Injectable Thermo-Sensitive Chitosan Hydrogel Containing CPT-11-Loaded EGFR-Targeted Graphene Oxide and SLP2 shRNA for Localized Drug/Gene Delivery in Glioblastoma Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
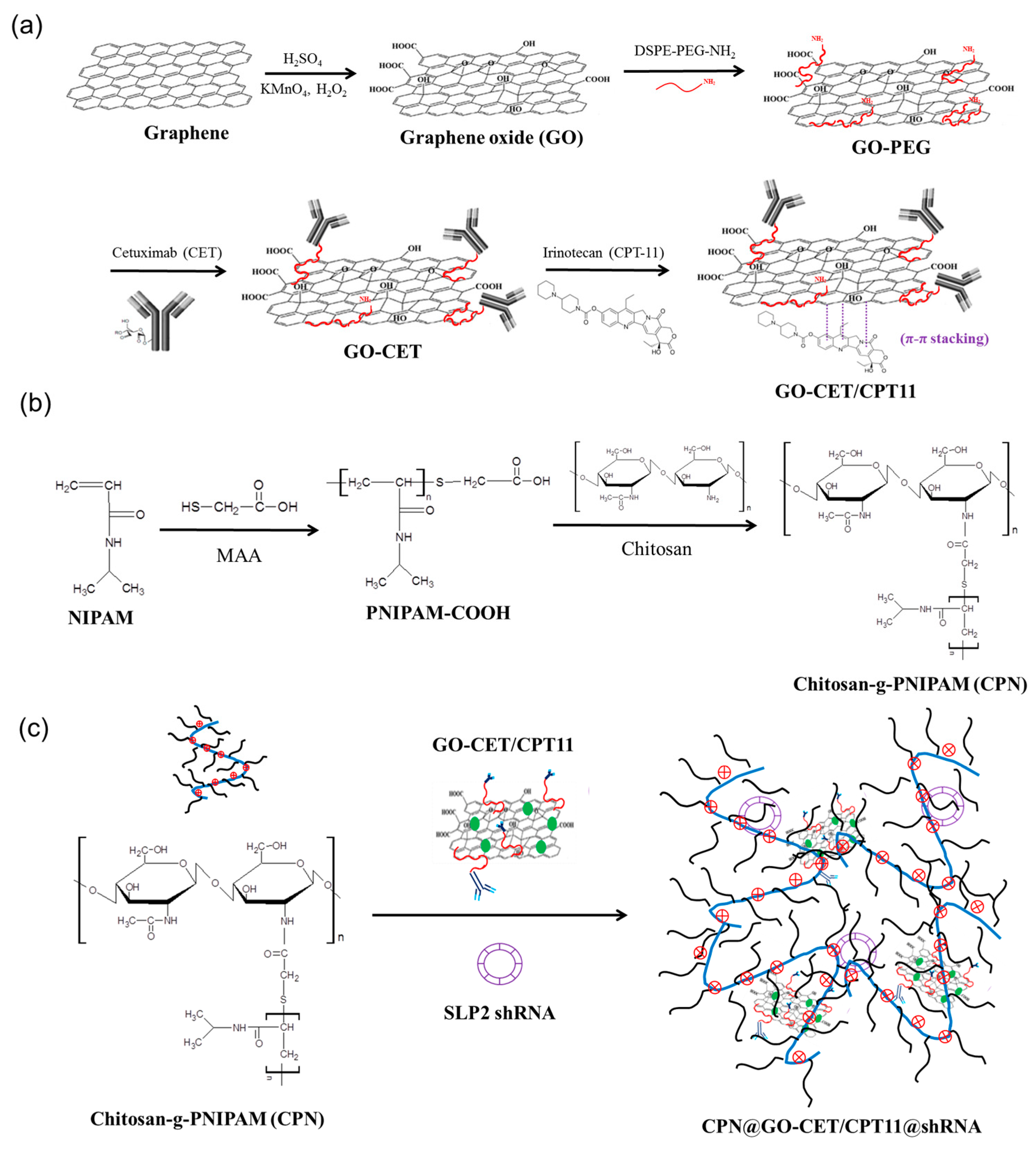
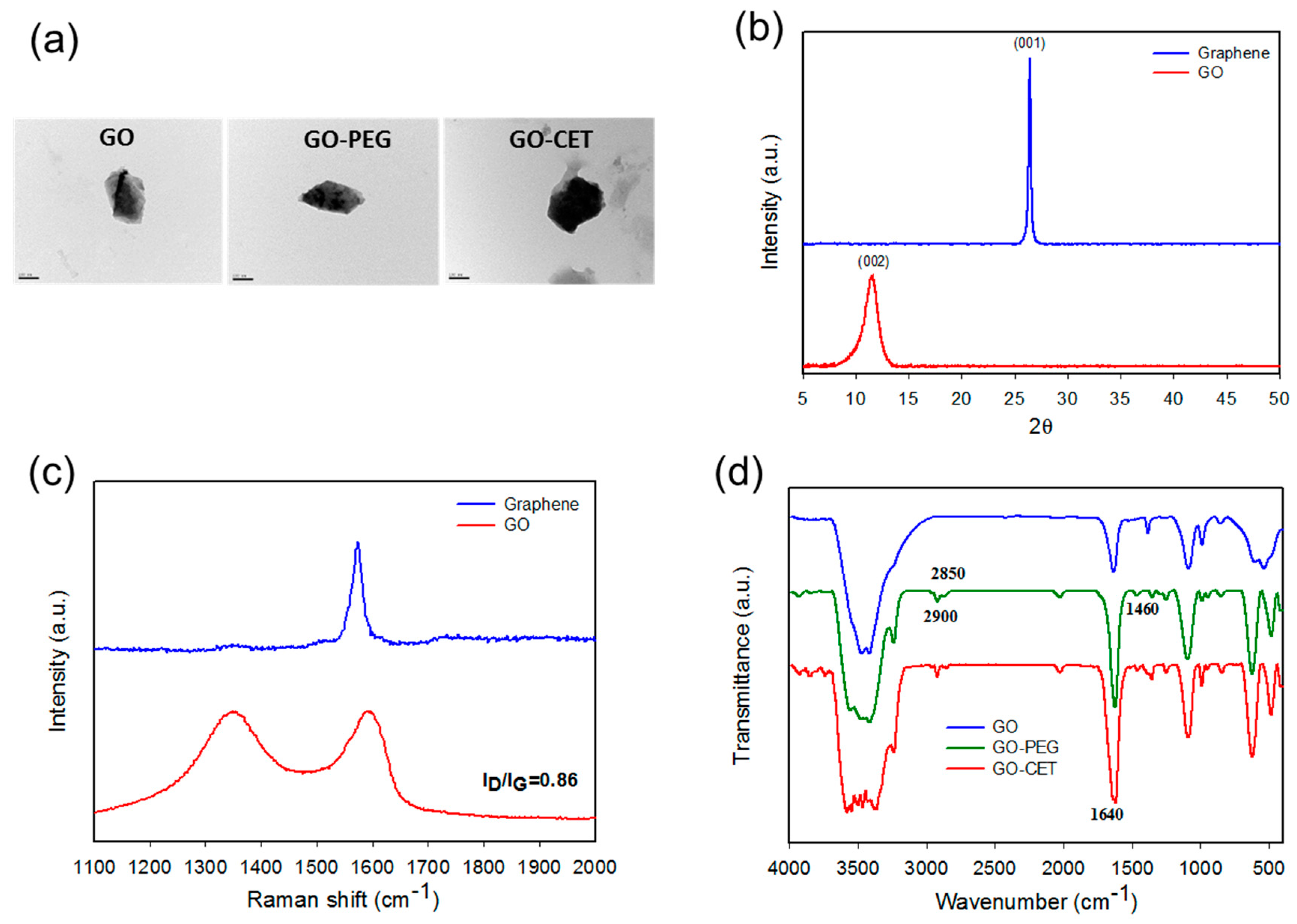
2.1. Synthesis and Characterization of GO, GO-PEG, GO-CET, and GO-CET/CPT11
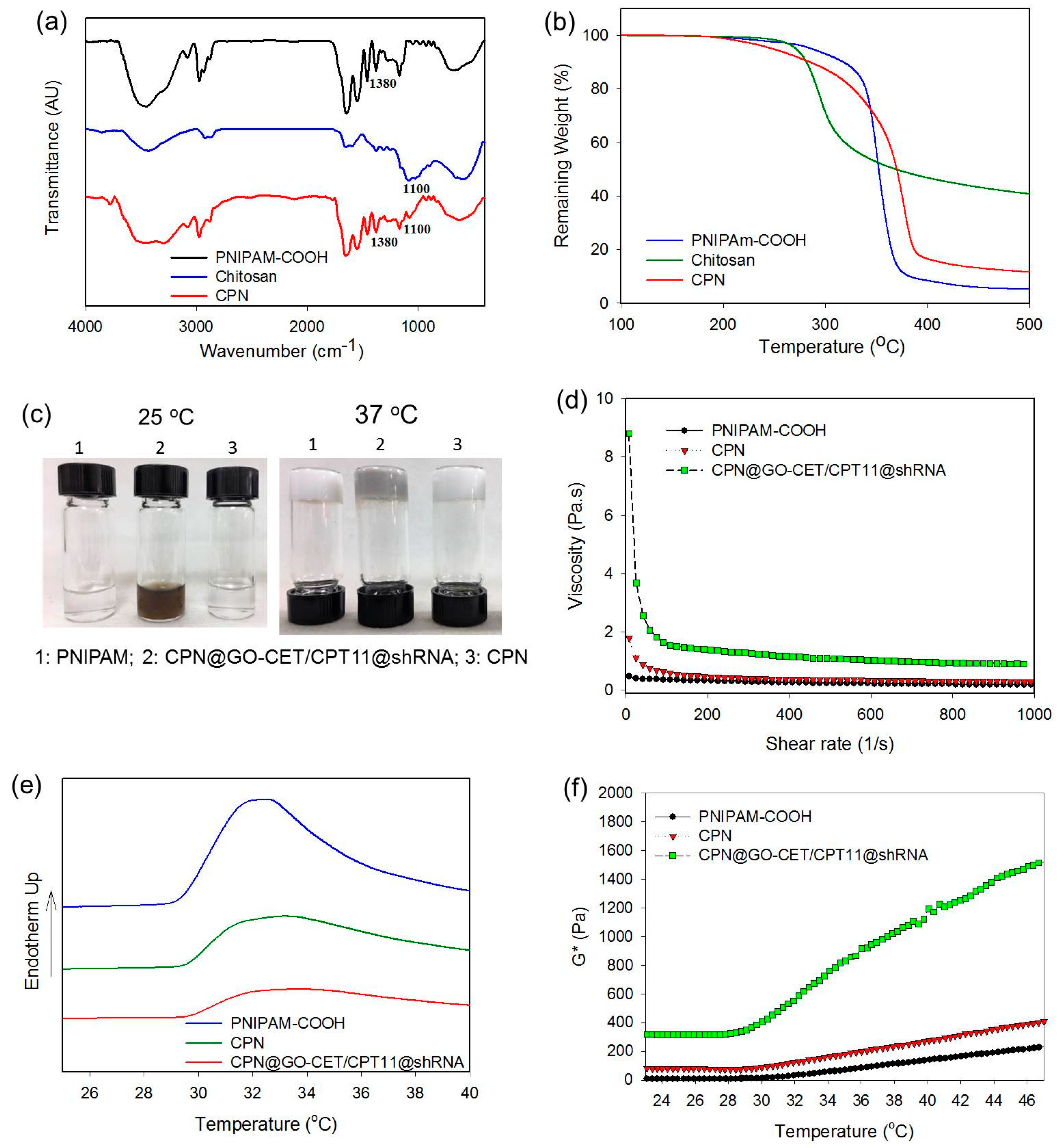
2.2. Synthesis and Characterization of CPN
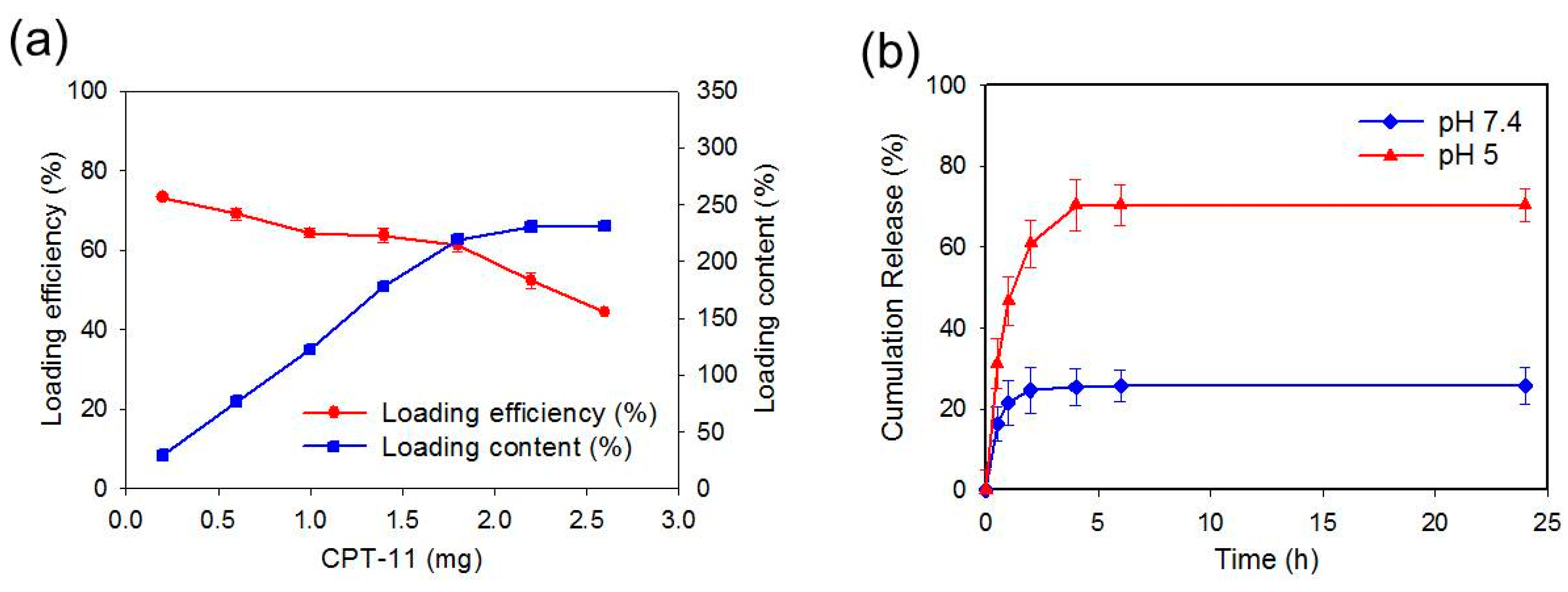
2.3. Loading and Release of CPT-11
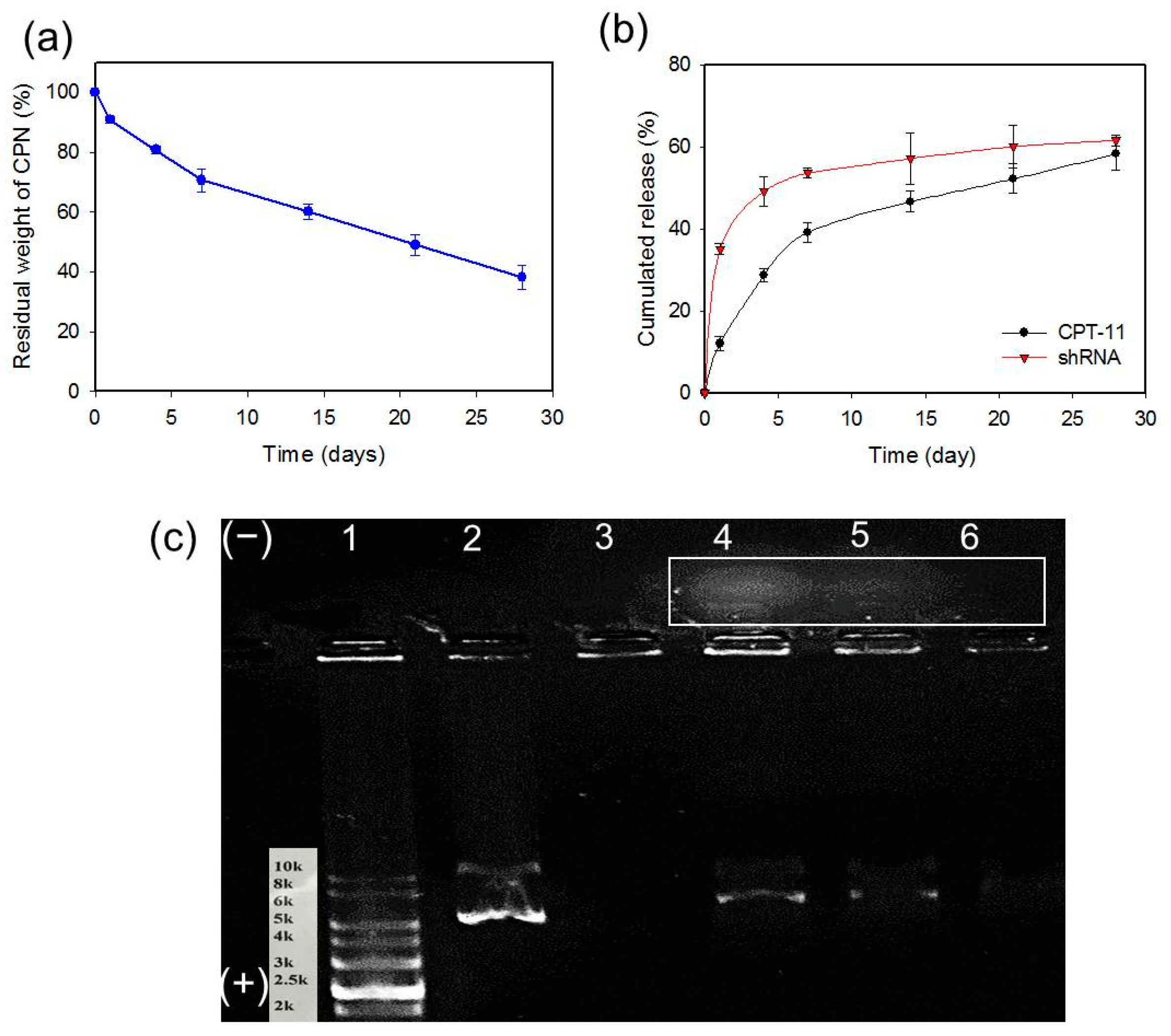
2.4. Degradation of CPN and Release of CPT-11 and shRNA from CPN
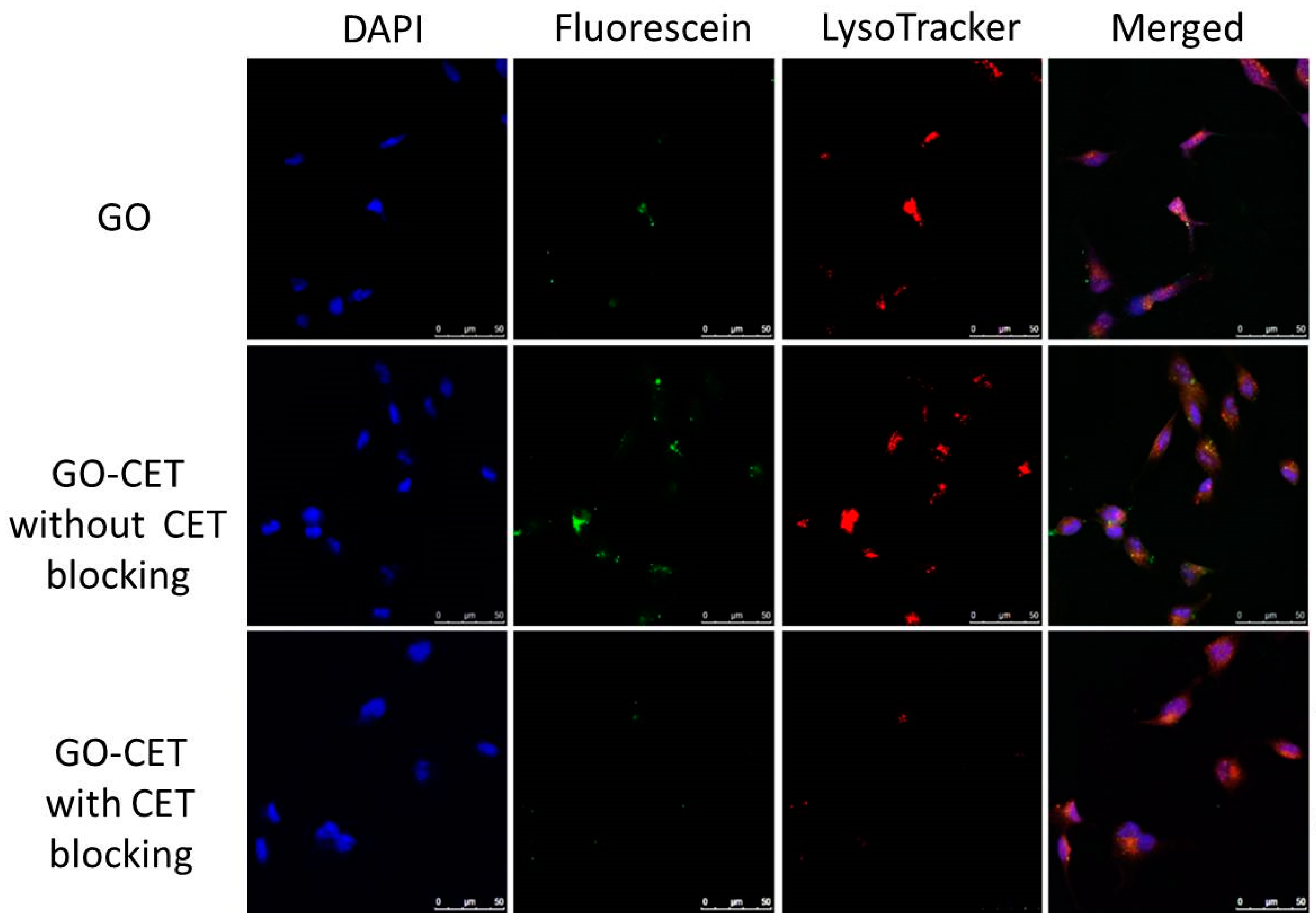
2.5. Intracellular Uptake
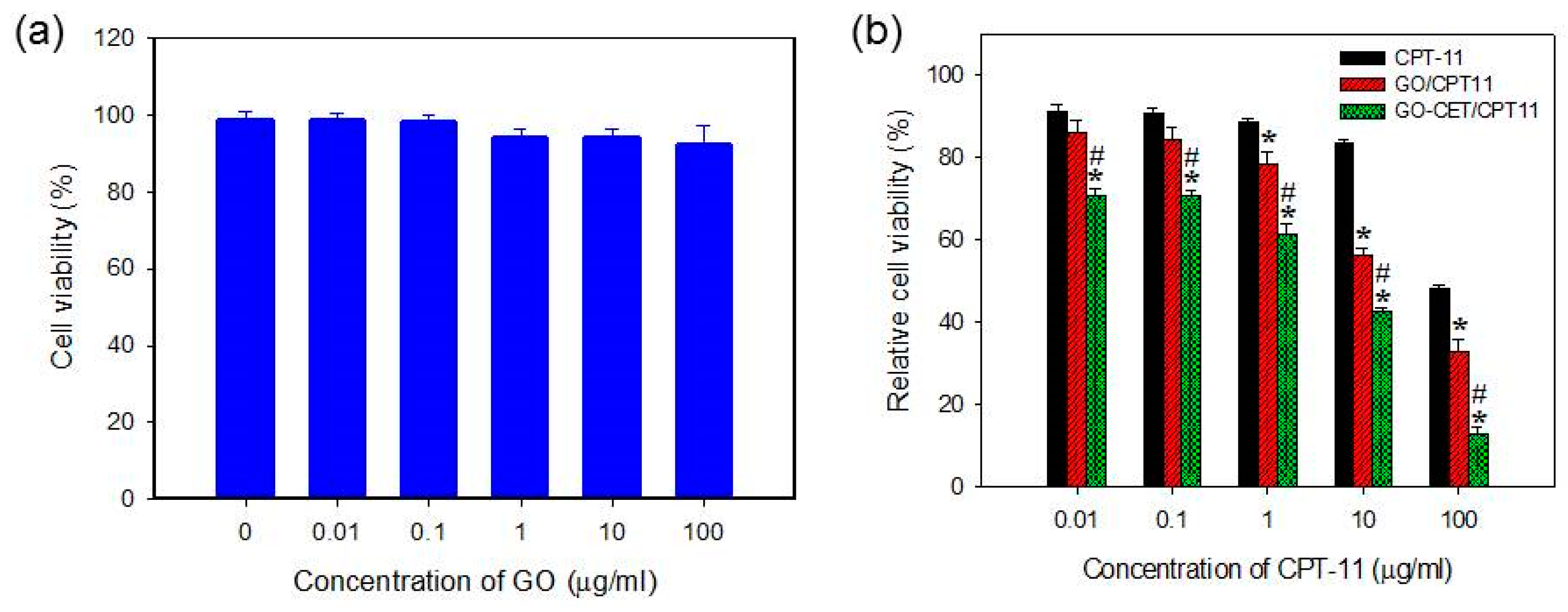
2.6. In Vitro Biocompatibility of GO and Cytotoxicity of CPT-11-Loaded GO
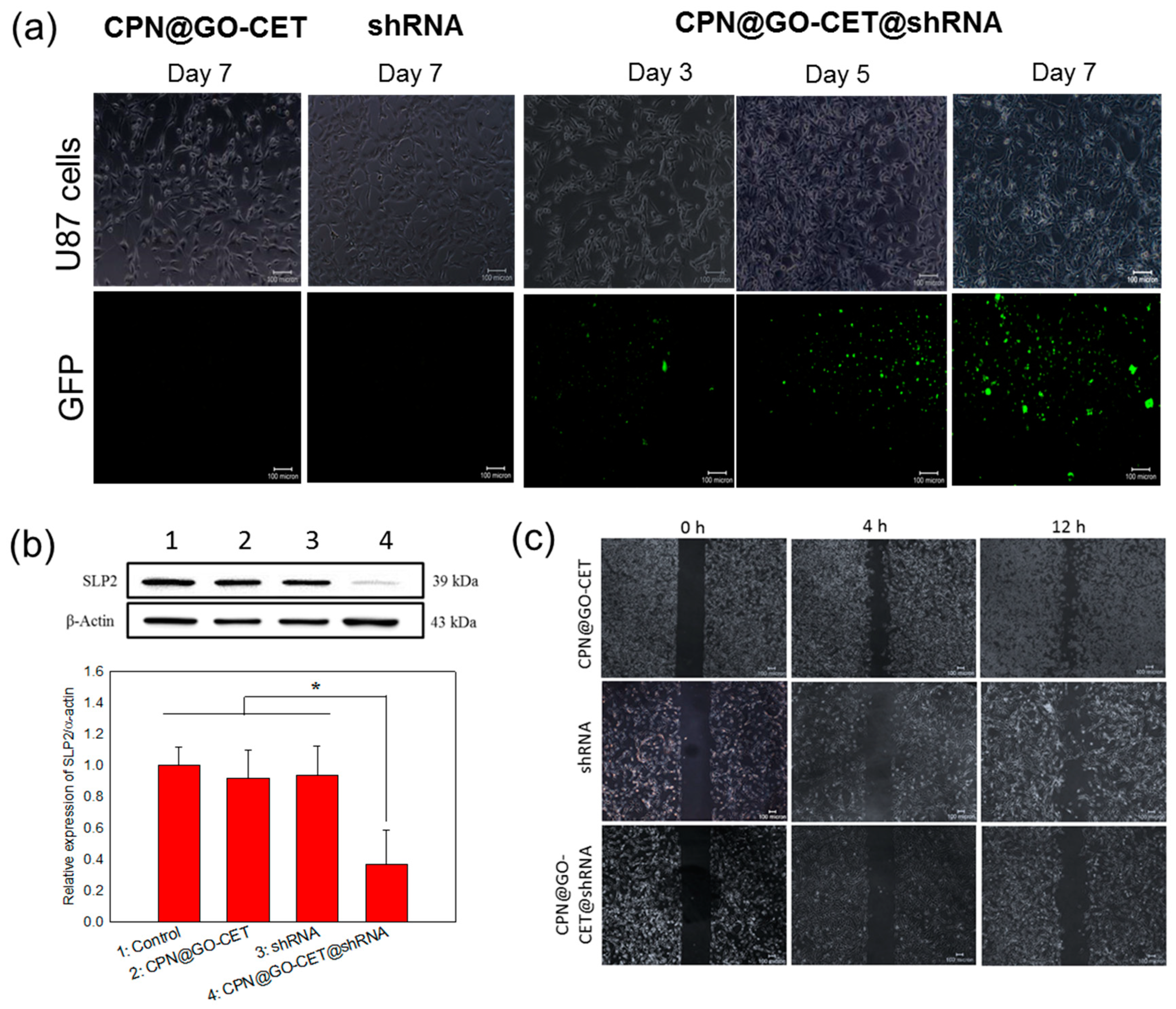
2.7. Gene Delivery, Gene Silencing, and Migration Inhibition of U87 Cells with SLP2 shRNA
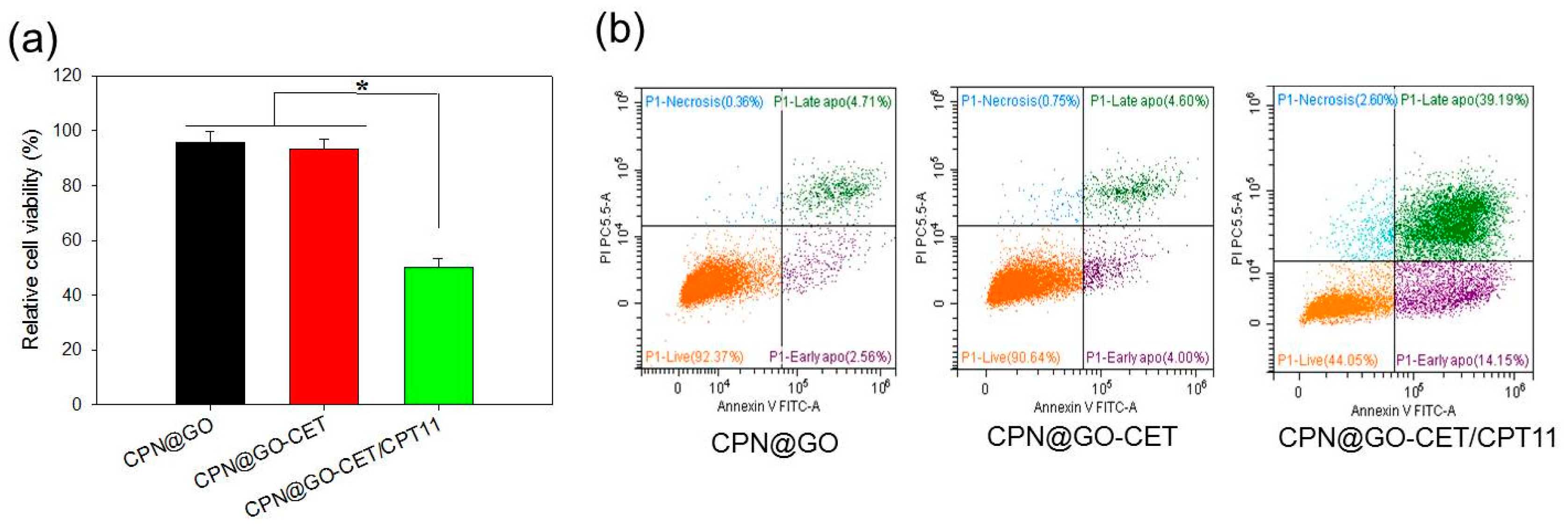
2.8. Cell Cytotoxicity and Apoptosis of CPT-11-Loaded GO in CPN
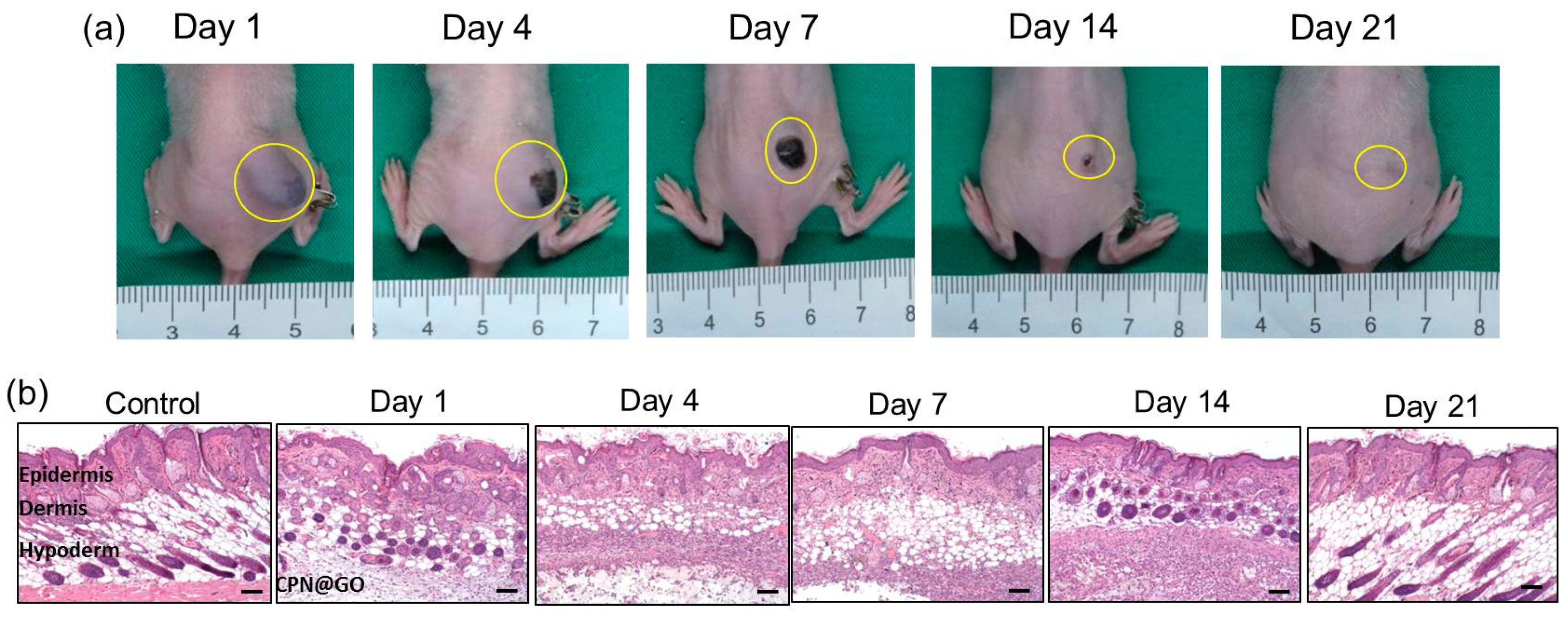
2.9. Degradation and Biocompatibility of CPN@GO In Vivo
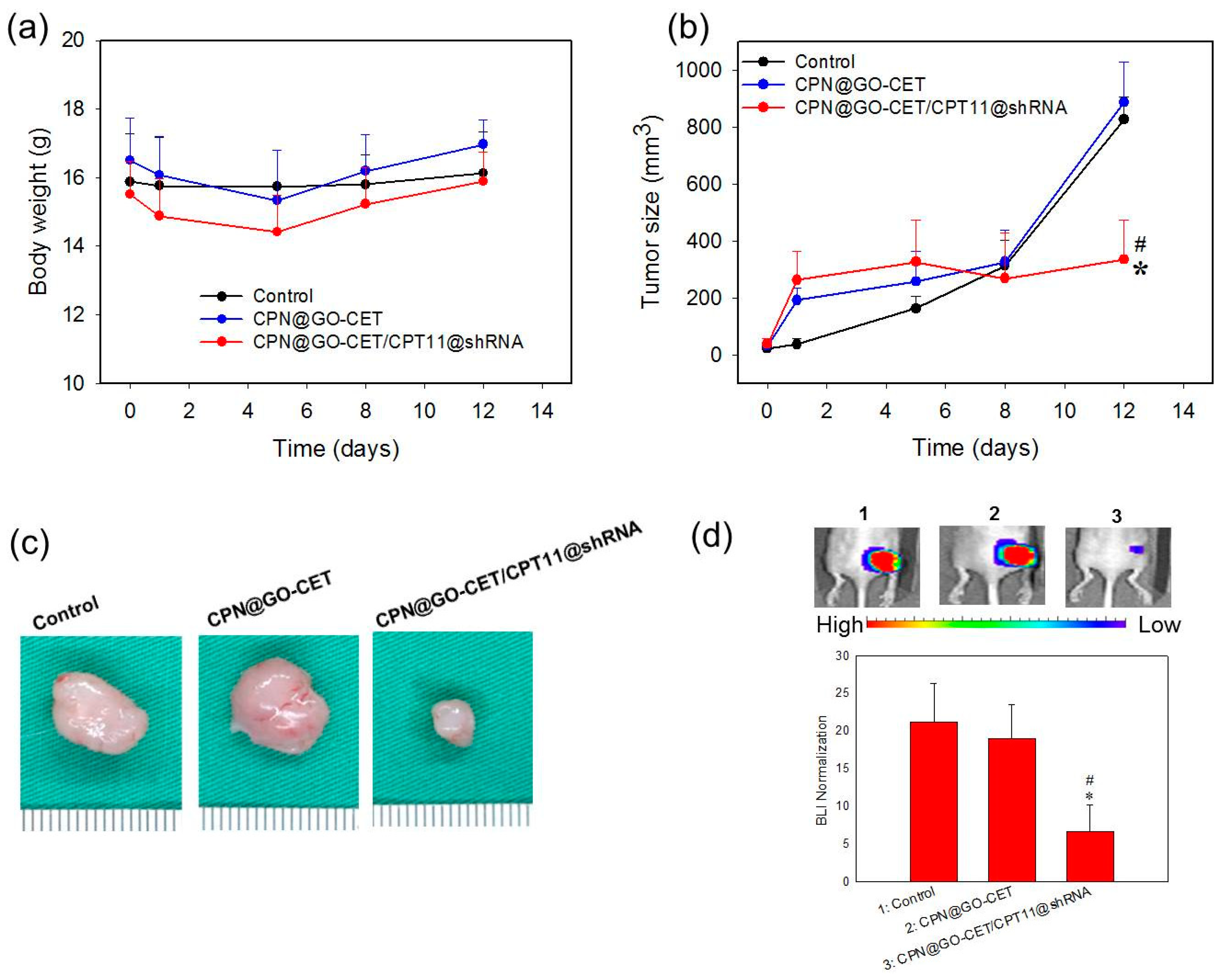
2.10. Anticancer Efficacy in Xenograft Nude Mice Animal Model
3. Materials and Methods
3.1. Materials
3.2. Preparation of GO, GO-PEG, GO-CET, and Fluorescently Labeled GO
3.3. Characterization of GO-Based Nanocarriers
3.4. Synthesis of CPN Copolymer
3.5. Characterization of Hydrogel
3.6. Drug Loading and Release
3.7. SLP2 shRNA Loading and Release
3.8. Cell Line and Cell Culture Condition
3.9. Intracellular Upake
3.10. Biocompatibility In Vitro
3.11. Cytotoxicity In Vitro
3.12. Transfection of U87 Cells
3.13. Western Blot Analysis
3.14. Xenograft Nude Mice Animal Model
3.15. In Vivo Anti-Tumor Efficacy
3.16. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GO | Graphene oxide |
| CET | Cetuximab |
| EGFR | Epidermal growth factor receptor |
| SLP2 | Stomatin-like protein 2 |
| shRNA | Short hairpin RNA |
| siRNA | Small interfering RNA |
| CPN | Chitosan-g-poly(N-isopropylacrylamide) |
| GBM | Glioblastoma multiforme |
| RNAi | RNA interference |
| WHO | World Health Organization |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| PNIPAM | Poly(N-isopropylacrylamide) |
| DSPE | 1,2-Distearoyl-sn-glycero-3-phosphorylethanolamine |
| PEG | Polyethylene glycol |
| PNIPAM-COOH | Carboxylic acid-ended poly(N-isopropylacrylamide) |
| NIPAM | N-isopropylacrylamide |
| MAA | Mercaptoacetic acid |
| DLS | Dynamic light scattering |
| XRD | X-ray diffraction |
| FTIR | Fourier transform infrared |
| TNBSA | 2,4,6-Trinitrobenzene sulfonic acid |
| DSC | Differential scanning calorimeter |
| GFP | Green fluorescent protein |
| PI | Propidium iodide |
| BLI | Bioluminescence imaging |
| IHC | Immunohistochemistry |
| pERK | Phosphorylated extracellular signal-regulated kinases |
| EDC | 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride |
| NHS | N-hydroxysuccinimide |
| DAPI | 4′,6-Diamidino-2-phenylindole dihydrochloride |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| FBS | Fetal bovine serum |
| MES | 2-(N-morpholino)ethanesulfonic acid |
| TGA | Thermogravimetric analysis |
| H&E | Hematoxylin–eosin |
| PBS | Phosphate buffered saline |
| HPLC | High-performance liquid chromatography |
| CLSM | Confocal laser scanning microscope |
| EDTA | Ethylenediaminetetraacetic acid |
References
- Koshy, M.; Villano, J.L.; Dolecek, T.A.; Howard, A.; Mahmood, U.; Chmura, S.J.; Weichselbaum, R.R.; McCarthy, B.J. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J. Neuro Oncol. 2011, 107, 207–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012-2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab Alone and in Combination With Irinotecan in Recurrent Glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740. [Google Scholar] [CrossRef] [Green Version]
- Prados, M.D.; Lamborn, K.; Yung, W.; Jaeckle, K.; Robins, H.I.; Mehta, M.; Fine, H.A.; Wen, P.Y.; Cloughesy, T.; Chang, S.; et al. A phase 2 trial of irinotecan (CPT-11) in patients with recurrent malignant glioma: A North American Brain Tumor Consortium study1. Neuro-Oncology 2006, 8, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.-J.; Chuang, E.-Y.; Cheng, Y.-H.; Anilkumar, T.; Chen, H.-A.; Chen, J.-P. Thermosensitive magnetic liposomes for alternating magnetic field-inducible drug delivery in dual targeted brain tumor chemotherapy. Chem. Eng. J. 2019, 373, 720–733. [Google Scholar] [CrossRef]
- Liu, J.; Cui, L.; Losic, D. Graphene and graphene oxide as new nanocarriers for drug delivery applications. Acta Biomater. 2013, 9, 9243–9257. [Google Scholar] [CrossRef]
- Huang, Y.-S.; Lu, Y.-J.; Chen, J.-P. Magnetic graphene oxide as a carrier for targeted delivery of chemotherapy drugs in cancer therapy. J. Magn. Magn. Mater. 2017, 427, 34–40. [Google Scholar] [CrossRef]
- Nguyen, K.; Dang, P.N.; Alsberg, E. Functionalized, biodegradable hydrogels for control over sustained and localized siRNA delivery to incorporated and surrounding cells. Acta Biomater. 2013, 9, 4487–4495. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.-J.; Lin, P.-Y.; Huang, P.-H.; Kuo, C.-Y.; Shalumon, K.T.; Chen, M.-Y.; Chen, J.-P. Magnetic Graphene Oxide for Dual Targeted Delivery of Doxorubicin and Photothermal Therapy. Nanomaterials 2018, 8, 193. [Google Scholar] [CrossRef] [Green Version]
- Attia, M.F.; Anton, H.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An overview of active and passive targeting strategies to improve the nanocarriers efficiency to tumour sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, T.E.; Furnari, F.; Cavenee, W.K. Targeting EGFR for treatment of glioblastoma: Molecular basis to overcome resistance. Curr. Cancer Drug Targets 2012, 12, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Akhter, H.; Madhav, N.S.; Ahmad, J. Epidermal growth factor receptor based active targeting: A paradigm shift towards advance tumor therapy. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1188–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Song, H.; Zhang, J.; Li, P.; Li, C.; Wang, C.; Kong, D.; Zhao, Q. An injectable, thermosensitive and multicompartment hydrogel for simultaneous encapsulation and independent release of a drug cocktail as an effective combination therapy platform. J. Control. Release 2015, 203, 57–66. [Google Scholar] [CrossRef]
- Dai, X.; Tan, C. Combination of microRNA therapeutics with small-molecule anticancer drugs: Mechanism of action and co-delivery nanocarriers. Adv. Drug Deliv. Rev. 2015, 81, 184–197. [Google Scholar] [CrossRef]
- Zhan, C.; Meng, Q.; Li, Q.; Feng, L.; Zhu, J.; Lu, W. Cyclic RGD-Polyethylene Glycol-Polyethylenimine for Intracranial Glioblastoma-Targeted Gene Delivery. Chem. Asian J. 2011, 7, 91–96. [Google Scholar] [CrossRef]
- Shen, H.; Sun, T.; Ferrari, M. Nanovector delivery of siRNA for cancer therapy. Cancer Gene Ther. 2012, 19, 367–373. [Google Scholar] [CrossRef] [Green Version]
- Kozielski, K.L.; Ruiz-Valls, A.; Tzeng, S.Y.; Guerrero-Cázares, H.; Rui, Y.; Li, Y.; Vaughan, H.J.; Gionet-Gonzales, M.; Vantucci, C.; Kim, J.; et al. Cancer-selective nanoparticles for combinatorial siRNA delivery to primary human GBM in vitro and in vivo. Biomaterials 2019, 209, 79–87. [Google Scholar] [CrossRef]
- Wang, J.; Lu, Z.; Wientjes, M.G.; Au, J.L.-S. Delivery of siRNA Therapeutics: Barriers and Carriers. AAPS J. 2010, 12, 492–503. [Google Scholar] [CrossRef]
- Dong, Y.; Siegwart, D.J.; Anderson, D.G. Strategies, design, and chemistry in siRNA delivery systems. Adv. Drug Deliv. Rev. 2019, 144, 133–147. [Google Scholar] [CrossRef]
- Zhang, L.; Elaraj, D.M.; Weinreich, D.M.; Varghese, S.; Puhlmann, M.; Hewitt, S.M.; Carroll, N.M.; Feldman, E.D.; Turner, E.M.; Alexander, H.R. Stomatin-like Protein 2 Is Overexpressed in Cancer and Involved in Regulating Cell Growth and Cell Adhesion in Human Esophageal Squamous Cell Carcinoma. Clin. Cancer Res. 2006, 12, 1639–1646. [Google Scholar] [CrossRef] [Green Version]
- Chang, D.; Ma, K.; Gong, M.; Cui, Y.; Liu, Z.-H.; Zhou, X.-G.; Zhou, C.-N.; Wang, T.-Y. SLP-2overexpression is associated with tumour distant metastasis and poor prognosis in pulmonary squamous cell carcinoma. Biomarkers 2009, 15, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Meleady, P.; Dowd, A.J.; Henry, M.; Glynn, S.A.; Clynes, M. Proteomic analysis of isolated membrane fractions from superinvasive cancer cells. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2007, 1774, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xie, J.; He, H.-Y.; Huang, E.-W.; Cao, Q.-H.; Luo, L.; Liao, Y.-S.; Guo, Y. Suppression of CLC-3 chloride channel reduces the aggressiveness of glioma through inhibiting nuclear factor-κB pathway. Oncotarget 2017, 8, 63788–63798. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Liu, L.; Wu, Z.; Lin, C.; Dai, T.; Yu, C.; Wang, X.; Wu, J.; Li, M.; Li, J. Knockdown of stomatin-like protein 2 (STOML2) reduces the invasive ability of glioma cells through inhibition of the NF-κB/MMP-9 pathway. J. Pathol. 2011, 226, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, M.; Nazari, B.; Miller, D.W. Injectable hydrogel-based drug delivery systems for local cancer therapy. Drug Discov. Today 2016, 21, 1835–1849. [Google Scholar] [CrossRef]
- Schild, H. Poly(N-isopropylacrylamide): Experiment, theory and application. Prog. Polym. Sci. 1992, 17, 163–249. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; Wu, W.; Li, H. Vaginal delivery of carboplatin-loaded thermosensitive hydrogel to prevent local cervical cancer recurrence in mice. Drug Deliv. 2016, 23, 1–22. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, H.; Jia, Y.; Guo, Q.; Qu, Y.; Su, J.; Lu, X.; Zhao, Y.; Qian, Z. A novel gene delivery composite system based on biodegradable folate-poly (ester amine) polymer and thermosensitive hydrogel for sustained gene release. Sci. Rep. 2016, 6, 21402. [Google Scholar] [CrossRef]
- Huang, P.; Song, H.; Zhang, Y.; Liu, J.; Zhang, J.; Wang, W.; Liu, J.; Li, C.; Kong, D. Bridging the Gap between Macroscale Drug Delivery Systems and Nanomedicines: A Nanoparticle-Assembled Thermosensitive Hydrogel for Peritumoral Chemotherapy. ACS Appl. Mater. Interfaces 2016, 8, 29323–29333. [Google Scholar] [CrossRef]
- Guo, D.-D.; Hong, S.-H.; Jiang, H.-L.; Kim, J.-H.; Minai-Tehrani, A.; Kim, J.-E.; Shin, J.-Y.; Jiang, T.; Kim, Y.-K.; Choi, Y.-J.; et al. Synergistic effects of Akt1 shRNA and paclitaxel-incorporated conjugated linoleic acid-coupled poloxamer thermosensitive hydrogel on breast cancer. Biomaterials 2012, 33, 2272–2281. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; He, C.; Cheng, Y.; Li, D.; Gong, Y.; Liu, J.; Tian, H.; Chen, X. PLK1shRNA and doxorubicin co-loaded thermosensitive PLGA-PEG-PLGA hydrogels for osteosarcoma treatment. Biomaterials 2014, 35, 8723–8734. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, N.; Gunn, J.; Zhang, M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv. Drug Deliv. Rev. 2010, 62, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Ragelle, H.; Vandermeulen, G.; Préat, V. Chitosan-based siRNA delivery systems. J. Control. Release 2013, 172, 207–218. [Google Scholar] [CrossRef]
- Afkham, A.; Aghebati-Maleki, L.; Siahmansouri, H.; Sadreddini, S.; Ahmadi, M.; Dolati, S.; Afkham, N.M.; Akbarzadeh, P.; Jadidi-Niaragh, F.; Younesi, V.; et al. Chitosan (CMD)-mediated co-delivery of SN38 and Snail-specific siRNA as a useful anticancer approach against prostate cancer. Pharmacol. Rep. 2018, 70, 418–425. [Google Scholar] [CrossRef]
- Chen, J.-P.; Cheng, T.-H. Thermo-Responsive Chitosan-graft-poly(N-isopropylacrylamide) Injectable Hydrogel for Cultivation of Chondrocytes and Meniscus Cells. Macromol. Biosci. 2006, 6, 1026–1039. [Google Scholar] [CrossRef]
- Fang, J.-Y.; Chen, J.-P.; Leu, Y.; Hu, J.-W. The Delivery of Platinum Drugs from Thermosensitive Hydrogels Containing Different Ratios of Chitosan. Drug Deliv. 2008, 15, 235–243. [Google Scholar] [CrossRef]
- Prabha, S.; Arya, G.; Chandra, R.; Ahmed, B.; Nimesh, S. Effect of size on biological properties of nanoparticles employed in gene delivery. Artif. Cells Nanomed. Biotechnol. 2014, 44, 83–91. [Google Scholar] [CrossRef]
- Chu, I.-M.; Tseng, S.-H.; Chou, M.-Y. Cetuximab-conjugated iron oxide nanoparticles for cancer imaging and therapy. Int. J. Nanomed. 2015, 10, 3663–3685. [Google Scholar] [CrossRef] [Green Version]
- Hassan, H.M.A.; Abdelsayed, V.; Khder, A.E.R.S.; Abouzeid, K.M.; Terner, J.; El-Shall, M.S.; Al-Resayes, S.I.; El-Azhary, A.A. Microwave synthesis of graphene sheets supporting metal nanocrystals in aqueous and organic media. J. Mater. Chem. 2009, 19, 3832–3837. [Google Scholar] [CrossRef]
- Perumbilavil, S.; Sankar, P.; Rose, T.P.; Philip, R. White light Z-scan measurements of ultrafast optical nonlinearity in reduced graphene oxide nanosheets in the 400–700 nm region. Appl. Phys. Lett. 2015, 107, 051104. [Google Scholar] [CrossRef]
- Kim, N.H.; Kuila, T.; Lee, J.H. Simultaneous reduction, functionalization and stitching of graphene oxide with ethylenediamine for composites application. J. Mater. Chem. A 2013, 1, 1349–1358. [Google Scholar] [CrossRef]
- Elias, E.R.; Poloukhtine, A.; Popik, V.V.; Tsourkas, A. Effect of ligand density, receptor density, and nanoparticle size on cell targeting. Nanomed. Nanotechnol. Biol. Med. 2012, 9, 194–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Villalona-Calero, M.A. Irinotecan: Mechanisms of tumor resistance and novel strategies for modulating its activity. Ann. Oncol. 2002, 13, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Shah, J.; Hein, S.; Misra, R. Controlled and extended drug release behavior of chitosan-based nanoparticle carrier. Acta Biomater. 2010, 6, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.-W.; Syu, W.-J.; Chen, W.-Y.; Ruaan, R.-C.; Cheng, Y.-C.; Chien, C.-C.; Li, C.; Chung, C.-A.; Tsao, C.-W. Use of Biotinylated Chitosan for Substrate-Mediated Gene Delivery. Bioconjugate Chem. 2012, 23, 1587–1599. [Google Scholar] [CrossRef] [PubMed]
- Santos-Carballal, B.; Fernández, E.F.; Goycoolea, F.M. Chitosan in Non-Viral Gene Delivery: Role of Structure, Characterization Methods, and Insights in Cancer and Rare Diseases Therapies. Polymers 2018, 10, 444. [Google Scholar] [CrossRef] [Green Version]
- Mu, Q.; Su, G.; Li, L.; Gilbertson, B.O.; Yu, L.H.; Zhang, Q.; Sun, Y.-P.; Yan, B. Size-Dependent Cell Uptake of Protein-Coated Graphene Oxide Nanosheets. ACS Appl. Mater. Interfaces 2012, 4, 2259–2266. [Google Scholar] [CrossRef]
- Bao, H.; Pan, Y.; Ping, Y.; Sahoo, N.G.; Wu, T.; Li, L.; Li, J.; Gan, L.H. Chitosan-Functionalized Graphene Oxide as a Nanocarrier for Drug and Gene Delivery. Small 2011, 7, 1569–1578. [Google Scholar] [CrossRef]
- Hannon, G.J. RNA interference. Nature 2002, 418, 244–251. [Google Scholar] [CrossRef]
- Rao, D.D.; Vorhies, J.S.; Senzer, N.N.; Nemunaitis, J. siRNA vs. shRNA: Similarities and differences. Adv. Drug Deliv. Rev. 2009, 61, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Klibanov, A.M. Enhancing polyethylenimine’s delivery of plasmid DNA into mammalian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14640–14645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, C.B.; Guthrie, E.H.; Huang, M.T.-H.; Taxman, D.J. Short Hairpin RNA (shRNA): Design, Delivery, and Assessment of Gene Knockdown. Methods Mol. Biol. 2010, 629, 139–156. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Tan, Y.F.; Wong, Y.S.; Liew, W.J.M.; Venkatraman, S.S. Recent Advances in Chitosan-Based Carriers for Gene Delivery. Mar. Drugs 2019, 17, 381. [Google Scholar] [CrossRef] [Green Version]
- Köping-Höggård, M.; Vårum, K.M.; Issa, M.; Danielsen, S.; Christensen, B.E.; Stokke, B.T.; Artursson, P. Improved chitosan-mediated gene delivery based on easily dissociated chitosan polyplexes of highly defined chitosan oligomers. Gene Ther. 2004, 11, 1441–1452. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, W.; Yu, Z.; Liu, Z. Downregulation of a mitochondria associated protein SLP-2 inhibits tumor cell motility, proliferation and enhances cell sensitivity to chemotherapeutic reagents. Cancer Biol. Ther. 2009, 8, 1651–1658. [Google Scholar] [CrossRef] [Green Version]
- Rivera, F.; Vega-Villegas, M.E.; López-Brea, M.F. Cetuximab, its clinical use and future perspectives. Anti-Cancer Drugs 2008, 19, 99–113. [Google Scholar] [CrossRef]
- Cusack, J.C.; Liu, R.; Baldwin, A.S. Inducible Chemoresistance to 7-Ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin (CPT-11) in Colorectal Cancer Cells and a Xenograft Model Is Overcome by Inhibition of Nuclear Factor-κB Activation. Cancer Res. 2000, 60, 2323–2330. [Google Scholar]
- Liao, H.-T.; Chen, C.-T.; Chen, J.-P. Osteogenic Differentiation and Ectopic Bone Formation of Canine Bone Marrow-Derived Mesenchymal Stem Cells in Injectable Thermo-Responsive Polymer Hydrogel. Tissue Eng. Part C Methods 2011, 17, 1139–1149. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Ding, J.; Xiao, C.; Zhuang, X.; Chen, X. Thermosensitive hydrogels based on polypeptides for localized and sustained delivery of anticancer drugs. Biomaterials 2013, 34, 10338–10347. [Google Scholar] [CrossRef]
- Jost, S.C.; Collins, L.; Travers, S.; Piwnica-Worms, D.; Garbow, J.R. Measuring Brain Tumor Growth: A Combined BLI / MRI Strategy. Mol. Imaging 2009, 8, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, Z.; Welsher, K.; Robinson, J.T.; Goodwin, A.; Zaric, S.; Dai, H. Nano-graphene oxide for cellular imaging and drug delivery. Nano Res. 2008, 1, 203–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cayot, P.; Tainturier, G. The Quantification of Protein Amino Groups by the Trinitrobenzenesulfonic Acid Method: A Reexamination. Anal. Biochem. 1997, 249, 184–200. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.-Y.; Chen, S.-H.; Chen, C.-H.; Fong, Y.T.; Chen, J.-P.; Chen, S.-H. Thermo-responsive in-situ forming hydrogels as barriers to prevent post-operative peritendinous adhesion. Acta Biomater. 2017, 63, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Fong, Y.T.; Chen, C.-H.; Chen, J.-P. Intratumoral Delivery of Doxorubicin on Folate-Conjugated Graphene Oxide by In-Situ Forming Thermo-Sensitive Hydrogel for Breast Cancer Therapy. Nanomaterials 2017, 7, 388. [Google Scholar] [CrossRef] [Green Version]
- Kojima, S.-I.; Vignjevic, D.M.; Borisy, G.G. Improved silencing vector co-expressing GFP and small hairpin RNA. Biotechniques 2004, 36, 74–79. [Google Scholar] [CrossRef] [Green Version]












© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.-J.; Lan, Y.-H.; Chuang, C.-C.; Lu, W.-T.; Chan, L.-Y.; Hsu, P.-W.; Chen, J.-P. Injectable Thermo-Sensitive Chitosan Hydrogel Containing CPT-11-Loaded EGFR-Targeted Graphene Oxide and SLP2 shRNA for Localized Drug/Gene Delivery in Glioblastoma Therapy. Int. J. Mol. Sci. 2020, 21, 7111. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197111
Lu Y-J, Lan Y-H, Chuang C-C, Lu W-T, Chan L-Y, Hsu P-W, Chen J-P. Injectable Thermo-Sensitive Chitosan Hydrogel Containing CPT-11-Loaded EGFR-Targeted Graphene Oxide and SLP2 shRNA for Localized Drug/Gene Delivery in Glioblastoma Therapy. International Journal of Molecular Sciences. 2020; 21(19):7111. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197111
Chicago/Turabian StyleLu, Yu-Jen, Yu-Hsiang Lan, Chi-Cheng Chuang, Wan-Ting Lu, Li-Yang Chan, Peng-Wei Hsu, and Jyh-Ping Chen. 2020. "Injectable Thermo-Sensitive Chitosan Hydrogel Containing CPT-11-Loaded EGFR-Targeted Graphene Oxide and SLP2 shRNA for Localized Drug/Gene Delivery in Glioblastoma Therapy" International Journal of Molecular Sciences 21, no. 19: 7111. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197111