Clearing of Foreign Episomal DNA from Human Cells by CRISPRa-Mediated Activation of Cytidine Deaminases

 , and
, and
Abstract
:1. Introduction
2. Results
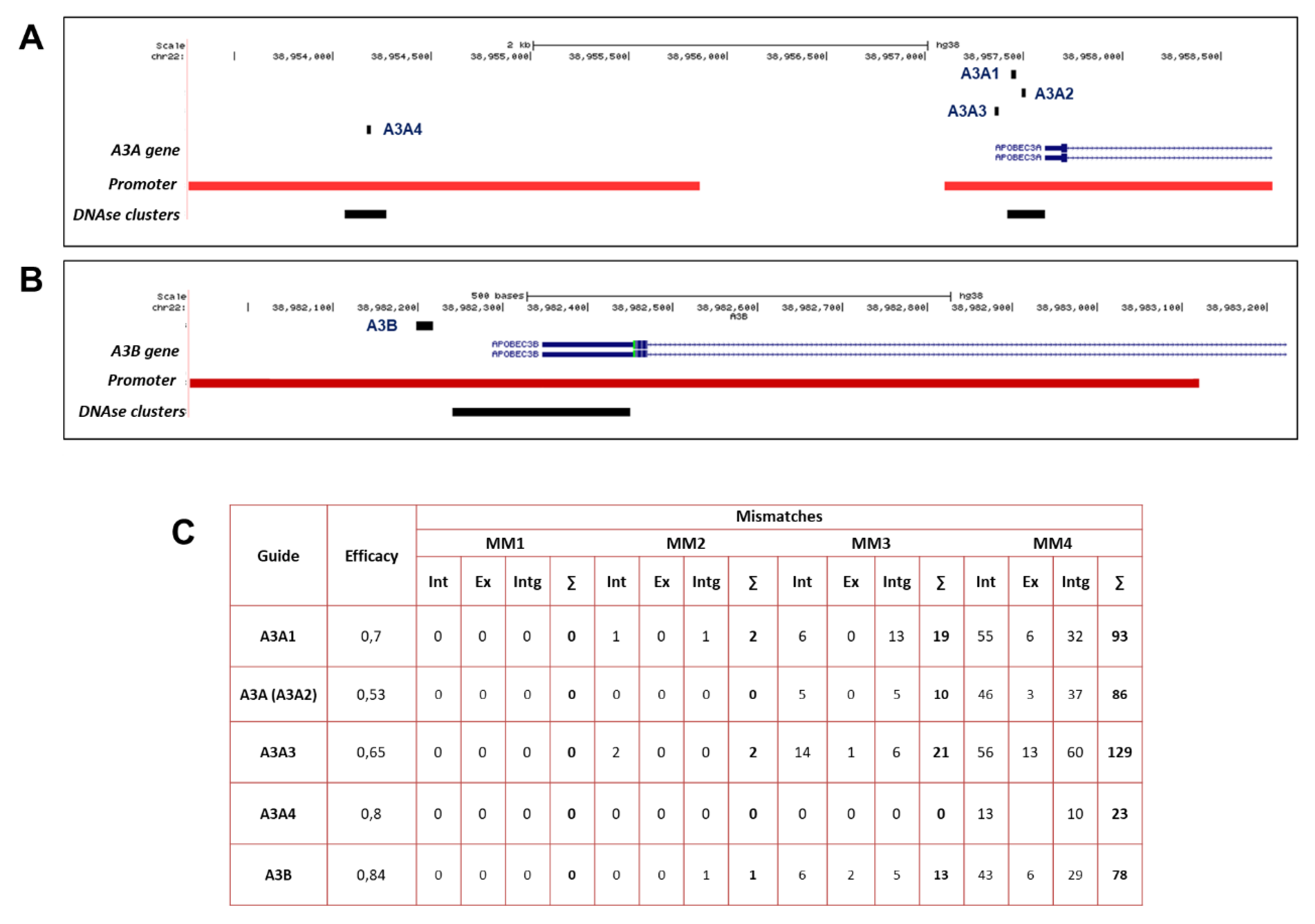
2.1. Design of sgRNAs for CRISPRa-Mediated Transactivation of A3A and A3B
2.2. Assessment of Single sgRNAs Targeting A3A and A3B Promoters
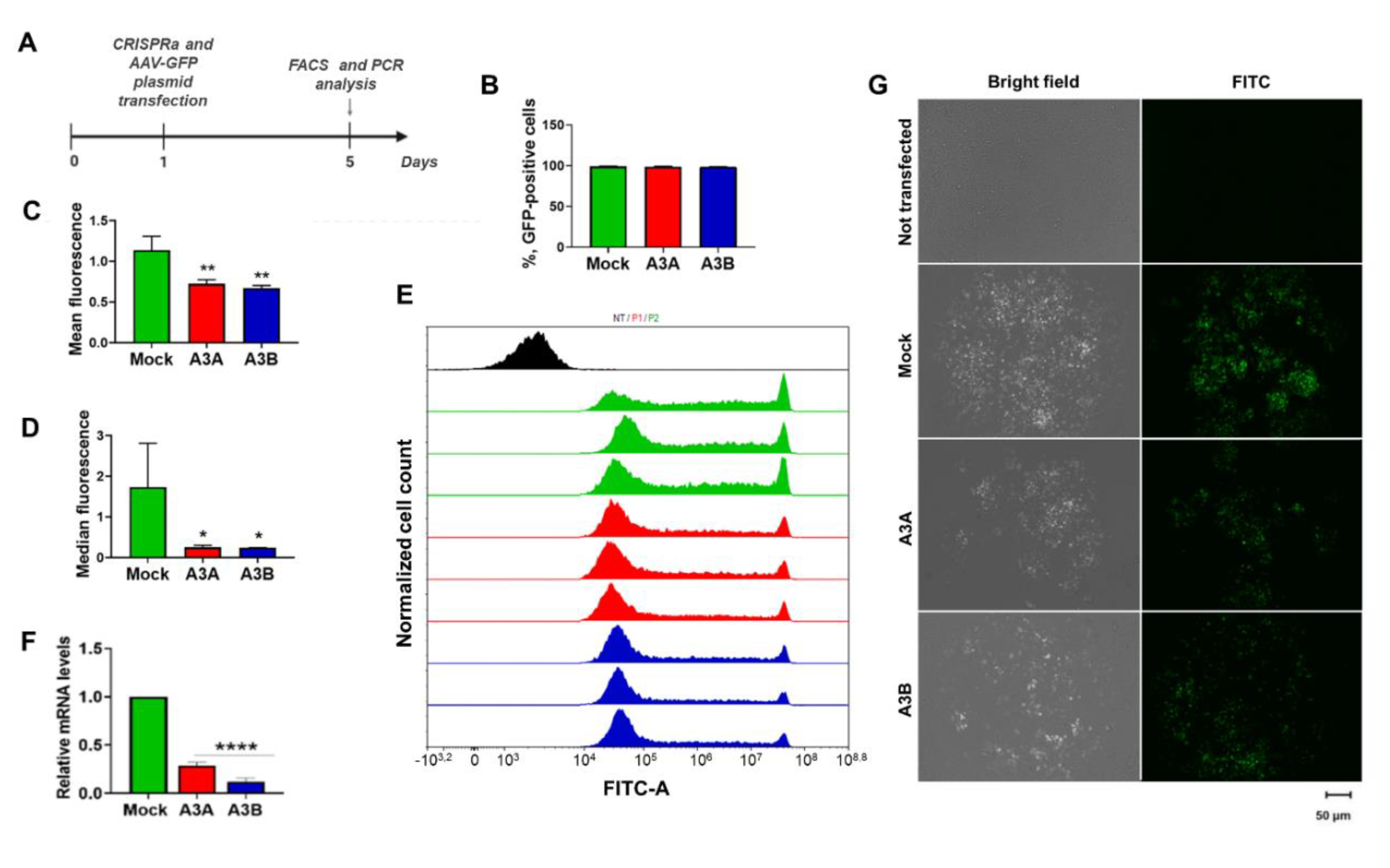
2.3. CRISPRa-Mediated A3A and A3B Overexpression Inhibits Transient Gene Expression
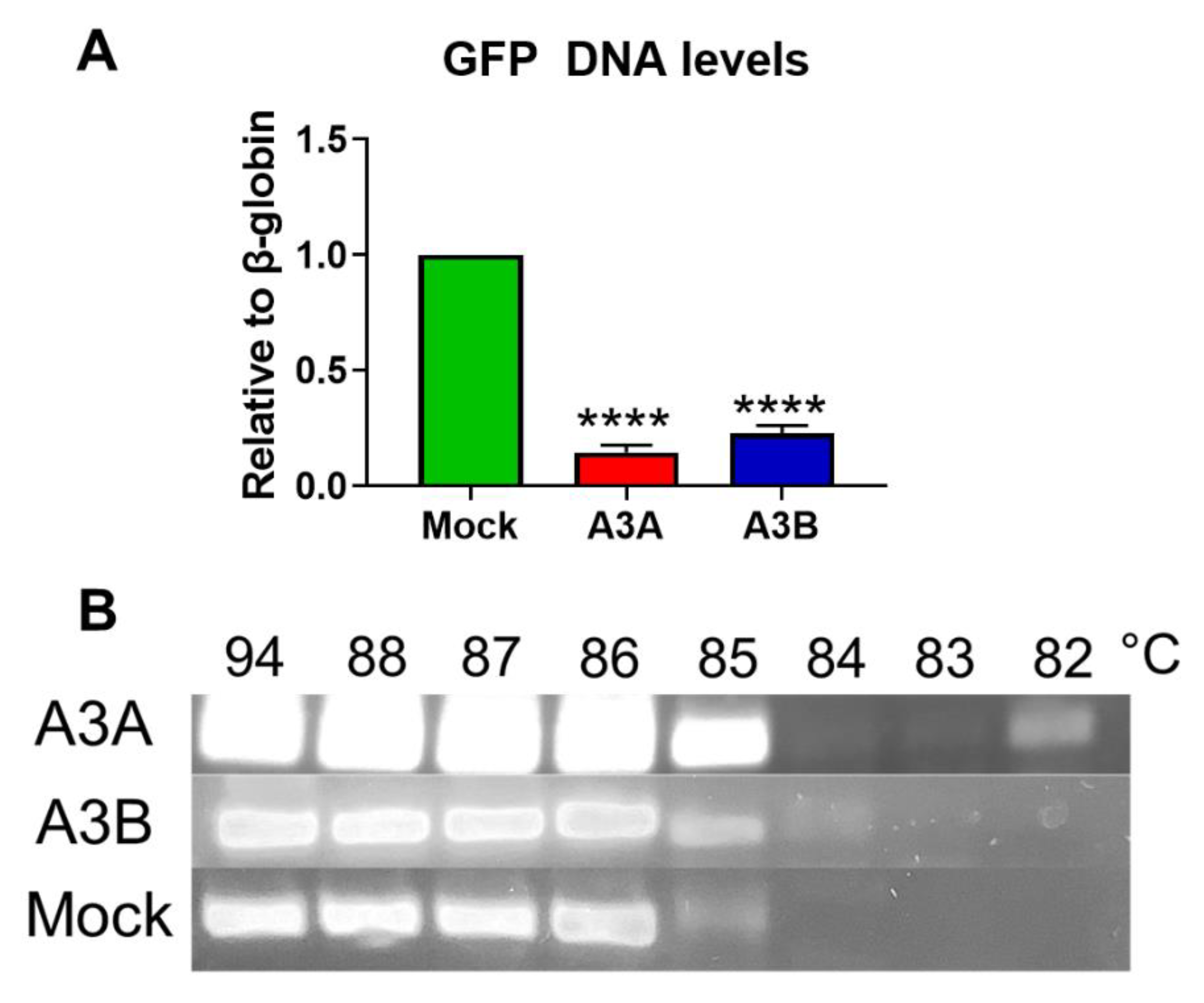
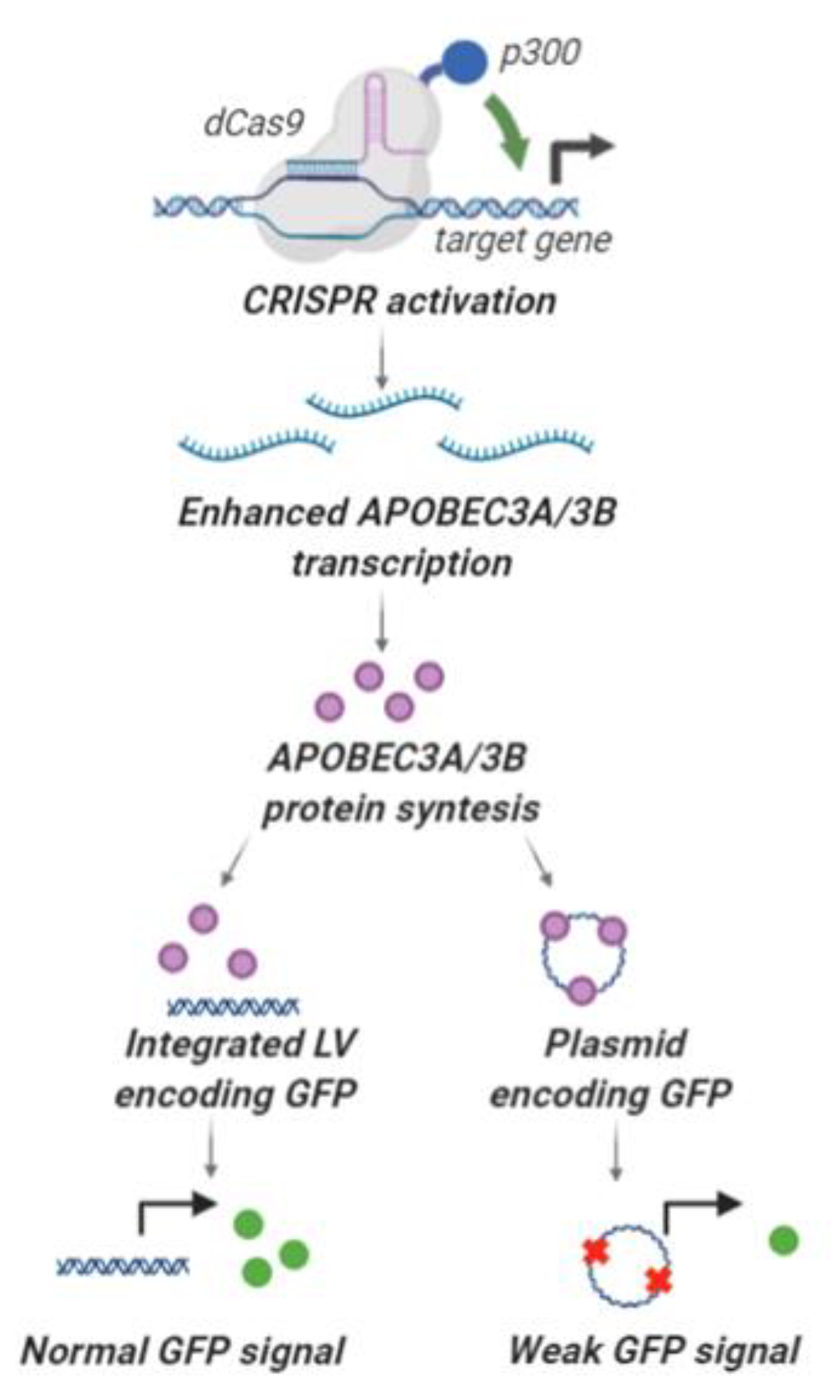
2.4. Deamination and Degradation of Foreign DNA Using the CRISPRa Approach
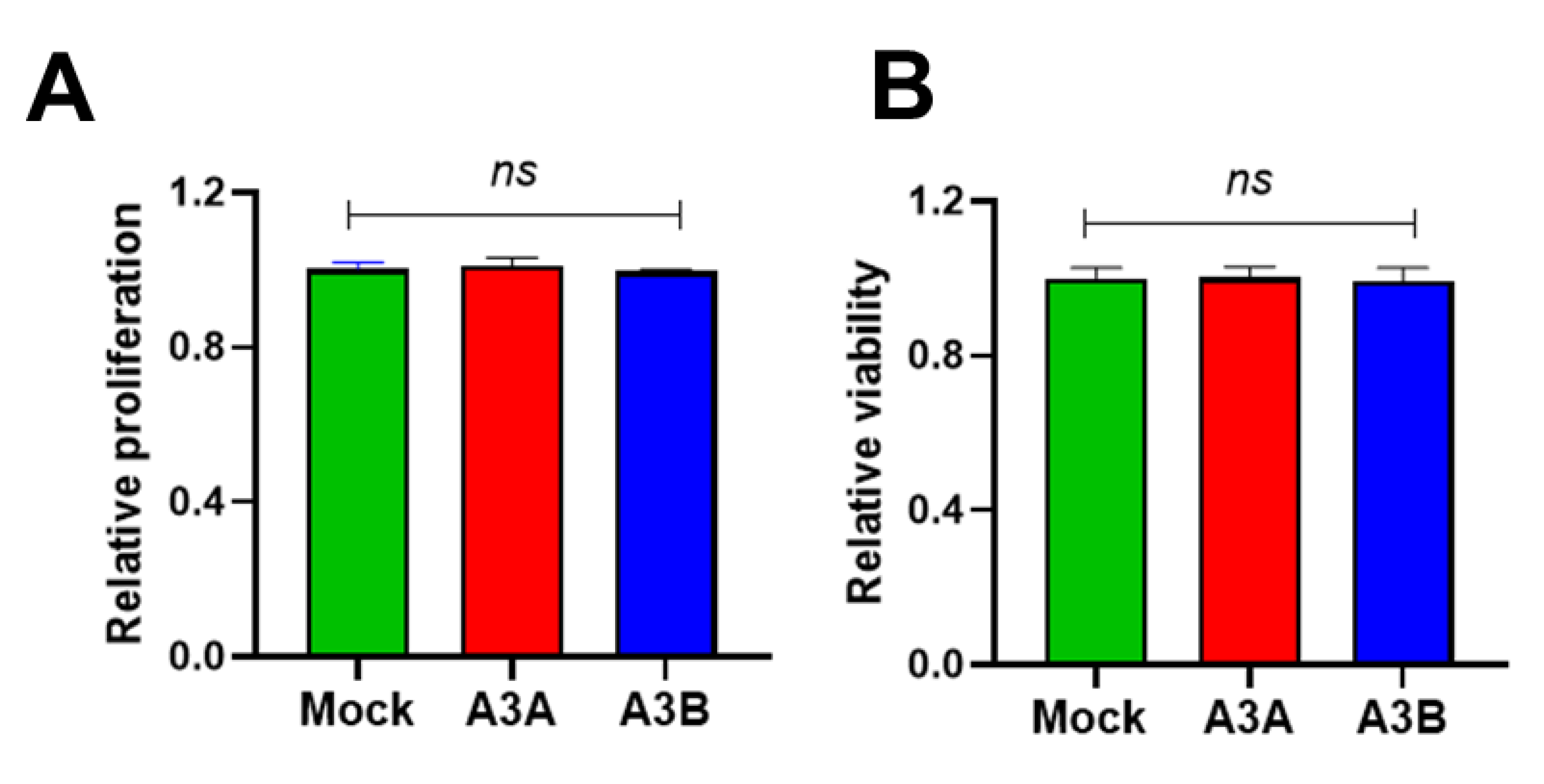
2.5. Short-Term Overexpression of A3A and A3B Is Not Toxic
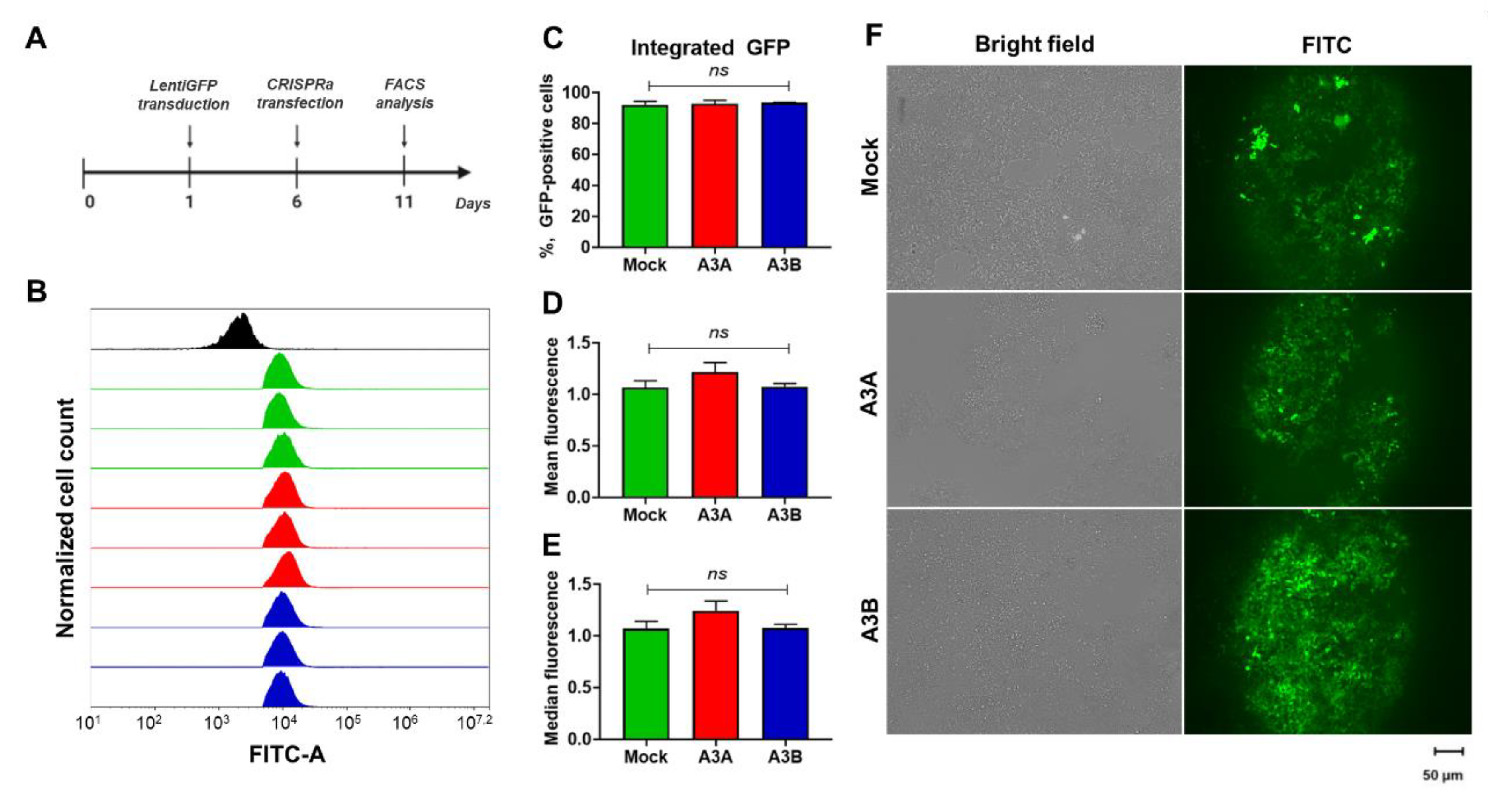
2.6. Expression of Integrated Foreign DNA Is Not Affected by CRISPRa Transactivated A3A and A3B
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. sgRNAs Design and Generation of sgRNA-Encoding PCR-Products Preparation
4.3. Isolation of Nucleic Acids and Semi-Quantitative PCR Analysis
4.4. 3D-PCR
4.5. Production of Lentiviruses
4.6. Lentiviral Transduction
4.7. Flow Cytometry and Fluorescent Microscopy
4.8. Toxicity Assays
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sparrer, K.M.J.; Gack, M.U. Intracellular detection of viral nucleic acids. Curr. Opin. Microbiol. 2015, 26, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paludan, S.R.; Bowie, A.G. Immune sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussil, B.; Suspène, R.; Caval, V.; Durandy, A.; Wain-Hobson, S.; Vartanian, J.-P. Genotoxic stress increases cytoplasmic mitochondrial DNA editing by human APOBEC3 mutator enzymes at a single cell level. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Motani, K.; Ito, S.; Nagata, S. DNA-mediated cyclic GMP–AMP synthase–dependent and–independent regulation of innate immune responses. J. Immunol. 2015, 194, 4914–4923. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, 6227. [Google Scholar] [CrossRef] [Green Version]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wen, M.; Cao, X. Nuclear hnRNPA2B1 initiates and amplifies the innate immune response to DNA viruses. Science 2019, 365, 6454. [Google Scholar] [CrossRef]
- Love, R.P.; Xu, H.; Chelico, L. Biochemical analysis of hypermutation by the deoxycytidine deaminase APOBEC3A. J. Biol. Chem. 2012, 287, 30812–30822. [Google Scholar] [CrossRef] [Green Version]
- Jarmuz, A.; Chester, A.; Bayliss, J.; Gisbourne, J.; Dunham, I.; Scott, J.; Navaratnam, N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 2002, 79, 285–296. [Google Scholar] [CrossRef]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and Nonhepatotoxic Degradation of Nuclear Hepatitis B Virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Stadler, D.; Lucifora, J.; Reisinger, F.; Webb, D.; Hoesel, M.; Michler, T.; Wisskirchen, K.; Cheng, X.; Zhang, K.; et al. Interferon-gamma and Tumor Necrosis Factor-alpha Produced by T Cells Reduce the HBV Persistence Form, cccDNA, Without Cytolysis. Gastroenterology 2016, 150, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, V.C.; Booiman, T.; De Taeye, S.W.; Van Dort, K.A.; Rits, M.A.N.; Hamann, J.; Kootstra, N.A. Differential expression of HIV-1 interfering factors in monocyte-derived macrophages stimulated with polarizing cytokines or interferons. Sci. Rep. 2012, 2, 1–7. [Google Scholar]
- Bockmann, J.-H.; Stadler, D.; Xia, Y.; Ko, C.; Wettengel, J.M.; Schulze zur Wiesch, J.; Dandri, M.; Protzer, U. Comparative Analysis of the Antiviral Effects Mediated by Type I and III Interferons in Hepatitis B Virus–Infected Hepatocytes. J. Infect. Dis. 2019, 220, 567–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, B.; McCann, J.L.; Starrett, G.J.; Kosyakovsky, L.; Luengas, E.M.; Molan, A.M.; Burns, M.B.; McDougle, R.M.; Parker, P.J.; Brown, W.L. The PKC/NF-κB signaling pathway induces APOBEC3B expression in multiple human cancers. Cancer Res. 2015, 75, 4538–4547. [Google Scholar] [CrossRef] [Green Version]
- Greenwell-Wild, T.; Vázquez, N.; Jin, W.; Rangel, Z.; Munson, P.J.; Wahl, S.M. Interleukin-27 inhibition of HIV-1 involves an intermediate induction of type I interferon. Blood J. Am. Soc. Hematol. 2009, 114, 1864–1874. [Google Scholar] [CrossRef] [Green Version]
- Tasker, C.; Ding, J.; Schmolke, M.; Rivera-Medina, A.; García-Sastre, A.; Chang, T.L. 17β-estradiol protects primary macrophages against HIV infection through induction of interferon-alpha. Viral Immunol. 2014, 27, 140–150. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wang, X.; Liu, M.; Hu, Q.; Song, L.; Ye, L.; Zhou, D.; Ho, W. A critical function of toll-like receptor-3 in the induction of anti-human immunodeficiency virus activities in macrophages. Immunology 2010, 131, 40–49. [Google Scholar] [CrossRef]
- Menendez, D.; Nguyen, T.-A.; Snipe, J.; Resnick, M.A. The cytidine deaminase APOBEC3 family is subject to transcriptional regulation by p53. Mol. Cancer Res. 2017, 15, 735–743. [Google Scholar] [CrossRef] [Green Version]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99. [Google Scholar] [CrossRef] [PubMed]
- Brezgin, S.; Kostyusheva, A.; Bayurova, E.; Gordeychuk, I.; Isaguliants, M.; Goptar, I.; Nikiforova, A.; Smirnov, V.; Volchkova, E.; Glebe, D. Replenishment of Hepatitis B Virus cccDNA Pool Is Restricted by Baseline Expression of Host Restriction Factors In Vitro. Microorganisms 2019, 7, 533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.-P.; Peng, Z.-G.; Wu, Z.-Y.; Li, J.-R.; Huang, M.-H.; Si, S.-Y.; Jiang, J.-D. Host APOBEC3G protein inhibits HCV replication through direct binding at NS3. PLoS ONE 2015, 10, e0121608. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.J.; Xu, T.; Guo, K.; Griffin, L.M.; Westrich, J.A.; Lee, D.; Lambert, P.F.; Santiago, M.L.; Pyeon, D. APOBEC3A functions as a restriction factor of human papillomavirus. J. Virol. 2015, 89, 688–702. [Google Scholar] [CrossRef] [Green Version]
- Suspène, R.; Aynaud, M.-M.; Koch, S.; Pasdeloup, D.; Labetoulle, M.; Gaertner, B.; Vartanian, J.-P.; Meyerhans, A.; Wain-Hobson, S. Genetic editing of herpes simplex virus 1 and Epstein-Barr herpesvirus genomes by human APOBEC3 cytidine deaminases in culture and in vivo. J. Virol. 2011, 85, 7594–7602. [Google Scholar] [CrossRef] [Green Version]
- Iwatani, Y.; Chan, D.S.B.; Wang, F.; Maynard, K.S.; Sugiura, W.; Gronenborn, A.M.; Rouzina, I.; Williams, M.C.; Musier-Forsyth, K.; Levin, J.G. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007, 35, 7096–7108. [Google Scholar] [CrossRef]
- Stenglein, M.D.; Burns, M.B.; Li, M.; Lengyel, J.; Harris, R.S. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 2010, 17, 222–229. [Google Scholar] [CrossRef]
- Brezgin, S.; Kostyusheva, A.; Kostyushev, D.; Chulanov, V. Dead Cas Systems: Types, Principles, and Applications. Int. J. Mol. Sci. 2019, 20, 6041. [Google Scholar] [CrossRef] [Green Version]
- Hilton, I.B.; Vockley, C.M.; Pratiksha, I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A.; Carolina, N.; States, U.; Biology, C.; Carolina, N.; et al. CRISPR Acetyltransferase Activates Genes From Promoters and Enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479, 131–145. [Google Scholar] [CrossRef] [Green Version]
- Knisbacher, B.A.; Gerber, D.; Levanon, E.Y. DNA editing by APOBECs: A genomic preserver and transformer. Trends Genet. 2016, 32, 16–28. [Google Scholar] [CrossRef]
- Bowie, A.G.; Unterholzner, L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008, 8, 911–922. [Google Scholar] [CrossRef]
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 2016, 14, 360. [Google Scholar] [CrossRef]
- Finlay, B.B.; McFadden, G. Anti-immunology: Evasion of the host immune system by bacterial and viral pathogens. Cell 2006, 124, 767–782. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Limsirichai, P.; Gaj, T.; Schaffer, D.V. CRISPR-mediated Activation of Latent HIV-1 Expression. Mol. Ther. 2016, 24, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Jiang, Z.; Lu, P.; Ma, L.; Liz, C.; Pan, H.; Fill, Z.; Qui, X.; Wang, P.; Deng, J.; et al. Specific Reactivation of Latent HIV-1 by dCas9-SunTag-VP64-mediated Guide RNA Targeting the HIV-1 Promoter. Mol. Ther. 2016, 24, 508–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogerd, H.P.; Kornepati, A.V.R.; Marshall, J.B.; Kennedy, E.M.; Cullen, B.R. Specific induction of endogenous viral restriction factors using CRISPR/Cas-derived transcriptional activators. Proc. Natl. Acad. Sci. USA 2015, 112, E7249–E7256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ozono, S.; Yao, W.; Tobiume, M.; Yamaoka, S.; Kishigami, S.; Fujita, H.; Tokunaga, K. CRISPR-mediated activation of endogenous BST-2/tetherin expression inhibits wild-type HIV-1 production. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez, A.; Tuttle, M.; Pruitt, B.W.; Ewen-Campen, B.; Chari, R.; Ter-Ovanesyan, D.; Haque, S.J.; Cecchi, R.J.; Kowal, E.J.K.; Buchthal, J.; et al. Comparative Analysis of Cas9 Activators Across Multiple Species. Nat. Methods 2016, 13, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostyusheva, A.; Kostyushev, D.; Brezgin, S.; Volchkova, E.; Chulanov, V. Clinical Implications of Hepatitis B Virus RNA and Covalently Closed Circular DNA in Monitoring Patients with Chronic Hepatitis B Today with a Gaze into the Future: The Field Is Unprepared for a Sterilizing Cure. Genes 2018, 9, 483. [Google Scholar] [CrossRef] [Green Version]
- Ko, C.; Chakraborty, A.; Chou, W.-M.; Hasreiter, J.; Wettengel, J.M.; Stadler, D.; Bester, R.; Asen, T.; Zhang, K.; Wisskirchen, K. Hepatitis B virus (HBV) genome recycling and de novo secondary infection events maintain stable cccDNA levels. J. Hepatol. 2018, 69, 1231–1241. [Google Scholar] [CrossRef]
- Nair, S.; Zlotnick, A. Asymmetric Modification of Hepatitis B Virus (HBV) Genomes by an Endogenous Cytidine Deaminase inside HBV Cores Informs a Model of Reverse Transcription. J. Virol. 2018, 92, 10. [Google Scholar] [CrossRef] [Green Version]
- Kostyushev, D.; Kostyusheva, A.; Brezgin, S.; Zarifyan, D.; Utkina, A.; Goptar, I.; Chulanov, V. Suppressing the NHEJ pathway by DNA-PKcs inhibitor NU7026 prevents degradation of HBV cccDNA cleaved by CRISPR/Cas9. Sci. Rep. 2019, 9, 1847. [Google Scholar] [CrossRef] [Green Version]
- Kostyusheva, A.; Brezgin, S.; Bayurova, E.; Gordeychuk, I.; Isaguliants, M.; Goptar, I.; Urusov, F.; Nikiforova, A.; Volchkova, E.; Kostyushev, D.; et al. ATM and ATR Expression Potentiates HBV Replication and Contributes to Reactivation of HBV Infection upon DNA Damage. Viruses 2019, 11, 997. [Google Scholar] [CrossRef] [Green Version]
- Kostyusheva, A.P.; Kostyushev, D.S.; Brezgin, S.A.; Zarifyan, D.N.; Volchkova, E.V.; Chulanov, V.P. Small Molecular Inhibitors of DNA Double Strand Break Repair Pathways Increase the ANTI-HBV Activity of CRISPR/Cas9. Mol. Biol. 2019, 53, 274–285. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | 5′–3′ Sequence |
|---|---|
| sgA3A1_fw | CAAAACCAAAGCTTGCCCAGGTTTTAGAGCTAGAAATAG |
| sgA3A1_rev | CTGGGCAAGCTTTGGTTTTGCGGTGTTTCGTCCTTTC |
| sgA3A2_fw | GCTAATGAGGGTGGCACACTGTTTTAGAGCTAGAAATAG |
| sgA3A2_rev | AGTGTGCCACCCTCATTAGCCGGTGTTTCGTCCTTTC |
| sgA3A3_fw | GGCCCACAGGGAGCAAAGTGGTTTTAGAGCTAGAAATAG |
| sgA3A3_rev | CACTTTGCTCCCTGTGGGCCCGGTGTTTCGTCCTTTC |
| sgA3A4_fw | ATTCTTACCGTGAAGAGTGCGTTTTAGAGCTAGAAATAG |
| sgA3A4_rev | GCACTCTTCACGGTAAGAATCGGTGTTTCGTCCTTTC |
| sgA3B fw | ATTGGAGGTTCCTCTGCCAGGTTTTAGAGCTAGAAATAG |
| sgA3B rev | CTGGCAGAGGAACCTCCAATCGGTGTTTCGTCCTTTC |
| sgNC_fw | CTGCCTGCCTCGTCAACACCGTTTTAGAGCTAGAAATAG |
| sgNC_rev | GGTGTTGACGAGGCAGGCAGCGGTGTTTCGTCCTTTC |
| pLX-sgRNA_U6 fw | TATATAGGATCCGAGGGCCTATTTCCCATGATTCCTTCATATTTG |
| pLX-sgRNA_SP rev | TATATAGCTAGCAAAAAAAGCACCGACTCGG |
| Primer Name | 5′–3′ Sequence |
|---|---|
| GAPDH fw | CAACGGATTTGGTCGTATTGG |
| GAPDH rev | GCAACAATATCCACTTTACCAGAGTTAA |
| GAPDH probe | (FAM)-CGCCTGGTCACCAGGGCTGC-(BHQ1) |
| β-globin fw | V31-FEP-CE-AmpliSens HPV HCR-Screen (CRIE) |
| β-globin rev | V31-FEP-CE-AmpliSens HPV HCR-Screen (CRIE) |
| β-globin probe | V31-FEP-CE-AmpliSens HPV HCR-Screen (CRIE) |
| A3A fw | AGATGGAGTCTGGTACTGTCG |
| A3A rev | GAGGCAGGAGAGTAGCGT |
| A3B fw | GAGCTACACTTGGCTGTGCT |
| A3B rev | TGACATTGGGGTGCTCAGAC |
| Primer Name | 5′-3′ Sequence |
|---|---|
| 3D/DNA/RNA-GFP fw | AAG TTC AGC GTG TCC GGC GA |
| 3D/DNA/RNA-GFP rev | GCG CTC CTG GAC GTA GCC TT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brezgin, S.; Kostyusheva, A.; Ponomareva, N.; Volia, V.; Goptar, I.; Nikiforova, A.; Shilovskiy, I.; Smirnov, V.; Kostyushev, D.; Chulanov, V. Clearing of Foreign Episomal DNA from Human Cells by CRISPRa-Mediated Activation of Cytidine Deaminases. Int. J. Mol. Sci. 2020, 21, 6865. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186865
Brezgin S, Kostyusheva A, Ponomareva N, Volia V, Goptar I, Nikiforova A, Shilovskiy I, Smirnov V, Kostyushev D, Chulanov V. Clearing of Foreign Episomal DNA from Human Cells by CRISPRa-Mediated Activation of Cytidine Deaminases. International Journal of Molecular Sciences. 2020; 21(18):6865. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186865
Chicago/Turabian StyleBrezgin, Sergey, Anastasiya Kostyusheva, Natalia Ponomareva, Viktoriia Volia, Irina Goptar, Anastasiya Nikiforova, Igor Shilovskiy, Valery Smirnov, Dmitry Kostyushev, and Vladimir Chulanov. 2020. "Clearing of Foreign Episomal DNA from Human Cells by CRISPRa-Mediated Activation of Cytidine Deaminases" International Journal of Molecular Sciences 21, no. 18: 6865. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21186865