
- Reiberger, T.; Mandorfer, M. Beta adrenergic blockade and decompensated cirrhosis. J. Hepatol. 2017, 66, 849–859. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, S.G.; Mendoza, Y.P.; Bosch, J. Beta-blockers in cirrhosis: Evidence-based indications and limitations. JHEP Rep. 2020, 2, 100063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrarese, A.; Zanetto, A.; Germani, G.; Burra, P.; Senzolo, M. Rethinking the role of non-selective beta blockers in patients with cirrhosis and portal hypertension. World J. Hepatol. 2016, 8, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Bhutta, A.Q.; Garcia-Tsao, G.; Reddy, K.R.; Tandon, P.; Wong, F.; O’Leary, J.G.; Acharya, C.; Banerjee, D.; Abraldes, J.G.; Jones, T.M.; et al. Beta-blockers in hospitalised patients with cirrhosis and ascites: Mortality and factors determining discontinuation and reinitiation. Aliment. Pharmacol. Ther. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moctezuma-Velazquez, C.; Kalainy, S.; Abraldes, J.G. Beta-blockers in patients with advanced liver disease: Has the dust settled? Liver Transplant. 2017, 23, 1058–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelico, M.; Lionetti, R. Long-acting nitrates in portal hypertension: To be or not to be? Dig. Liver Dis. 2001, 33, 205–211. [Google Scholar] [CrossRef]
- Merkel, C.; Sacerdoti, D.; Bolognesi, M.; Buonamico, P.; Sticca, A.; Amodio, P.; Angeli, P.; Micotti, L.; Gatta, A. Effect of chronic treatment with nadolol plus isosorbide mononitrate on liver blood flow and liver metabolic activity in cirrhosis. Eur. J. Gastroenterol. Hepatol. 1999, 11, 1221–1225. [Google Scholar] [CrossRef]
- Villanueva, C.; López-Balaguer, J.M.; Aracil, C.; Kolle, L.; González, B.; Miñana, J.; Soriano, G.; Guarner, C.; Balanzó, J. Maintenance of hemodynamic response to treatment for portal hypertension and influence on complications of cirrhosis. J. Hepatol. 2004, 40, 757–765. [Google Scholar] [CrossRef]
- Grose, R.D.; Plevris, J.N.; Redhead, D.N.; Bouchier, I.A.D.; Hayes, P.C. The acute and chronic effects of isosorbide-5-mononitrate on portal haemodynamics in cirrhosis. J. Hepatol. 1994, 20, 542–547. [Google Scholar] [CrossRef]
- Gluud, L.L.; Langholz, E.; Krag, A. Meta-analysis: Isosorbide-mononitrate alone or with either beta-blockers or endoscopic therapy for the management of oesophageal varices: Meta-analysis: Isosorbide-mononitrate for oesophageal varices. Aliment. Pharmacol. Ther. 2010, 32, 859–871. [Google Scholar] [CrossRef]
- Kakumitsu, S.; Shijo, H.; Yokoyama, M.; Kim, T.; Akiyoshi, N.; Ota, K.; Kubara, K.; Okumura, M.; Inoue, K. Effects ofl-arginine on the systemic, mesenteric, and Hepatic circulation in patients with cirrhosis. Hepatology 1998, 27, 377–382. [Google Scholar] [CrossRef]
- Baiges, A.; Hernández-Gea, V.; Bosch, J. Pharmacologic prevention of variceal bleeding and rebleeding. Hepatol. Int. 2018, 12, 68–80. [Google Scholar] [CrossRef]
- Fiorucci, S.; Antonelli, E.; Morelli, O.; Mencarelli, A.; Casini, A.; Mello, T.; Palazzetti, B.; Tallet, D.; del Soldato, P.; Morelli, A. NCX-1000, a NO-releasing derivative of ursodeoxycholic acid, selectively delivers NO to the liver and protects against development of portal hypertension. Proc. Natl. Acad. Sci. USA 2001, 98, 8897–8902. [Google Scholar] [CrossRef] [Green Version]
- Fiorucci, S.; Antonelli, E.; Tocchetti, P.; Morelli, A. Treatment of portal hypertension with NCX-1000, a liver-specific NO donor. A review of its current status. Cardiovasc. Drug Rev. 2004, 22, 135–146. [Google Scholar] [CrossRef]
- Berzigotti, A.; Bellot, P.; De Gottardi, A.; Garcia-Pagan, J.C.; Gagnon, C.; Spénard, J.; Bosch, J. NCX-1000, a nitric oxide-releasing derivative of UDCA, does not decrease portal pressure in patients with cirrhosis: Results of a randomized, double-blind, dose-escalating study. Am. J. Gastroenterol. 2010, 105, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Abraldes, J.G.; Rodríguez-Vilarrupla, A.; Graupera, M.; Zafra, C.; García-Calderó, H.; García-Pagán, J.C.; Bosch, J. Simvastatin treatment improves liver sinusoidal endothelial dysfunction in CCl4 cirrhotic rats. J. Hepatol. 2007, 46, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Abraldes, J.G.; Albillos, A.; Bañares, R.; Turnes, J.; González, R.; García-Pagán, J.C.; Bosch, J. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: A randomized controlled trial. Gastroenterology 2009, 136, 1651–1658. [Google Scholar] [CrossRef]
- Abraldes, J.G.; Villanueva, C.; Aracil, C.; Turnes, J.; Hernandez-Guerra, M.; Genesca, J.; Rodriguez, M.; Castellote, J.; García-Pagán, J.C.; Torres, F.; et al. Addition of Simvastatin to Standard Therapy for the Prevention of Variceal Rebleeding Does Not Reduce Rebleeding but Increases Survival in Patients With Cirrhosis. Gastroenterology 2016, 150, 1160–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, U.C.; Benfield, T.; Bendtsen, F. Reduced risk of decompensation and death associated with use of statins in patients with alcoholic cirrhosis. A nationwide case-cohort study. Aliment. Pharmacol. Ther. 2017, 46, 673–680. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Grace, N.D.; Qamar, A.A. Statin use in patients with cirrhosis: A retrospective cohort study. Dig. Dis. Sci. 2014, 59, 1958–1965. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, A.; Tate, J.P.; Garcia-Tsao, G. Statins Are Associated With a Decreased Risk of Decompensation and Death in Veterans with Hepatitis C–Related Compensated Cirrhosis. Gastroenterology 2016, 150, 430–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bays, H.; Cohen, D.E.; Chalasani, N.; Harrison, S.A. An assessment by the Statin Liver Safety Task Force: 2014 update. J. Clin. Lipidol. 2014, 8, S47–S57. [Google Scholar] [CrossRef] [Green Version]
- Garbuzenko, D.V. Contemporary concepts of the medical therapy of portal hypertension under liver cirrhosis. World J. Gastroenterol. 2015, 21, 6117–6126. [Google Scholar] [CrossRef] [PubMed]
- Garbuzenko, D.V.; Arefyev, N.O.; Kazachkov, E.L. Antiangiogenic therapy for portal hypertension in liver cirrhosis: Current progress and perspectives. World J. Gastroenterol. 2018, 24, 3738–3748. [Google Scholar] [CrossRef]
- Schwabl, P.; Laleman, W. Novel treatment options for portal hypertension. Gastroenterol. Rep. 2017, 5, 90–103. [Google Scholar] [CrossRef]
- Huang, S.A.; Lie, J.D. Phosphodiesterase-5 (PDE5) Inhibitors in the Management of Erectile Dysfunction. Pharm. Therap. 2013, 38, 407–419. [Google Scholar]
- Andersson, K. PDE5 inhibitors—Pharmacology and clinical applications 20 years after sildenafil discovery. Br. J. Pharmacol. 2018, 175, 2554–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Compound | Dosage and Route | ΔMAP | ΔPVP | Remarks | |
|---|---|---|---|---|---|---|
| Colle 2004 [154] | Wistar rats, BDL | Sildenafil | 0.01–10 mg/kg, i.v. | −1–20% even more in sham rats | +2–+6% even more in sham rats | |
| Halverscheid 2009 [155] | Sprague Dawley rats, non-cirrhotic | Sildenafil Vardenafil | 1, 10, or 100 µg/kg, i.v. 1, 10, or 100 µg/kg, i.v. | 1.1; −3.9; −2.6% −11.0; −8.7; −7.4% | In all groups no increase, but decrease over time | 3.3; 24.1; 18.3% 15.9; 29.2; 23.9% increase in portal flow |
| Schaffner 2018 [88] | Wistar rats, TAA | Sildenafil | 0.1–1.0 mg/kg, i.v. | −14–−17% | −13–−19% | |
| Uschner 2020 [97] | Sprague Dawley rats, BDL or CCL4 | Udenafil Udenafil/propranolol Udenafil 1 or 5 mg/kg | 1 or 5 mg/kg 1 mg/kg | 1 mg/kg: −20%; 5 mg/kg: −22% −7.5% 1 mg/kg: −31%; 5 mg/kg: −34% | −30–−23% −40% −30–−0% | |
| Schaffner 2018 [88] | Wistar rats, TAA | Sildenafil | 0.1–1.0 mg/kg, i.v. | −14–−17% | −13–−19% | |
| Lee 2010 [100] | Sprague Dawley rats, BDL | Sildenafil, 1 week | 0.25 mg/kg twice daily p.o. | −25% | ||
| Choi 2009 [157] | Sprague Dawley rats, BDL | Udenafil for 3 weeks | 1, 5, or 25 mg/kg p.o. | −14, −13, −31% | ||
| Deibert 2006 [159] | Human, cirrhotic (n = 18) | Vardenafil | 10 mg, p.o. | −19% (n = 5) | Hepatic arterial resistance and portal flow increased significantly | |
| Bremer 2007 [160] | Human, cirrhotic PPHTN (n = 1) | Tadalafil | 10 mg, p.o. | −30% | PAP −25% | |
| Lee 2008 [161] | Human, cirrhotic (n = 7) | Sildenafil | 50 mg, p.o. | Unchanged | +1% | MPAP and sinusoidal resistance significantly reduced |
| Clemmesen 2008 [162] | Human, cirrhotic (n = 10) | Sildenafil | 50 mg, p.o. | −14% | −11% | |
| Tandon 2010 [163] | Human, cirrhotic (n = 12) | Sildenafil | 25 mg, p.o. | −8% | −4% n.s. | |
| Kreisel 2015 [164] | Human cirrhotic (n = 30) | Udenafil | 12.5; 25; 50; 75; 100 mg p.o. acute 6 days | Significant reduction with ≥75 mg in the acute setting and after 6 days | −3.5; −4.5; −7.5; −25.1; −17.3% −14.4; 3.1; −14.0; −13.5; −16.8% | |
| Deibert 2018 [165] | Human, cirrhotic (n = 1) | Vardenafil Tadalafil | 10 mg 5 mg | −11% | −14% −15% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kreisel, W.; Schaffner, D.; Lazaro, A.; Trebicka, J.; Merfort, I.; Schmitt-Graeff, A.; Deibert, P. Phosphodiesterases in the Liver as Potential Therapeutic Targets of Cirrhotic Portal Hypertension. Int. J. Mol. Sci. 2020, 21, 6223. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176223
Kreisel W, Schaffner D, Lazaro A, Trebicka J, Merfort I, Schmitt-Graeff A, Deibert P. Phosphodiesterases in the Liver as Potential Therapeutic Targets of Cirrhotic Portal Hypertension. International Journal of Molecular Sciences. 2020; 21(17):6223. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176223
Chicago/Turabian StyleKreisel, Wolfgang, Denise Schaffner, Adhara Lazaro, Jonel Trebicka, Irmgard Merfort, Annette Schmitt-Graeff, and Peter Deibert. 2020. "Phosphodiesterases in the Liver as Potential Therapeutic Targets of Cirrhotic Portal Hypertension" International Journal of Molecular Sciences 21, no. 17: 6223. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21176223