Targeting the SphK-S1P-SIPR Pathway as a Potential Therapeutic Approach for COVID-19


Abstract
:1. Introduction
2. Coronaviruses from Animals to Humans
3. SARS-CoV-2 Infectivity, Symptoms, and Complications
3.1. SARS-CoV-2 Infectivity
3.2. COVID-19 and Respiratory Failure
3.3. COVID-19 Pulmonary Vascular Disorders
3.4. COVID-19, the Heart and Cardiovascular System
3.5. Vascular Endotheliitis Contribution to Multiorgan Failure in COVID-19 Patients
3.6. Neurological Disease and COVID-19
3.7. Rare Inflammatory Diseases Associated with COVID-19
3.8. The Cytokine Storm (Hyperinflammation, Morbidity and Mortality)
4. Current Strategies for the Prevention and Treatment of COVID-19
4.1. COVID-19—Vaccine Development Targets
4.2. Current COVID-19 Vaccine Clinical Trials
4.3. COVID-19 Vaccine Limitations
4.4. Current COVID-19 Treatment
COVID-19 Treatment—Targeting Vasculature Failure
5. Targeting SphK/S1P/S1PR in Viral Infection and Alleviation of COVID-19 Symptoms
5.1. The Sphingosine Kinase Rheostat
5.2. SphK-S1P-S1PR1 Autocrine and Paracrine Inflammatory Actions
5.3. Autocrine SphK-S1P Response in Systemic Inflammation and the Immune Response
5.4. The S1P/S1PR Paracrine “Outside–Inside” Response
5.5. The Sphingolipid Pathway in Coronavirus Infection and Replication
5.6. The S1P/S1PR1 Response to Inflammatory Lung Viral Infections
5.7. SphK/S1P/S1PRs in Maintaining Vascular Integrity
Differential Roles of S1P1-3 Receptors in Vascular Function and Regulation
5.8. SphK/S1P/S1PR1 Role in Thrombosis
5.9. SphK/S1P/S1PR and Sepsis
5.10. SphK/S1P/S1PR and Cardioprotection
5.11. SphK/S1P in Neuroinflammation and Neurodegeneration
6. Repurposing Anti-SphK-S1P-S1PR Compounds in Curtailing COVID-19 Symptoms
6.1. FTY720 in the Prevention of SARS-CoV-2 Infection and Therapy for COVID-19 Patients
6.2. Ozanimod—A safer COVID-19 Alternative S1PR Therapy
6.3. Opaganib—A SphK2 Specific Inhibitor in COVID-19 Therapy
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Shereen, M.A.; Khan, S.; Kazmi, A.; Bashir, N.; Siddique, R. COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020, 24, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Homes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Xia, S.; Ying, T.; Lu, L. A novel coronavirus (2019-nCoV) causing pneumonia-associated respiratory syndrome. Cell. Mol. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.; Mehra, M.R.; Scholkmann, F.; Schupbach, R.; Ruschitzka, F.; et al. Electron microscopy of SARS-CoV-2: A challenging task—Authors’ reply. Lancet 2020, 395, e100. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb. Res. 2020, 191, 148–150. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef]
- Willyard, C. Coronavirus blood-clot mystery intensifies. Nature 2020, 581, 250. [Google Scholar] [CrossRef]
- Reichard, R.R.; Kashani, K.B.; Boire, N.A.; Constantopoulos, E.; Guo, Y.; Lucchinetti, C.F. Neuropathology of COVID-19: A spectrum of vascular and acute disseminated encephalomyelitis (ADEM)-like pathology. Acta Neuropathol. 2020, 140, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Varatharaj, A.; Thomas, N.; Ellul, M.A.; Davies, N.W.S.; Pollak, T.A.; Tenorio, E.L.; Sultan, M.; Easton, A.; Breen, G.; Zandi, M.; et al. Neurological and neuropsychiatric complications of COVID-19 in 153 patients: A UK-wide surveillance study. Lancet Psychiatry 2020. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; Hlh Across Speciality Collaboration, U.K. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Stebbing, J.; Phelan, A.; Griffin, I.; Tucker, C.; Oechsle, O.; Smith, D.; Richardson, P. COVID-19: Combining antiviral and anti-inflammatory treatments. Lancet Infect. Dis. 2020, 20, 400–402. [Google Scholar] [CrossRef]
- Gaborit, B.J.; Bergmann, J.F.; Mussini, C.; Arribas, J.R.; Behrens, G.; Walmsley, S.; Pozniak, A.; Raffi, F. Plea for multitargeted interventions for severe COVID-19. Lancet Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Xiong, Y.; Hla, T. S1P control of endothelial integrity. Curr. Top. Microbiol. Immunol. 2014, 378, 85–105. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, S.; Milstien, S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011, 11, 403–415. [Google Scholar] [CrossRef]
- Tyrrell, D.A.; Bynoe, M.L. Cultivation of a Novel Type of Common-Cold Virus in Organ Cultures. Br. Med. J. 1965, 1, 1467–1470. [Google Scholar] [CrossRef] [Green Version]
- Monto, A.S. Medical reviews. Coronaviruses. Yale J. Biol. Med. 1974, 47, 234–251. [Google Scholar]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292 e286. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.R.; Navas-Martin, S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntosh, K. Coronaviruses in the limelight. J. Infect. Dis. 2005, 191, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Tsui, P.T.; Kwok, M.L.; Yuen, H.; Lai, S.T. Severe acute respiratory syndrome: Clinical outcome and prognostic correlates. Emerg. Infect. Dis. 2003, 9, 1064–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Q.H.; Luo, X.D.; Zhang, J.Z.; Su, Q. Current status of severe acute respiratory syndrome in China. World J. Gastroenterol. 2003, 9, 1635–1645. [Google Scholar] [CrossRef]
- Chafekar, A.; Fielding, B.C. MERS-CoV: Understanding the Latest Human Coronavirus Threat. Viruses 2018, 10, 93. [Google Scholar] [CrossRef]
- Mackay, I.M.; Arden, K.E. MERS coronavirus: Diagnostics, epidemiology and transmission. Virol. J. 2015, 12, 222. [Google Scholar] [CrossRef] [Green Version]
- Chowell, G.; Bettencourt, L.M.; Johnson, N.; Alonso, W.J.; Viboud, C. The 1918-1919 influenza pandemic in England and Wales: Spatial patterns in transmissibility and mortality impact. Proc. Biol. Sci. 2008, 275, 501–509. [Google Scholar] [CrossRef] [Green Version]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta Biomed. 2020, 91, 157–160. [Google Scholar] [CrossRef]
- Mahase, E. Covid-19: WHO declares pandemic because of “alarming levels” of spread, severity, and inaction. BMJ 2020, 368, m1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Wu, Q.; Zhang, Z. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr. Biol. 2020, 30, 1346–1351.e1342. [Google Scholar] [CrossRef] [PubMed]
- Morawska, L.; Cao, J. Airborne transmission of SARS-CoV-2: The world should face the reality. Environ. Int. 2020, 139, 105730. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.C.; Shih, T.P.; Ko, W.C.; Tang, H.J.; Hsueh, P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Zheng, Z.; Peng, F.; Xu, B.; Zhao, J.; Liu, H.; Peng, J.; Li, Q.; Jiang, C.; Zhou, Y.; Liu, S.; et al. Risk factors of critical & mortal COVID-19 cases: A systematic literature review and meta-analysis. J. Infect. 2020. [Google Scholar] [CrossRef]
- Zheng, Y.Y.; Ma, Y.T.; Zhang, J.Y.; Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef] [Green Version]
- Zheng, F.; Tang, W.; Li, H.; Huang, Y.X.; Xie, Y.L.; Zhou, Z.G. Clinical characteristics of 161 cases of corona virus disease 2019 (COVID-19) in Changsha. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3404–3410. [Google Scholar] [CrossRef] [PubMed]
- Koralnik, I.J.; Tyler, K.L. COVID-19: A Global Threat to the Nervous System. Ann. Neurol. 2020, 88, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.B.; Zhong, C.K.; Zhang, K.X.; Dong, C.; Peng, H.; Xu, T.; Wang, A.L.; Guo, Z.R.; Zhang, Y.H. [Epidemic trend of corona virus disease 2019 (COVID-19) in mainland China]. Zhonghua Yu Fang Yi Xue Za Zhi 2020, 54, E022. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Chen, J. Pathogenicity and transmissibility of 2019-nCoV-A quick overview and comparison with other emerging viruses. Microbes Infect. 2020, 22, 69–71. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Mentzer, S.J.; Konerding, M.A. Intussusceptive angiogenesis: Expansion and remodeling of microvascular networks. Angiogenesis 2014, 17, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Teuwen, L.A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- South, A.M.; Diz, D.I.; Chappell, M.C. COVID-19, ACE2, and the cardiovascular consequences. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1084–H1090. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.P.; Blet, A.; Smyth, D.; Li, H. The Science Underlying COVID-19: Implications for the Cardiovascular System. Circulation 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, T.J.; Mohiddin, S.A.; Dimarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D’Acquisto, F.; et al. COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Gross, S.; Jahn, C.; Cushman, S.; Bar, C.; Thum, T. SARS-CoV-2 receptor ACE2-dependent implications on the cardiovascular system: From basic science to clinical implications. J. Mol. Cell. Cardiol. 2020. [Google Scholar] [CrossRef]
- Yang, C.; Jin, Z. An Acute Respiratory Infection Runs Into the Most Common Noncommunicable Epidemic-COVID-19 and Cardiovascular Diseases. JAMA Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Chen, C.; Yan, J.T.; Zhou, N.; Zhao, J.P.; Wang, D.W. Analysis of myocardial injury in patients with COVID-19 and association between concomitant cardiovascular diseases and severity of COVID-19. Zhonghua Xin Xue Guan Bing Za Zhi 2020, 48, E008. [Google Scholar] [CrossRef]
- Aggarwal, G.; Cheruiyot, I.; Aggarwal, S.; Wong, J.; Lippi, G.; Lavie, C.J.; Henry, B.M.; Sanchis-Gomar, F. Association of Cardiovascular Disease With Coronavirus Disease 2019 (COVID-19) Severity: A Meta-Analysis. Curr. Probl. Cardiol. 2020, 45, 100617. [Google Scholar] [CrossRef] [PubMed]
- Kow, C.S.; Zaidi, S.T.R.; Hasan, S.S. Cardiovascular Disease and Use of Renin-Angiotensin System Inhibitors in COVID-19. Am. J. Cardiovasc. Drugs 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Premer, C.; Kanelidis, A.J.; Hare, J.M.; Schulman, I.H. Rethinking Endothelial Dysfunction as a Crucial Target in Fighting Heart Failure. Mayo Clin. Proc. Innov. Qual. Outcomes 2019, 3, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Pons, S.; Fodil, S.; Azoulay, E.; Zafrani, L. The vascular endothelium: The cornerstone of organ dysfunction in severe SARS-CoV-2 infection. Crit. Care 2020, 24, 353. [Google Scholar] [CrossRef]
- Flammer, A.J.; Anderson, T.; Celermajer, D.S.; Creager, M.A.; Deanfield, J.; Ganz, P.; Hamburg, N.M.; Luscher, T.F.; Shechter, M.; Taddei, S.; et al. The assessment of endothelial function: From research into clinical practice. Circulation 2012, 126, 753–767. [Google Scholar] [CrossRef]
- Mosleh, W.; Chen, K.; Pfau, S.E.; Vashist, A. Endotheliitis and Endothelial Dysfunction in Patients with COVID-19: Its Role in Thrombosis and Adverse Outcomes. J. Clin. Med. 2020, 9, 1862. [Google Scholar] [CrossRef]
- Steardo, L.; Steardo, L., Jr.; Zorec, R.; Verkhratsky, A. Neuroinfection may contribute to pathophysiology and clinical manifestations of COVID-19. Acta Physiol. 2020, 229, e13473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.C.; Bai, W.Z.; Hashikawa, T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 2020, 92, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020. [Google Scholar] [CrossRef]
- Cavalcanti, D.D.; Raz, E.; Shapiro, M.; Dehkharghani, S.; Yaghi, S.; Lillemoe, K.; Nossek, E.; Torres, J.; Jain, R.; Riina, H.A.; et al. Cerebral Venous Thrombosis Associated with COVID-19. AJNR Am. J. Neuroradiol. 2020, 41, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Poillon, G.; Obadia, M.; Perrin, M.; Savatovsky, J.; Lecler, A. Cerebral venous thrombosis associated with COVID-19 infection: Causality or coincidence? J. Neuroradiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Merkler, A.E.; Parikh, N.S.; Mir, S.; Gupta, A.; Kamel, H.; Lin, E.; Lantos, J.; Schenck, E.J.; Goyal, P.; Bruce, S.S.; et al. Risk of Ischemic Stroke in Patients With Coronavirus Disease 2019 (COVID-19) vs Patients With Influenza. JAMA Neurol. 2020. [Google Scholar] [CrossRef]
- Cohen, M.E.; Eichel, R.; Steiner-Birmanns, B.; Janah, A.; Ioshpa, M.; Bar-Shalom, R.; Paul, J.J.; Gaber, H.; Skrahina, V.; Bornstein, N.M.; et al. A case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol. 2020, 19, 804–805. [Google Scholar] [CrossRef]
- Fifi, J.T.; Mocco, J. COVID-19 related stroke in young individuals. Lancet Neurol. 2020, 19, 713–715. [Google Scholar] [CrossRef]
- Mehta, N.S.; Mytton, O.T.; Mullins, E.W.S.; Fowler, T.A.; Falconer, C.L.; Murphy, O.B.; Langenberg, C.; Jayatunga, W.J.P.; Eddy, D.H.; Nguyen-Van-Tam, J.S. SARS-CoV-2 (COVID-19): What do we know about children? A systematic review. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Ludvigsson, J.F. Systematic review of COVID-19 in children shows milder cases and a better prognosis than adults. Acta Paediatr. 2020, 109, 1088–1095. [Google Scholar] [CrossRef]
- Jones, V.G.; Mills, M.; Suarez, D.; Hogan, C.A.; Yeh, D.; Bradley Segal, J.; Nguyen, E.L.; Barsh, G.R.; Maskatia, S.; Mathew, R. COVID-19 and Kawasaki Disease: Novel Virus and Novel Case. Hosp. Pediatr. 2020. [Google Scholar] [CrossRef] [PubMed]
- Viner, R.M.; Whittaker, E. Kawasaki-like disease: Emerging complication during the COVID-19 pandemic. Lancet 2020. [Google Scholar] [CrossRef]
- Esper, F.; Shapiro, E.D.; Weibel, C.; Ferguson, D.; Landry, M.L.; Kahn, J.S. Association between a novel human coronavirus and Kawasaki disease. J. Infect. Dis. 2005, 191, 499–502. [Google Scholar] [CrossRef]
- Chang, L.Y.; Chiang, B.L.; Kao, C.L.; Wu, M.H.; Chen, P.J.; Berkhout, B.; Yang, H.C.; Huang, L.M.; Kawasaki Disease Research, G. Lack of association between infection with a novel human coronavirus (HCoV), HCoV-NH, and Kawasaki disease in Taiwan. J. Infect. Dis. 2006, 193, 283–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belay, E.D.; Erdman, D.D.; Anderson, L.J.; Peret, T.C.; Schrag, S.J.; Fields, B.S.; Burns, J.C.; Schonberger, L.B. Kawasaki disease and human coronavirus. J. Infect. Dis. 2005, 192, 352–353. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Chovatiya, R.; Medzhitov, R. Stress, inflammation, and defense of homeostasis. Mol. Cell 2014, 54, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Vaninov, N. In the eye of the COVID-19 cytokine storm. Nat. Rev. Immunol. 2020, 20, 277. [Google Scholar] [CrossRef]
- Kuppalli, K.; Rasmussen, A.L. A glimpse into the eye of the COVID-19 cytokine storm. EBioMedicine 2020, 55, 102789. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.; Lin, Q.; Jin, S.; You, L. Coronavirus 2019-nCoV: A brief perspective from the front line. J. Infect. 2020, 80, 373–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, T.T.; Cramer, J.P.; Chen, R.; Mayhew, S. Evolution of the COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yamey, G.; Schaferhoff, M.; Hatchett, R.; Pate, M.; Zhao, F.; McDade, K.K. Ensuring global access to COVID-19 vaccines. Lancet 2020, 395, 1405–1406. [Google Scholar] [CrossRef]
- Lundstrom, K. Coronavirus Pandemic-Therapy and Vaccines. Biomedicines 2020, 8, 109. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Bodiga, S.; Das, S.K.; Lo, J.; Patel, V.; Oudit, G.Y. Role of ACE2 in diastolic and systolic heart failure. Heart Fail. Rev. 2012, 17, 683–691. [Google Scholar] [CrossRef]
- Young, P.R. Disease X ver1.0: COVID-19. Microbiol. Aust. 2020, 41, 109–112. [Google Scholar] [CrossRef]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.X.; et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020, 382, 2327–2336. [Google Scholar] [CrossRef]
- Liang, C.; Tian, L.; Liu, Y.; Hui, N.; Qiao, G.; Li, H.; Shi, Z.; Tang, Y.; Zhang, D.; Xie, X.; et al. A promising antiviral candidate drug for the COVID-19 pandemic: A mini-review of remdesivir. Eur. J. Med. Chem. 2020, 201, 112527. [Google Scholar] [CrossRef]
- Zampino, R.; Mele, F.; Florio, L.L.; Bertolino, L.; Andini, R.; Galdo, M.; De Rosa, R.; Corcione, A.; Durante-Mangoni, E. Liver injury in remdesivir-treated COVID-19 patients. Hepatol. Int. 2020. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov 2019, 18, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; Gazzaniga, V.; Bragazzi, N.L.; Barberis, I. The Spanish Influenza Pandemic: A lesson from history 100 years after 1918. J. Prev. Med. Hyg. 2019, 60, E64–E67. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.C.; Goh, H.P.; Koh, D. Treatment of COVID-19: Old tricks for new challenges. Crit. Care 2020, 24, 91. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.C.H.; Sewa, D.W.; Phua, G.C. Potential role of statins in COVID-19. Int. J. Infect. Dis. 2020, 96, 615–617. [Google Scholar] [CrossRef] [PubMed]
- Bifulco, M.; Gazzerro, P. Statin therapy in COVID-19 infection: Much more than a single pathway. Eur. Heart J. Cardiovasc. Pharmacother. 2020. [Google Scholar] [CrossRef]
- Castiglione, V.; Chiriaco, M.; Emdin, M.; Taddei, S.; Vergaro, G. Statin therapy in COVID-19 infection. Eur. Heart J. Cardiovasc. Pharmacother. 2020, 6, 258–259. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: Signaling inside and out. FEBS Lett 2000, 476, 55–57. [Google Scholar] [CrossRef] [Green Version]
- Takabe, K.; Paugh, S.W.; Milstien, S.; Spiegel, S. “Inside-out” signaling of sphingosine-1-phosphate: Therapeutic targets. Pharmacol. Rev. 2008, 60, 181–195. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, S.E.; Milstien, S.; Spiegel, S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol. Metab. 2007, 18, 300–307. [Google Scholar] [CrossRef]
- Schneider-Schaulies, J.; Schneider-Schaulies, S. Sphingolipids in viral infection. Biol. Chem. 2015, 396, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Hatoum, D.; Haddadi, N.; Lin, Y.; Nassif, N.T.; McGowan, E.M. Mammalian sphingosine kinase (SphK) isoenzymes and isoform expression: Challenges for SphK as an oncotarget. Oncotarget 2017, 8, 36898–36929. [Google Scholar] [CrossRef] [Green Version]
- Haddadi, N.; Lin, Y.; Simpson, A.M.; Nassif, N.T.; McGowan, E.M. “Dicing and Splicing” Sphingosine Kinase and Relevance to Cancer. Int. J. Mol. Sci. 2017, 18, 1891. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, K.; Thangada, S.; Michaud, J.; Oo, M.L.; Ai, Y.; Lee, Y.M.; Wu, M.; Parikh, N.S.; Khan, F.; Proia, R.L.; et al. Extracellular export of sphingosine kinase-1a contributes to the vascular S1P gradient. Biochem. J. 2006, 397, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ancellin, N.; Colmont, C.; Su, J.; Li, Q.; Mittereder, N.; Chae, S.S.; Stefansson, S.; Liau, G.; Hla, T. Extracellular export of sphingosine kinase-1 enzyme. Sphingosine 1-phosphate generation and the induction of angiogenic vascular maturation. J. Biol. Chem. 2002, 277, 6667–6675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takuwa, Y.; Takuwa, N.; Sugimoto, N. The Edg family G protein-coupled receptors for lysophospholipids: Their signaling properties and biological activities. J. Biochem. 2002, 131, 767–771. [Google Scholar] [CrossRef]
- Hla, T.; Lee, M.J.; Ancellin, N.; Thangada, S.; Liu, C.H.; Kluk, M.; Chae, S.S.; Wu, M.T. Sphingosine-1-phosphate signaling via the EDG-1 family of G-protein-coupled receptors. Ann. N. Y. Acad. Sci. 2000, 905, 16–24. [Google Scholar] [CrossRef]
- Spiegel, S. Sphingosine 1-phosphate: A ligand for the EDG-1 family of G-protein-coupled receptors. Ann. N. Y. Acad. Sci. 2000, 905, 54–60. [Google Scholar] [CrossRef]
- Hla, T.; Brinkmann, V. Sphingosine 1-phosphate (S1P): Physiology and the effects of S1P receptor modulation. Neurology 2011, 76, S3–S8. [Google Scholar] [CrossRef]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366. [Google Scholar] [CrossRef]
- Blaho, V.A.; Hla, T. An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 2014, 55, 1596–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelson, K.; Evans, T.; Hla, T. Sphingosine 1-phosphate signalling. Development 2014, 141, 5–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, M.; Mi, Y.; Liu, Y.; Sasaki, T.; Allende, M.L.; Wu, Y.P.; Yamashita, T.; Proia, R.L. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J. Biol. Chem. 2004, 279, 29367–29373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obinata, H.; Hla, T. Sphingosine 1-phosphate and inflammation. Int. Immunol. 2019, 31, 617–625. [Google Scholar] [CrossRef]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Bryan, A.M.; Del Poeta, M. Sphingosine-1-phosphate receptors and innate immunity. Cell. Microbiol. 2018, 20, e12836. [Google Scholar] [CrossRef] [Green Version]
- Maceyka, M.; Sankala, H.; Hait, N.C.; Le Stunff, H.; Liu, H.; Toman, R.; Collier, C.; Zhang, M.; Satin, L.S.; Merrill, A.H., Jr.; et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J. Biol. Chem. 2005, 280, 37118–37129. [Google Scholar] [CrossRef] [Green Version]
- Lai, W.Q.; Irwan, A.W.; Goh, H.H.; Howe, H.S.; Yu, D.T.; Valle-Onate, R.; McInnes, I.B.; Melendez, A.J.; Leung, B.P. Anti-inflammatory effects of sphingosine kinase modulation in inflammatory arthritis. J. Immunol. 2008, 181, 8010–8017. [Google Scholar] [CrossRef] [Green Version]
- Michaud, J.; Kohno, M.; Proia, R.L.; Hla, T. Normal acute and chronic inflammatory responses in sphingosine kinase 1 knockout mice. FEBS Lett. 2006, 580, 4607–4612. [Google Scholar] [CrossRef] [Green Version]
- Pyne, N.J.; Adams, D.R.; Pyne, S. Sphingosine Kinase 2 in Autoimmune/Inflammatory Disease and the Development of Sphingosine Kinase 2 Inhibitors. Trends Pharmacol. Sci. 2017, 38, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizugishi, K.; Yamashita, T.; Olivera, A.; Miller, G.F.; Spiegel, S.; Proia, R.L. Essential role for sphingosine kinases in neural and vascular development. Mol. Cell Biol 2005, 25, 11113–11121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, L.F. Immune Response, Inflammation, and the Clinical Spectrum of COVID-19. Front. Immunol. 2020, 11, 1441. [Google Scholar] [CrossRef] [PubMed]
- McFadden, G.; Mohamed, M.R.; Rahman, M.M.; Bartee, E. Cytokine determinants of viral tropism. Nat. Rev. Immunol. 2009, 9, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Gamble, J.R.; Rye, K.A.; Wang, L.; Hii, C.S.; Cockerill, P.; Khew-Goodall, Y.; Bert, A.G.; Barter, P.J.; Vadas, M.A. Tumor necrosis factor-alpha induces adhesion molecule expression through the sphingosine kinase pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 14196–14201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, P.; Wang, L.; Moretti, P.A.; Albanese, N.; Chai, F.; Pitson, S.M.; D’Andrea, R.J.; Gamble, J.R.; Vadas, M.A. Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J. Biol. Chem. 2002, 277, 7996–8003. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef] [Green Version]
- Pchejetski, D.; Bohler, T.; Stebbing, J.; Waxman, J. Therapeutic potential of targeting sphingosine kinase 1 in prostate cancer. Nat. Rev. Urol. 2011, 8, 569–678. [Google Scholar] [CrossRef]
- Ebenezer, D.L.; Fu, P.; Suryadevara, V.; Zhao, Y.; Natarajan, V. Epigenetic regulation of pro-inflammatory cytokine secretion by sphingosine 1-phosphate (S1P) in acute lung injury: Role of S1P lyase. Adv. Biol. Regul. 2017, 63, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Ihlefeld, K.; Claas, R.F.; Koch, A.; Pfeilschifter, J.M.; Meyer Zu Heringdorf, D. Evidence for a link between histone deacetylation and Ca(2)+ homoeostasis in sphingosine-1-phosphate lyase-deficient fibroblasts. Biochem. J. 2012, 447, 457–464. [Google Scholar] [CrossRef] [Green Version]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigert, A.; von Knethen, A.; Thomas, D.; Faria, I.; Namgaladze, D.; Zezina, E.; Fuhrmann, D.; Petcherski, A.; Heringdorf, D.M.Z.; Radeke, H.H.; et al. Sphingosine kinase 2 is a negative regulator of inflammatory macrophage activation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Ji, C.; Zhou, Y.; Huang, W.; Ni, W.; Tong, X.; Wei, J.F. Sphingosine kinase inhibitors: A patent review. Int. J. Mol. Med. 2018, 41, 2450–2460. [Google Scholar] [CrossRef] [PubMed]
- Haass, N.K.; Nassif, N.; McGowan, E.M. Switching the sphingolipid rheostat in the treatment of diabetes and cancer comorbidity from a problem to an advantage. Biomed. Res. Int. 2015, 2015, 165105. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef]
- Saddoughi, S.A.; Gencer, S.; Peterson, Y.K.; Ward, K.E.; Mukhopadhyay, A.; Oaks, J.; Bielawski, J.; Szulc, Z.M.; Thomas, R.J.; Selvam, S.P.; et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol. Med. 2013, 5, 105–121. [Google Scholar] [CrossRef]
- Mandala, S.; Hajdu, R.; Bergstrom, J.; Quackenbush, E.; Xie, J.; Milligan, J.; Thornton, R.; Shei, G.J.; Card, D.; Keohane, C.; et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 2002, 296, 346–349. [Google Scholar] [CrossRef]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. Available online: http://0-www-nature-com.brum.beds.ac.uk/nm/journal/v16/n12/abs/nm.2250.html#supplementary-information (accessed on 21 November 2010). [CrossRef] [Green Version]
- Scuto, A.; Kujawski, M.; Kowolik, C.; Krymskaya, L.; Wang, L.; Weiss, L.M.; Digiusto, D.; Yu, H.; Forman, S.; Jove, R. STAT3 inhibition is a therapeutic strategy for ABC-like diffuse large B-cell lymphoma. Cancer Res. 2011, 71, 3182–3188. [Google Scholar] [CrossRef] [Green Version]
- Nishi, T.; Miyazaki, S.; Takemoto, T.; Suzuki, K.; Iio, Y.; Nakajima, K.; Ohnuki, T.; Kawase, Y.; Nara, F.; Inaba, S.; et al. Discovery of CS-0777: A Potent, Selective, and Orally Active S1P1 Agonist. ACS Med. Chem. Lett. 2011, 2, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Pitman, M.R.; Costabile, M.; Pitson, S.M. Recent advances in the development of sphingosine kinase inhibitors. Cell. Signal. 2016, 28, 1349–1363. [Google Scholar] [CrossRef] [PubMed]
- Sanada, Y.; Mizushima, T.; Kai, Y.; Nishimura, J.; Hagiya, H.; Kurata, H.; Mizuno, H.; Uejima, E.; Ito, T. Therapeutic effects of novel sphingosine-1-phosphate receptor agonist W-061 in murine DSS colitis. PLoS ONE 2011, 6, e23933. [Google Scholar] [CrossRef] [PubMed]
- Komiya, T.; Sato, K.; Shioya, H.; Inagaki, Y.; Hagiya, H.; Kozaki, R.; Imai, M.; Takada, Y.; Maeda, T.; Kurata, H.; et al. Efficacy and immunomodulatory actions of ONO-4641, a novel selective agonist for sphingosine 1-phosphate receptors 1 and 5, in preclinical models of multiple sclerosis. Clin. Exp. Immunol. 2013, 171, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gergely, P.; Nuesslein-Hildesheim, B.; Guerini, D.; Brinkmann, V.; Traebert, M.; Bruns, C.; Pan, S.; Gray, N.S.; Hinterding, K.; Cooke, N.G.; et al. The selective sphingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br. J. Pharmacol. 2012, 167, 1035–1047. [Google Scholar] [CrossRef]
- Lien, Y.H.; Yong, K.C.; Cho, C.; Igarashi, S.; Lai, L.W. S1P(1)-selective agonist, SEW2871, ameliorates ischemic acute renal failure. Kidney Int. 2006, 69, 1601–1608. [Google Scholar] [CrossRef] [Green Version]
- Jo, E.; Sanna, M.G.; Gonzalez-Cabrera, P.J.; Thangada, S.; Tigyi, G.; Osborne, D.A.; Hla, T.; Parrill, A.L.; Rosen, H. S1P1-selective in vivo-active agonists from high-throughput screening: Off-the-shelf chemical probes of receptor interactions, signaling, and fate. Chem. Biol. 2005, 12, 703–715. [Google Scholar] [CrossRef] [Green Version]
- Pepin, E.; Jalinier, T.; Lemieux, G.L.; Massicotte, G.; Cyr, M. Sphingosine-1-Phosphate Receptors Modulators Decrease Signs of Neuroinflammation and Prevent Parkinson’s Disease Symptoms in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model. Front. Pharmacol. 2020, 11, 77. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Y.; Zhang, Z.; Zug, C.; Nuesslein-Hildesheim, B.; Leppert, D.; Schluesener, H.J. AUY954, a selective S1P(1) modulator, prevents experimental autoimmune neuritis. J. Neuroimmunol. 2009, 216, 59–65. [Google Scholar] [CrossRef]
- Galicia-Rosas, G.; Pikor, N.; Schwartz, J.A.; Rojas, O.; Jian, A.; Summers-Deluca, L.; Ostrowski, M.; Nuesslein-Hildesheim, B.; Gommerman, J.L. A sphingosine-1-phosphate receptor 1-directed agonist reduces central nervous system inflammation in a plasmacytoid dendritic cell-dependent manner. J. Immunol. 2012, 189, 3700–3706. [Google Scholar] [CrossRef] [Green Version]
- Sandborn, W.J.; Peyrin-Biroulet, L.; Zhang, J.; Chiorean, M.; Vermeire, S.; Lee, S.D.; Kühbacher, T.; Yacyshyn, B.; Cabell, C.H.; Naik, S.U.; et al. Efficacy and Safety of Etrasimod in a Phase 2 Randomized Trial of Patients With Ulcerative Colitis. Gastroenterology 2020, 158, 550–561. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, R.; Aoki, T.; Koseki, H.; Fukuda, M.; Hirose, J.; Tsuji, K.; Takizawa, K.; Nakamura, S.; Miyata, H.; Hamakawa, N.; et al. A sphingosine-1-phosphate receptor type 1 agonist, ASP4058, suppresses intracranial aneurysm through promoting endothelial integrity and blocking macrophage transmigration. Br. J. Pharmacol. 2017, 174, 2085–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, R.; Okada, Y.; Hirose, J.; Koshika, T.; Kawato, Y.; Maeda, M.; Saito, R.; Hattori, K.; Harada, H.; Nagasaka, Y.; et al. ASP4058, a novel agonist for sphingosine 1-phosphate receptors 1 and 5, ameliorates rodent experimental autoimmune encephalomyelitis with a favorable safety profile. PLoS ONE 2014, 9, e110819. [Google Scholar] [CrossRef] [PubMed]
- Poti, F.; Costa, S.; Bergonzini, V.; Galletti, M.; Pignatti, E.; Weber, C.; Simoni, M.; Nofer, J.R. Effect of sphingosine 1-phosphate (S1P) receptor agonists FTY720 and CYM5442 on atherosclerosis development in LDL receptor deficient (LDL-R(-)/(-)) mice. Vasc. Pharmacol. 2012, 57, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Takahashi, M.; Kaneko, T.; Murakami, T.; Hakamata, Y.; Kudou, S.; Kishi, T.; Fukuchi, K.; Iwanami, S.; Kuriyama, K.; et al. KRP-203, a novel synthetic immunosuppressant, prolongs graft survival and attenuates chronic rejection in rat skin and heart allografts. Circulation 2005, 111, 222–229. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Davis, M.D.; Heise, C.E.; Albert, R.; Cottens, S.; Hof, R.; Bruns, C.; Prieschl, E.; Baumruker, T.; Hiestand, P.; et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 2002, 277, 21453–21457. [Google Scholar] [CrossRef] [Green Version]
- Marsolais, D.; Hahm, B.; Walsh, K.B.; Edelmann, K.H.; McGavern, D.; Hatta, Y.; Kawaoka, Y.; Rosen, H.; Oldstone, M.B. A critical role for the sphingosine analog AAL-R in dampening the cytokine response during influenza virus infection. Proc. Natl. Acad. Sci. USA 2009, 106, 1560–1565. [Google Scholar] [CrossRef] [Green Version]
- Mathews, T.P.; Kennedy, A.J.; Kharel, Y.; Kennedy, P.C.; Nicoara, O.; Sunkara, M.; Morris, A.J.; Wamhoff, B.R.; Lynch, K.R.; Macdonald, T.L. Discovery, biological evaluation, and structure-activity relationship of amidine based sphingosine kinase inhibitors. J. Med. Chem. 2010, 53, 2766–2778. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Cabrera, P.J.; Jo, E.; Sanna, M.G.; Brown, S.; Leaf, N.; Marsolais, D.; Schaeffer, M.T.; Chapman, J.; Cameron, M.; Guerrero, M.; et al. Full pharmacological efficacy of a novel S1P1 agonist that does not require S1P-like headgroup interactions. Mol. Pharmacol. 2008, 74, 1308–1318. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Cabrera, P.J.; Cahalan, S.M.; Nguyen, N.; Sarkisyan, G.; Leaf, N.B.; Cameron, M.D.; Kago, T.; Rosen, H. S1P(1) receptor modulation with cyclical recovery from lymphopenia ameliorates mouse model of multiple sclerosis. Mol. Pharmacol. 2012, 81, 166–174. [Google Scholar] [CrossRef] [Green Version]
- Clemens, J.J.; Davis, M.D.; Lynch, K.R.; Macdonald, T.L. Synthesis of benzimidazole based analogues of sphingosine-1-phosphate: Discovery of potent, subtype-selective S1P4 receptor agonists. Bioorg. Med. Chem. Lett. 2004, 14, 4903–4906. [Google Scholar] [CrossRef]
- Ota, H.; Beutz, M.A.; Ito, M.; Abe, K.; Oka, M.; McMurtry, I.F. S1P(4) receptor mediates S1P-induced vasoconstriction in normotensive and hypertensive rat lungs. Pulm. Circ. 2011, 1, 399–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Hagiya, H.; Kurata, H.; Mizuno, H.; Ito, T. Prevention of GVHD and graft rejection by a new S1P receptor agonist, W-061, in rat small bowel transplantation. Transplant. Immunol. 2012, 26, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Piali, L.; Birker-Robaczewska, M.; Lescop, C.; Froidevaux, S.; Schmitz, N.; Morrison, K.; Kohl, C.; Rey, M.; Studer, R.; Vezzali, E.; et al. Cenerimod, a novel selective S1P(1) receptor modulator with unique signaling properties. Pharmacol. Res. Perspect. 2017, 5, e00370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herr, D.R.; Reolo, M.J.; Peh, Y.X.; Wang, W.; Lee, C.W.; Rivera, R.; Paterson, I.C.; Chun, J. Sphingosine 1-phosphate receptor 2 (S1P2) attenuates reactive oxygen species formation and inhibits cell death: Implications for otoprotective therapy. Sci. Rep. 2016, 6, 24541. [Google Scholar] [CrossRef] [Green Version]
- Waters, C.M.; Long, J.; Gorshkova, I.; Fujiwara, Y.; Connell, M.; Belmonte, K.E.; Tigyi, G.; Natarajan, V.; Pyne, S.; Pyne, N.J. Cell migration activated by platelet-derived growth factor receptor is blocked by an inverse agonist of the sphingosine 1-phosphate receptor-1. FASEB J. 2006, 20, 509–511. [Google Scholar] [CrossRef] [Green Version]
- Pyne, N.; Pyne, S. Selectivity and Specificity of Sphingosine 1-Phosphate Receptor Ligands: “Off-Targets” or Complex Pharmacology? Front. Pharmacol. 2011, 2. [Google Scholar] [CrossRef] [Green Version]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate, lysophosphatidic acid and growth factor signaling and termination. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2008, 1781, 467–476. [Google Scholar] [CrossRef]
- Davis, M.D.; Clemens, J.J.; Macdonald, T.L.; Lynch, K.R. Sphingosine 1-phosphate analogs as receptor antagonists. J. Biol. Chem. 2005, 280, 9833–9841. [Google Scholar] [CrossRef] [Green Version]
- Fujii, Y.; Hirayama, T.; Ohtake, H.; Ono, N.; Inoue, T.; Sakurai, T.; Takayama, T.; Matsumoto, K.; Tsukahara, N.; Hidano, S.; et al. Amelioration of collagen-induced arthritis by a novel S1P1 antagonist with immunomodulatory activities. J. Immunol. 2012, 188, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.K.; Drabkin, H.A.; Reeves, J.A.; Hainsworth, J.D.; Hazel, S.E.; Paggiarino, D.A.; Wojciak, J.; Woodnutt, G.; Bhatt, R.S. A phase 2 study of the sphingosine-1-phosphate antibody sonepcizumab in patients with metastatic renal cell carcinoma. Cancer 2017, 123, 576–582. [Google Scholar] [CrossRef]
- Quancard, J.; Bollbuck, B.; Janser, P.; Angst, D.; Berst, F.; Buehlmayer, P.; Streiff, M.; Beerli, C.; Brinkmann, V.; Guerini, D.; et al. A potent and selective S1P(1) antagonist with efficacy in experimental autoimmune encephalomyelitis. Chem. Biol. 2012, 19, 1142–1151. [Google Scholar] [CrossRef] [Green Version]
- Tarrason, G.; Auli, M.; Mustafa, S.; Dolgachev, V.; Domenech, M.T.; Prats, N.; Dominguez, M.; Lopez, R.; Aguilar, N.; Calbet, M.; et al. The sphingosine-1-phosphate receptor-1 antagonist, W146, causes early and short-lasting peripheral blood lymphopenia in mice. Int. Immunopharmacol. 2011, 11, 1773–1779. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Satoh, H.; Yanase, M.; Inoue, Y.; Tomiya, T.; Arai, M.; Tejima, K.; Nagashima, K.; Maekawa, H.; Yahagi, N.; et al. Antiproliferative property of sphingosine 1-phosphate in rat hepatocytes involves activation of Rho via Edg-5. Gastroenterology 2003, 124, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Skoura, A.; Michaud, J.; Im, D.S.; Thangada, S.; Xiong, Y.; Smith, J.D.; Hla, T. Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 81–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunkel, G.T.; Maceyka, M.; Milstien, S.; Spiegel, S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discov. 2013, 12, 688–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyne, N.J.; McNaughton, M.; Boomkamp, S.; MacRitchie, N.; Evangelisti, C.; Martelli, A.M.; Jiang, H.R.; Ubhi, S.; Pyne, S. Role of sphingosine 1-phosphate receptors, sphingosine kinases and sphingosine in cancer and inflammation. Adv. Biol. Regul. 2016, 60, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Pyne, S.; Adams, D.R.; Pyne, N.J. Sphingosine 1-phosphate and sphingosine kinases in health and disease: Recent advances. Prog Lipid Res. 2016, 62, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Stepanovska, B.; Huwiler, A. Targeting the S1P receptor signaling pathways as a promising approach for treatment of autoimmune and inflammatory diseases. Pharmacol. Res. 2020, 154, 104170. [Google Scholar] [CrossRef]
- Gao, P.; Peterson, Y.K.; Smith, R.A.; Smith, C.D. Characterization of isoenzyme-selective inhibitors of human sphingosine kinases. PLoS ONE 2012, 7, e44543. [Google Scholar] [CrossRef]
- Neubauer, H.A.; Pham, D.H.; Zebol, J.R.; Moretti, P.A.; Peterson, A.L.; Leclercq, T.M.; Chan, H.; Powell, J.A.; Pitman, M.R.; Samuel, M.S.; et al. An oncogenic role for sphingosine kinase 2. Oncotarget 2016. [Google Scholar] [CrossRef]
- Dickson, M.A.; Carvajal, R.D.; Merrill, A.H., Jr.; Gonen, M.; Cane, L.M.; Schwartz, G.K. A phase I clinical trial of safingol in combination with cisplatin in advanced solid tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 2484–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, A.; Casas, J.; Llebaria, A.; Abad, J.L.; Fabrias, G. Inhibitors of sphingolipid metabolism enzymes. Biochim. Biophys. Acta 2006, 1758, 1957–1977. [Google Scholar] [CrossRef] [PubMed]
- Lynch, K.R.; Thorpe, S.B.; Santos, W.L. Sphingosine kinase inhibitors: A review of patent literature (2006–2015). Expert Opin. Ther. Pat. 2016, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, D.; Weber, J.; Ihlefeld, K.; Bruggerhoff, A.; Proschak, E.; Stark, H. Design, synthesis and evaluation of 2-aminothiazole derivatives as sphingosine kinase inhibitors. Bioorg. Med. Chem. 2014, 22, 5354–5367. [Google Scholar] [CrossRef]
- French, K.J.; Upson, J.J.; Keller, S.N.; Zhuang, Y.; Yun, J.K.; Smith, C.D. Antitumor activity of sphingosine kinase inhibitors. J. Pharmacol. Exp. Ther. 2006, 318, 596–603. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.F.; Gao, Z.H.; Liu, H.P.; Yuan, Y.; Qu, X.J. Sphingosine 1-phosphate antagonizes the effect of all-trans retinoic acid (ATRA) in a human colon cancer cell line by modulation of RARbeta expression. Cancer Lett. 2012, 319, 182–189. [Google Scholar] [CrossRef]
- Gustin, D.J.; Li, Y.; Brown, M.L.; Min, X.; Schmitt, M.J.; Wanska, M.; Wang, X.; Connors, R.; Johnstone, S.; Cardozo, M.; et al. Structure guided design of a series of sphingosine kinase (SphK) inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 4608–4616. [Google Scholar] [CrossRef]
- Kapitonov, D.; Allegood, J.C.; Mitchell, C.; Hait, N.C.; Almenara, J.A.; Adams, J.K.; Zipkin, R.E.; Dent, P.; Kordula, T.; Milstien, S.; et al. Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res. 2009, 69, 6915–6923. [Google Scholar] [CrossRef] [Green Version]
- Nagahashi, M.; Ramachandran, S.; Kim, E.Y.; Allegood, J.C.; Rashid, O.M.; Yamada, A.; Zhao, R.; Milstien, S.; Zhou, H.; Spiegel, S.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 1 promotes breast cancer progression by stimulating angiogenesis and lymphangiogenesis. Cancer Res. 2012, 72, 726–735. [Google Scholar] [CrossRef] [Green Version]
- Paugh, S.W.; Paugh, B.S.; Rahmani, M.; Kapitonov, D.; Almenara, J.A.; Kordula, T.; Milstien, S.; Adams, J.K.; Zipkin, R.E.; Grant, S.; et al. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood 2008, 112, 1382–1391. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.K. Sphingo-guanidines and their use as inhibitors of sphingosine kinase (WO2010078247). Expert Opin. Ther. Pat. 2011, 21, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Hirth, B.; Kane, J.L., Jr.; Liao, J.; Noson, K.D.; Yee, C.; Asmussen, G.; Fitzgerald, M.; Klaus, C.; Booker, M. Discovery of novel sphingosine kinase-1 inhibitors. Part 2. Bioorg. Med. Chem. Lett. 2010, 20, 4550–4554. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.G.; Gray, A.I.; Pyne, S.; Pyne, N.J. Resveratrol dimers are novel sphingosine kinase 1 inhibitors and affect sphingosine kinase 1 expression and cancer cell growth and survival. Br. J. Pharmacol. 2012, 166, 1605–1616. [Google Scholar] [CrossRef] [Green Version]
- Byun, H.S.; Pyne, S.; Macritchie, N.; Pyne, N.J.; Bittman, R. Novel sphingosine-containing analogues selectively inhibit sphingosine kinase (SK) isozymes, induce SK1 proteasomal degradation and reduce DNA synthesis in human pulmonary arterial smooth muscle cells. Medchemcomm 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Baek, D.J.; MacRitchie, N.; Pyne, N.J.; Pyne, S.; Bittman, R. Synthesis of selective inhibitors of sphingosine kinase 1. Chem. Commun. 2013, 49, 2136–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmonds, Y.; Milstien, S.; Spiegel, S. Development of small-molecule inhibitors of sphingosine-1-phosphate signaling. Pharmacol. Ther. 2011, 132, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Santos, W.L.; Lynch, K.R. Drugging sphingosine kinases. ACS Chem. Biol. 2015, 10, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Okada, H.; Takabatake, R.; Honda, M.; Takegoshi, K.; Yamashita, T.; Nakamura, M.; Shirasaki, T.; Sakai, Y.; Shimakami, T.; Nagata, N.; et al. Peretinoin, an acyclic retinoid, suppresses steatohepatitis and tumorigenesis by activating autophagy in mice fed an atherogenic high-fat diet. Oncotarget 2017, 8, 39978–39993. [Google Scholar] [CrossRef] [Green Version]
- Funaki, M.; Kitabayashi, J.; Shimakami, T.; Nagata, N.; Sakai, Y.; Takegoshi, K.; Okada, H.; Murai, K.; Shirasaki, T.; Oyama, T.; et al. Peretinoin, an acyclic retinoid, inhibits hepatocarcinogenesis by suppressing sphingosine kinase 1 expression in vitro and in vivo. Sci. Rep. 2017, 7, 16978. [Google Scholar] [CrossRef] [Green Version]
- French, K.J.; Zhuang, Y.; Maines, L.W.; Gao, P.; Wang, W.; Beljanski, V.; Upson, J.J.; Green, C.L.; Keller, S.N.; Smith, C.D. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J. Pharmacol. Exp. Ther. 2010, 333, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Kim, Y.W.; Inagaki, Y.; Hwang, Y.A.; Mitsutake, S.; Ryu, Y.W.; Lee, W.K.; Ha, H.J.; Park, C.S.; Igarashi, Y. Synthesis and evaluation of sphingoid analogs as inhibitors of sphingosine kinases. Bioorg. Med. Chem. 2005, 13, 3475–3485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharel, Y.; Morris, E.A.; Congdon, M.D.; Thorpe, S.B.; Tomsig, J.L.; Santos, W.L.; Lynch, K.R. Sphingosine Kinase 2 Inhibition and Blood Sphingosine 1-Phosphate Levels. J. Pharmacol. Exp. Ther. 2015, 355, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raje, M.R.; Knott, K.; Kharel, Y.; Bissel, P.; Lynch, K.R.; Santos, W.L. Design, synthesis and biological activity of sphingosine kinase 2 selective inhibitors. Bioorg. Med. Chem. 2012, 20, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Yoshimitsu, Y.; Oishi, S.; Miyagaki, J.; Inuki, S.; Ohno, H.; Fujii, N. Pachastrissamine (jaspine B) and its stereoisomers inhibit sphingosine kinases and atypical protein kinase C. Bioorg. Med. Chem. 2011, 19, 5402–5408. [Google Scholar] [CrossRef] [Green Version]
- Aurelio, L.; Scullino, C.V.; Pitman, M.R.; Sexton, A.; Oliver, V.; Davies, L.; Rebello, R.J.; Furic, L.; Creek, D.J.; Pitson, S.M.; et al. From Sphingosine Kinase to Dihydroceramide Desaturase: A Structure-Activity Relationship (SAR) Study of the Enzyme Inhibitory and Anticancer Activity of 4-((4-(4-Chlorophenyl)thiazol-2-yl)amino)phenol (SKI-II). J. Med. Chem 2016, 59, 965–984. [Google Scholar] [CrossRef]
- Pitson, S.M.; Moretti, P.A.; Zebol, J.R.; Lynn, H.E.; Xia, P.; Vadas, M.A.; Wattenberg, B.W. Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. EMBO J. 2003, 22, 5491–5500. [Google Scholar] [CrossRef] [Green Version]
- Jarman, K.E.; Moretti, P.A.; Zebol, J.R.; Pitson, S.M. Translocation of sphingosine kinase 1 to the plasma membrane is mediated by calcium- and integrin-binding protein 1. J. Biol. Chem. 2010, 285, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Wilkerson, B.A.; Grass, G.D.; Wing, S.B.; Argraves, W.S.; Argraves, K.M. Sphingosine 1-phosphate (S1P) carrier-dependent regulation of endothelial barrier: High density lipoprotein (HDL)-S1P prolongs endothelial barrier enhancement as compared with albumin-S1P via effects on levels, trafficking, and signaling of S1P1. J. Biol. Chem. 2012, 287, 44645–44653. [Google Scholar] [CrossRef] [Green Version]
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnstrom, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B.; et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 2011, 108, 9613–9618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyne, N.J.; Pyne, S. Sphingosine 1-Phosphate Receptor 1 Signaling in Mammalian Cells. Molecules 2017, 22, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, Y.J.; Blake, C.; Alexander, S.; Hahm, B. Sphingosine 1-phosphate-metabolizing enzymes control influenza virus propagation and viral cytopathogenicity. J. Virol. 2010, 84, 8124–8131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, Y.J.; Pritzl, C.J.; Vijayan, M.; Bomb, K.; McClain, M.E.; Alexander, S.; Hahm, B. Sphingosine kinase 1 serves as a pro-viral factor by regulating viral RNA synthesis and nuclear export of viral ribonucleoprotein complex upon influenza virus infection. PLoS ONE 2013, 8, e75005. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, M.; Seo, Y.J.; Pritzl, C.J.; Squires, S.A.; Alexander, S.; Hahm, B. Sphingosine kinase 1 regulates measles virus replication. Virology 2014, 450–451, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, C.; Seo, Y.J.; Studstill, C.J.; Vijayan, M.; Wolf, J.J.; Hahm, B. Transient inhibition of sphingosine kinases confers protection to influenza A virus infected mice. Antivir. Res. 2018, 158, 171–177. [Google Scholar] [CrossRef]
- Yager, E.J.; Konan, K.V. Sphingolipids as Potential Therapeutic Targets against Enveloped Human RNA Viruses. Viruses 2019, 11, 912. [Google Scholar] [CrossRef] [Green Version]
- Bezgovsek, J.; Gulbins, E.; Friedrich, S.K.; Lang, K.S.; Duhan, V. Sphingolipids in early viral replication and innate immune activation. Biol. Chem. 2018, 399, 1115–1123. [Google Scholar] [CrossRef]
- Zhao, J.; Zhu, M.; Jiang, H.; Shen, S.; Su, X.; Shi, Y. Combination of sphingosine-1-phosphate receptor 1 (S1PR1) agonist and antiviral drug: A potential therapy against pathogenic influenza virus. Sci. Rep. 2019, 9, 5272. [Google Scholar] [CrossRef]
- Lu, Z.P.; Xiao, Z.L.; Yang, Z.; Li, J.; Feng, G.X.; Chen, F.Q.; Li, Y.H.; Feng, J.Y.; Gao, Y.E.; Ye, L.H.; et al. Hepatitis B virus X protein promotes human hepatoma cell growth via upregulation of transcription factor AP2alpha and sphingosine kinase 1. Acta Pharmacol. Sin. 2015, 36, 1228–1236. [Google Scholar] [CrossRef] [Green Version]
- Aloia, A.L.; Calvert, J.K.; Clarke, J.N.; Davies, L.T.; Helbig, K.J.; Pitson, S.M.; Carr, J.M. Investigation of sphingosine kinase 1 in interferon responses during dengue virus infection. Clin. Transl. Immunol. 2017, 6, e151. [Google Scholar] [CrossRef]
- Wolf, J.J.; Studstill, C.J.; Hahm, B. Emerging Connections of S1P-Metabolizing Enzymes with Host Defense and Immunity During Virus Infections. Viruses 2019, 11, 1097. [Google Scholar] [CrossRef] [Green Version]
- Pleschka, S.; Wolff, T.; Ehrhardt, C.; Hobom, G.; Planz, O.; Rapp, U.R.; Ludwig, S. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat. Cell Biol. 2001, 3, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Grafen, A.; Schumacher, F.; Chithelen, J.; Kleuser, B.; Beyersdorf, N.; Schneider-Schaulies, J. Use of Acid Ceramidase and Sphingosine Kinase Inhibitors as Antiviral Compounds Against Measles Virus Infection of Lymphocytes in vitro. Front. Cell Dev. Biol. 2019, 7, 218. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.; Qian, C.; Luo, Z.; Li, Q.; Wang, F. Invasive mechanical ventilation in COVID-19 patient management: The experience with 469 patients in Wuhan. Crit. Care 2020, 24, 348. [Google Scholar] [CrossRef] [PubMed]
- Suryadevara, V.; Fu, P.; Ebenezer, D.L.; Berdyshev, E.; Bronova, I.A.; Huang, L.S.; Harijith, A.; Natarajan, V. Sphingolipids in Ventilator Induced Lung Injury: Role of Sphingosine-1-Phosphate Lyase. Int. J. Mol. Sci. 2018, 19, 114. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.S.; Berdyshev, E.V.; Tran, J.T.; Xie, L.; Chen, J.; Ebenezer, D.L.; Mathew, B.; Gorshkova, I.; Zhang, W.; Reddy, S.P.; et al. Sphingosine-1-phosphate lyase is an endogenous suppressor of pulmonary fibrosis: Role of S1P signalling and autophagy. Thorax 2015, 70, 1138–1148. [Google Scholar] [CrossRef] [Green Version]
- Ebenezer, D.L.; Fu, P.; Natarajan, V. Targeting sphingosine-1-phosphate signaling in lung diseases. Pharmacol. Ther. 2016, 168, 143–157. [Google Scholar] [CrossRef] [Green Version]
- Walsh, K.B.; Teijaro, J.R.; Rosen, H.; Oldstone, M.B. Quelling the storm: Utilization of sphingosine-1-phosphate receptor signaling to ameliorate influenza virus-induced cytokine storm. Immunol. Res. 2011, 51, 15–25. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Walsh, K.B.; Long, J.P.; Tordoff, K.P.; Stark, G.V.; Eisfeld, A.J.; Kawaoka, Y.; Rosen, H.; Oldstone, M.B. Protection of ferrets from pulmonary injury due to H1N1 2009 influenza virus infection: Immunopathology tractable by sphingosine-1-phosphate 1 receptor agonist therapy. Virology 2014, 452–453, 152–157. [Google Scholar] [CrossRef] [Green Version]
- De Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.; Chau, T.N.; Hoang, D.M.; Chau, N.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Walsh, K.B.; Cahalan, S.; Fremgen, D.M.; Roberts, E.; Scott, F.; Martinborough, E.; Peach, R.; Oldstone, M.B.; Rosen, H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011, 146, 980–991. [Google Scholar] [CrossRef] [Green Version]
- Naz, F.; Arish, M. Battling COVID-19 Pandemic: Sphingosine-1-Phosphate Analogs as an Adjunctive Therapy? Front. Immunol. 2020, 11, 1102. [Google Scholar] [CrossRef] [PubMed]
- Mehra, M.R.; Ruschitzka, F. COVID-19 Illness and Heart Failure: A Missing Link? JACC Heart Fail. 2020, 8, 512–514. [Google Scholar] [CrossRef] [PubMed]
- Dimasi, D.P.; Pitson, S.M.; Bonder, C.S. Examining the Role of Sphingosine Kinase-2 in the Regulation of Endothelial Cell Barrier Integrity. Microcirculation 2016, 23, 248–265. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wada, R.; Yamashita, T.; Mi, Y.; Deng, C.X.; Hobson, J.P.; Rosenfeldt, H.M.; Nava, V.E.; Chae, S.S.; Lee, M.J.; et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J. Clin. Investig. 2000, 106, 951–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaengel, K.; Niaudet, C.; Hagikura, K.; Lavina, B.; Muhl, L.; Hofmann, J.J.; Ebarasi, L.; Nystrom, S.; Rymo, S.; Chen, L.L.; et al. The sphingosine-1-phosphate receptor S1PR1 restricts sprouting angiogenesis by regulating the interplay between VE-cadherin and VEGFR2. Dev. Cell 2012, 23, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Orban, M.; Lorenz, M.; Barocke, V.; Braun, D.; Urtz, N.; Schulz, C.; von Bruhl, M.L.; Tirniceriu, A.; Gaertner, F.; et al. A novel role of sphingosine 1-phosphate receptor S1pr1 in mouse thrombopoiesis. J. Exp. Med. 2012, 209, 2165–2181. [Google Scholar] [CrossRef] [PubMed]
- Cantalupo, A.; Di Lorenzo, A. S1P Signaling and De Novo Biosynthesis in Blood Pressure Homeostasis. J. Pharmacol. Exp. Ther. 2016, 358, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Takuwa, Y.; Du, W.; Qi, X.; Okamoto, Y.; Takuwa, N.; Yoshioka, K. Roles of sphingosine-1-phosphate signaling in angiogenesis. World J. Biol. Chem. 2010, 1, 298–306. [Google Scholar] [CrossRef]
- Venkataraman, K.; Lee, Y.M.; Michaud, J.; Thangada, S.; Ai, Y.; Bonkovsky, H.L.; Parikh, N.S.; Habrukowich, C.; Hla, T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res. 2008, 102, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.H.; Baluk, P.; Xu, Y.; Grigorova, I.; Bankovich, A.J.; Pappu, R.; Coughlin, S.R.; McDonald, D.M.; Schwab, S.R.; Cyster, J.G. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J. Exp. Med. 2010, 207, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A.; Clare, S.; Hale, C.; Chen, J.; Raisen, C.; Mottram, L.; Lucas, M.; Estabel, J.; Ryder, E.; Adissu, H.; et al. The role of sphingosine-1-phosphate transporter Spns2 in immune system function. J. Immunol. 2012, 189, 102–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tukijan, F.; Chandrakanthan, M.; Nguyen, L.N. The signalling roles of sphingosine-1-phosphate derived from red blood cells and platelets. Br. J. Pharmacol. 2018, 175, 3741–3746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tauseef, M.; Kini, V.; Knezevic, N.; Brannan, M.; Ramchandaran, R.; Fyrst, H.; Saba, J.; Vogel, S.M.; Malik, A.B.; Mehta, D. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ. Res. 2008, 103, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Olesch, C.; Ringel, C.; Brune, B.; Weigert, A. Beyond Immune Cell Migration: The Emerging Role of the Sphingosine-1-phosphate Receptor S1PR4 as a Modulator of Innate Immune Cell Activation. Mediat. Inflamm. 2017, 2017, 6059203. [Google Scholar] [CrossRef] [PubMed]
- Camerer, E.; Regard, J.B.; Cornelissen, I.; Srinivasan, Y.; Duong, D.N.; Palmer, D.; Pham, T.H.; Wong, J.S.; Pappu, R.; Coughlin, S.R. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J. Clin. Investig. 2009, 119, 1871–1879. [Google Scholar] [CrossRef] [Green Version]
- Cartier, A.; Leigh, T.; Liu, C.H.; Hla, T. Endothelial sphingosine 1-phosphate receptors promote vascular normalization and antitumor therapy. Proc. Natl. Acad. Sci. USA 2020, 117, 3157–3166. [Google Scholar] [CrossRef] [Green Version]
- Allende, M.L.; Yamashita, T.; Proia, R.L. G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood 2003, 102, 3665–3667. [Google Scholar] [CrossRef]
- Green, J.A.; Suzuki, K.; Cho, B.; Willison, L.D.; Palmer, D.; Allen, C.D.; Schmidt, T.H.; Xu, Y.; Proia, R.L.; Coughlin, S.R.; et al. The sphingosine 1-phosphate receptor S1P(2) maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat. Immunol. 2011, 12, 672–680. [Google Scholar] [CrossRef]
- Zhao, Y.; Gorshkova, I.A.; Berdyshev, E.; He, D.; Fu, P.; Ma, W.; Su, Y.; Usatyuk, P.V.; Pendyala, S.; Oskouian, B.; et al. Protection of LPS-induced murine acute lung injury by sphingosine-1-phosphate lyase suppression. Am. J. Respir. Cell Mol. Biol. 2011, 45, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Yang, L.; Kim, G.S.; Ryan, K.; Lu, S.; O’Donnell, R.K.; Spokes, K.; Shapiro, N.; Aird, W.C.; Kluk, M.J.; et al. Critical role of sphingosine-1-phosphate receptor 2 (S1PR2) in acute vascular inflammation. Blood 2013, 122, 443–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sammani, S.; Moreno-Vinasco, L.; Mirzapoiazova, T.; Singleton, P.A.; Chiang, E.T.; Evenoski, C.L.; Wang, T.; Mathew, B.; Husain, A.; Moitra, J.; et al. Differential effects of sphingosine 1-phosphate receptors on airway and vascular barrier function in the murine lung. Am. J. Respir. Cell Mol. Biol. 2010, 43, 394–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, E.; Shapiro, D. Sphingosine as an inhibitor of blood clotting. Science 1957, 125, 1041–1042. [Google Scholar] [CrossRef] [PubMed]
- Vardon Bounes, F.; Mujalli, A.; Cenac, C.; Severin, S.; Le Faouder, P.; Chicanne, G.; Gaits-Iacovoni, F.; Minville, V.; Gratacap, M.P.; Payrastre, B. The importance of blood platelet lipid signaling in thrombosis and in sepsis. Adv. Biol. Regul. 2018, 67, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Mahajan-Thakur, S.; Bohm, A.; Jedlitschky, G.; Schror, K.; Rauch, B.H. Sphingosine-1-Phosphate and Its Receptors: A Mutual Link between Blood Coagulation and Inflammation. Mediat. Inflamm. 2015, 2015, 831059. [Google Scholar] [CrossRef] [Green Version]
- Yatomi, Y. Sphingosine 1-phosphate in vascular biology: Possible therapeutic strategies to control vascular diseases. Curr. Pharm. Des. 2006, 12, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Takeya, H.; Gabazza, E.C.; Aoki, S.; Ueno, H.; Suzuki, K. Synergistic effect of sphingosine 1-phosphate on thrombin-induced tissue factor expression in endothelial cells. Blood 2003, 102, 1693–1700. [Google Scholar] [CrossRef]
- Gazit, S.L.; Mariko, B.; Therond, P.; Decouture, B.; Xiong, Y.; Couty, L.; Bonnin, P.; Baudrie, V.; Le Gall, S.M.; Dizier, B.; et al. Platelet and Erythrocyte Sources of S1P Are Redundant for Vascular Development and Homeostasis, but Both Rendered Essential After Plasma S1P Depletion in Anaphylactic Shock. Circ. Res. 2016, 119, e110–e126. [Google Scholar] [CrossRef]
- Winkler, M.S.; Nierhaus, A.; Holzmann, M.; Mudersbach, E.; Bauer, A.; Robbe, L.; Zahrte, C.; Geffken, M.; Peine, S.; Schwedhelm, E.; et al. Decreased serum concentrations of sphingosine-1-phosphate in sepsis. Crit Care 2015, 19, 372. [Google Scholar] [CrossRef] [Green Version]
- Coldewey, S.M.; Benetti, E.; Collino, M.; Pfeilschifter, J.; Sponholz, C.; Bauer, M.; Huwiler, A.; Thiemermann, C. Elevation of serum sphingosine-1-phosphate attenuates impaired cardiac function in experimental sepsis. Sci. Rep. 2016, 6, 27594. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Chen, Q.; Zhang, K.; Cheng, B.; Xie, G.; Wu, X.; Luo, C.; Chen, L.; Liu, H.; Zhao, B.; et al. Sphingosine 1-phosphate Receptor 2 Signaling Suppresses Macrophage Phagocytosis and Impairs Host Defense against Sepsis. Anesthesiology 2015, 123, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Means, C.K.; Brown, J.H. Sphingosine-1-phosphate receptor signalling in the heart. Cardiovasc. Res. 2009, 82, 193–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Xia, Y.; Yan, W.; Zhang, H.; Zhou, F.; Zhao, S.; Wang, W.; Zhu, D.; Xin, C.; Lee, Y.; et al. Sphingosine 1-phosphate signaling contributes to cardiac inflammation, dysfunction, and remodeling following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H250–H261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karunakaran, I.; van Echten-Deckert, G. Sphingosine 1-phosphate - A double edged sword in the brain. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1573–1582. [Google Scholar] [CrossRef]
- Alirezaei, M.; Kemball, C.C.; Whitton, J.L. Autophagy, inflammation and neurodegenerative disease. Eur. J. Neurosci. 2011, 33, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grassi, S.; Mauri, L.; Prioni, S.; Cabitta, L.; Sonnino, S.; Prinetti, A.; Giussani, P. Sphingosine 1-Phosphate Receptors and Metabolic Enzymes as Druggable Targets for Brain Diseases. Front. Pharmacol. 2019, 10, 807. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.S.; Yang, L.; Zhang, G.; Zhao, H.; Selim, M.; McCullough, L.D.; Kluk, M.J.; Sanchez, T. Critical role of sphingosine-1-phosphate receptor-2 in the disruption of cerebrovascular integrity in experimental stroke. Nat. Commun. 2015, 6, 7893. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.J.; Shi, S.X.; Liu, Q.; Shi, F.D.; Gonzales, R.J. Targeted role for sphingosine-1-phosphate receptor 1 in cerebrovascular integrity and inflammation during acute ischemic stroke. Neurosci. Lett. 2020, 735, 135160. [Google Scholar] [CrossRef]
- Billich, A.; Bornancin, F.; Devay, P.; Mechtcheriakova, D.; Urtz, N.; Baumruker, T. Phosphorylation of the immunomodulatory drug FTY720 by sphingosine kinases. J. Biol. Chem. 2003, 278, 47408–47415. [Google Scholar] [CrossRef] [Green Version]
- Paugh, S.W.; Payne, S.G.; Barbour, S.E.; Milstien, S.; Spiegel, S. The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS Lett. 2003, 554, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Bigaud, M.; Dincer, Z.; Bollbuck, B.; Dawson, J.; Beckmann, N.; Beerli, C.; Fishli-Cavelti, G.; Nahler, M.; Angst, D.; Janser, P.; et al. Pathophysiological Consequences of a Break in S1P1-Dependent Homeostasis of Vascular Permeability Revealed by S1P1 Competitive Antagonism. PLoS ONE 2016, 11, e0168252. [Google Scholar] [CrossRef] [PubMed]
- Huwiler, A.; Zangemeister-Wittke, U. The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: Recent findings and new perspectives. Pharmacol. Ther. 2018, 185, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Karliner, J.S. Sphingosine kinase and sphingosine 1-phosphate in the heart: A decade of progress. Biochim. Biophys. Acta 2013, 1831, 203–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemdan, N.Y.; Weigel, C.; Reimann, C.M.; Graler, M.H. Modulating sphingosine 1-phosphate signaling with DOP or FTY720 alleviates vascular and immune defects in mouse sepsis. Eur. J. Immunol. 2016, 46, 2767–2777. [Google Scholar] [CrossRef] [PubMed]
- Shea, B.S.; Brooks, S.F.; Fontaine, B.A.; Chun, J.; Luster, A.D.; Tager, A.M. Prolonged exposure to sphingosine 1-phosphate receptor-1 agonists exacerbates vascular leak, fibrosis, and mortality after lung injury. Am. J. Respir. Cell Mol. Biol. 2010, 43, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Copland, D.A.; Liu, J.; Schewitz-Bowers, L.P.; Brinkmann, V.; Anderson, K.; Nicholson, L.B.; Dick, A.D. Therapeutic dosing of fingolimod (FTY720) prevents cell infiltration, rapidly suppresses ocular inflammation, and maintains the blood-ocular barrier. Am. J. Pathol. 2012, 180, 672–681. [Google Scholar] [CrossRef] [Green Version]
- Cipriani, R.; Chara, J.C.; Rodriguez-Antiguedad, A.; Matute, C. FTY720 attenuates excitotoxicity and neuroinflammation. J. Neuroinflamm. 2015, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Tan, L.; Wang, Q.; Zhang, D.; Ding, J.; Huang, Q.; Tang, Y.Q.; Wang, Q.; Miao, H. Lymphopenia predicts disease severity of COVID-19: A descriptive and predictive study. Signal. Transduct. Target. Ther. 2020, 5, 33. [Google Scholar] [CrossRef]
- Henry, B.M. COVID-19, ECMO, and lymphopenia: A word of caution. Lancet Respir. Med. 2020, 8, e24. [Google Scholar] [CrossRef]
- Tsai, H.C.; Han, M.H. Sphingosine-1-Phosphate (S1P) and S1P Signaling Pathway: Therapeutic Targets in Autoimmunity and Inflammation. Drugs 2016, 76, 1067–1079. [Google Scholar] [CrossRef]
- Scott, F.L.; Clemons, B.; Brooks, J.; Brahmachary, E.; Powell, R.; Dedman, H.; Desale, H.G.; Timony, G.A.; Martinborough, E.; Rosen, H.; et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1 ) and receptor-5 (S1P5 ) agonist with autoimmune disease-modifying activity. Br. J. Pharmacol. 2016, 173, 1778–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, Y.N. Ozanimod: First Approval. Drugs 2020, 80, 841–848. [Google Scholar] [CrossRef] [PubMed]


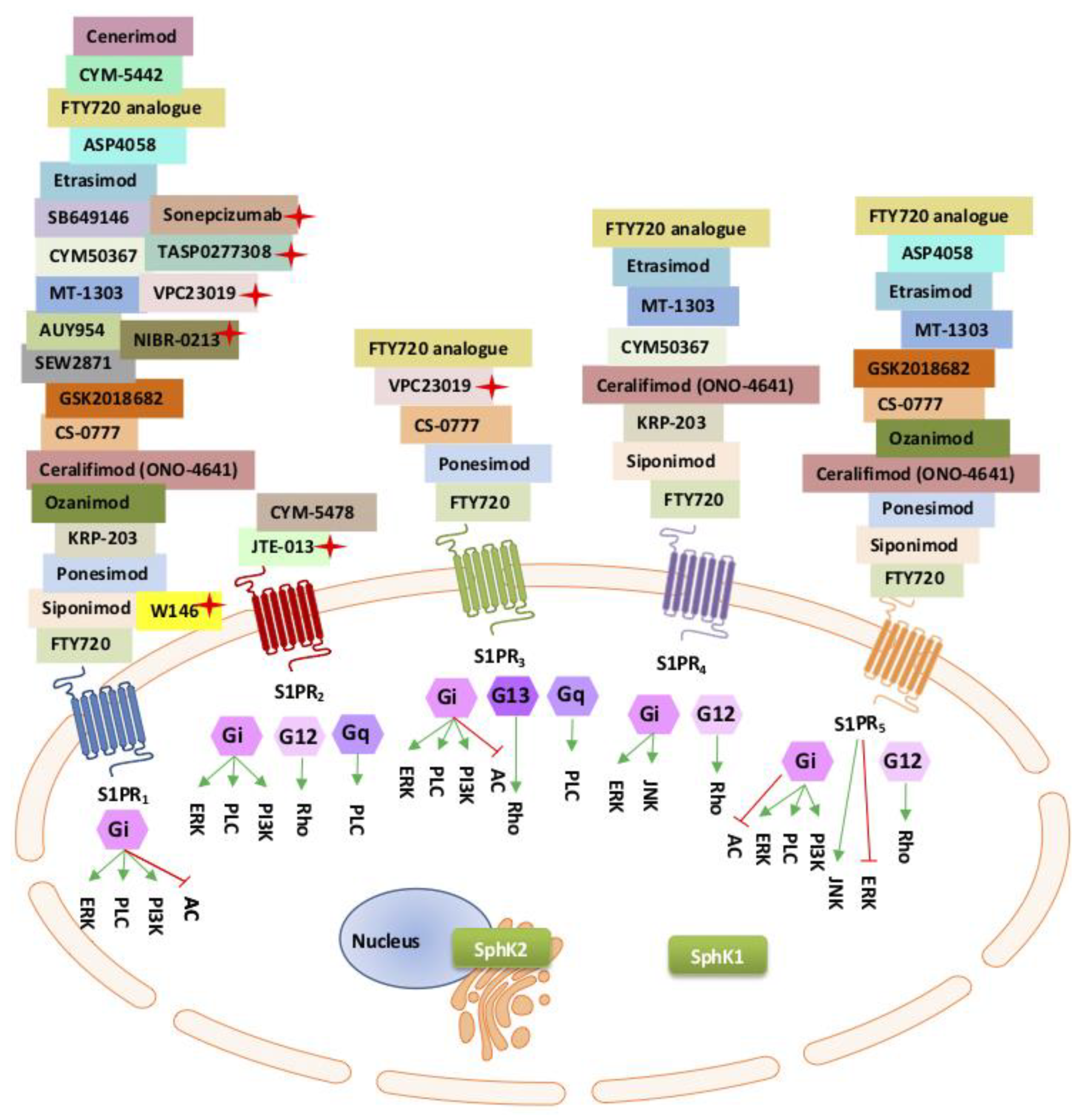{kind=link}
| S1P Modulator | S1PR Selectivity | References |
|---|---|---|
| Agonists | ||
| # FTY720 *# Fingolimod and phosphorylated fingolimod (Trade name: Gilenya) | S1P1 > S1P5 > S1P4 > S1P3 | [127,135,136,137,138,139] |
| S1P-specific antibody | Depletion of S1P | [112] |
| *# CS-0777 | S1P1 > S1P5 > S1P3 | [140] |
| * Ponesimod (ACT-128800) | S1P1 > S1P5 > S1P3 | [141] |
| *# Ozanimod (RPC1063) | S1P1 > S1P5 | [141] |
| * Ceralifimod (ONO-4641) | S1P1>S1P5>S1P4 | [142,143] |
| * Siponimod (BAF312) | S1P1 > S1P5 > S1P4 | [144] |
| * GSK2018682 | S1P1 > S1P5 | [141] |
| SEW2871 | S1P1 | [145,146,147] |
| AUY954 | S1P1 | [148,149] |
| * Amiselimod (MT-1303) | S1P1, S1P4, S1P5 | [141] |
| * Etrasimod (APD334) | S1P1, S1P4, S1P5 | [150] |
| * ASP4058 | S1P1, S1P5 | [151,152] |
| * Mocravimod (KRP-203) | S1P1>S1P4 | [153,154] |
| AAL(R) and phosphorylated AAL(R) (FTY720 analogue) | S1PR1, S1PR3, S1PR4 S1PR5 | [155,156,157] |
| CYM-5442 | S1P1 | [158,159] |
| VPC23153 | S1P4 | [160,161] |
| W-061 | S1P1 > S1P5 > S1P4 > S1P3 | [142,162] |
| * Cenerimod | S1P1 | [163] |
| # CYM-5478 | S1P2 | [164] |
| SB649146 | S1P1 | [165,166,167] |
| Antagonists | ||
| VPC44116 VPC23019 VPC25239 | S1P1 and/or S1P3 | [112] [168] |
| TASP0277308 | S1P1 | [169] |
| ** Sonepcizumab (Mab) | S1P1 | [170] |
| W146 | S1P1 | [171,172] |
| JTE-013 | S1P2 | [173,174] |
| NIBR-0213 | S1P1 | [171] |
| SphK Inhibitor | SphK Selectivity | References |
|---|---|---|
| SKi (2-(p-hydroxyanilino)- 4-(p-chlorophenyl)thiazole) or SK1-II | SphK1 and SphK2 | [112,179,180] |
| Safingol | SphK1 and SphK2 | [181] |
| L-threo-dihydrosphingosine (DHS) | SphK1 and SphK2 | [182] |
| N,N-dimethyl-D-erythro-sphingosine (DMS) | SphK1 and SphK2 | [112] |
| B-5354c, F-12509A (Natural products) | SphK1 and SphK2 | [112] |
| ABC294735 | SphK1 and SphK2 | [179] |
| Amgen 82 | SphK1 and SphK2 | [183] |
| Amidine-based range of sphingosine analogues | SphK1 and SphK2 | [112] |
| MP-A08 | SphK1 and SphK2 | [141] |
| ST-1083 | SphK1 and SphK2 | [184] |
| S-15183a and S-15183b (Natural product) | Not specified | [112] |
| SKI-V | Noncompetitive? | [185] |
| PF-543 ((R)-(1-(4-((3-methyl-5-(phenylsulfonylmethyl)phenoxy) methyl)benzyl)pyrrolidin-2-yl)methanol), SK1-5c (CAY10621), SK1-178, VPC96091 (36a), CB5468139 | SphK1 | [186] [180,187] |
| SKI-I | SphK1 | [188,189,190] |
| LCL351 | SphK1 | [191] |
| Compound inhibitors 51 and 54 | SphK1 | [141,192] |
| Balanocarpol | SphK1 | [193] |
| VPC94075 | SphK1 | [157] |
| 1-deoxysphinganines 55-21 and 77-7 (induces proteasomal degradation -SK1) | SphK1 | [194] |
| RB-005 | SphK1 | [195] |
| (S)-FTY720 vinylphosphonate | SphK1 | [196] |
| Genzyme | SphK1 | [183,197] |
| Peretinoin (NIK333) | SphK1 | [198,199] |
| ABC294640 | SphK2 | [112,200] |
| SG-12 and SG14 (sphingosine analogue) | SphK2 | [201] |
| SLC5111312 and SLM6041434 | SphK2 | [202] |
| F02 thiourea adduct of sphinganine | SphK2 | [194] |
| VT-ME6 | SphK2 | [203] |
| (2S,3S,4R)-Pachastrissamine | SphK2 | [204] |
| Trans-12a and Trans-12b | SphK2 | [203] |
| SLR080811, SLP120701 | SphK2 | [180] |
| K145 | SphK2 | [180] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGowan, E.M.; Haddadi, N.; Nassif, N.T.; Lin, Y. Targeting the SphK-S1P-SIPR Pathway as a Potential Therapeutic Approach for COVID-19. Int. J. Mol. Sci. 2020, 21, 7189. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197189
McGowan EM, Haddadi N, Nassif NT, Lin Y. Targeting the SphK-S1P-SIPR Pathway as a Potential Therapeutic Approach for COVID-19. International Journal of Molecular Sciences. 2020; 21(19):7189. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197189
Chicago/Turabian StyleMcGowan, Eileen M, Nahal Haddadi, Najah T. Nassif, and Yiguang Lin. 2020. "Targeting the SphK-S1P-SIPR Pathway as a Potential Therapeutic Approach for COVID-19" International Journal of Molecular Sciences 21, no. 19: 7189. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197189