Amphiphilic Aminoglycosides as Medicinal Agents


1
Molecular Pharmacochemistry Department, University Grenoble Alpes, CNRS, 470 Rue de la Chimie, F-38000 Grenoble, France
2
Cellular and Molecular Pharmacology Unit, Louvain Drug Research Institute, Catholic University of Louvain, Avenue E. Mounier 73, UCL B1.73.05, 1200 Brussels, Belgium
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(19), 7411; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197411
Submission received: 6 September 2020
/
Revised: 27 September 2020
/
Accepted: 2 October 2020
/
Published: 8 October 2020
(This article belongs to the Special Issue Structure–Activity Studies of Antibacterials and Potentiators of Antibiotics)
Abstract
:The conjugation of hydrophobic group(s) to the polycationic hydrophilic core of the antibiotic drugs aminoglycosides (AGs), targeting ribosomal RNA, has led to the development of amphiphilic aminoglycosides (AAGs). These drugs exhibit numerous biological effects, including good antibacterial effects against susceptible and multidrug-resistant bacteria due to the targeting of bacterial membranes. In the first part of this review, we summarize our work in identifying and developing broad-spectrum antibacterial AAGs that constitute a new class of antibiotic agents acting on bacterial membranes. The target-shift strongly improves antibiotic activity against bacterial strains that are resistant to the parent AG drugs and to antibiotic drugs of other classes, and renders the emergence of resistant Pseudomonas aeruginosa strains highly difficult. Structure–activity and structure–eukaryotic cytotoxicity relationships, specificity and barriers that need to be crossed in their development as antibacterial agents are delineated, with a focus on their targets in membranes, lipopolysaccharides (LPS) and cardiolipin (CL), and the corresponding mode of action against Gram-negative bacteria. At the end of the first part, we summarize the other recent advances in the field of antibacterial AAGs, mainly published since 2016, with an emphasis on the emerging AAGs which are made of an AG core conjugated to an adjuvant or an antibiotic drug of another class (antibiotic hybrids). In the second part, we briefly illustrate other biological and biochemical effects of AAGs, i.e., their antifungal activity, their use as delivery vehicles of nucleic acids, of short peptide (polyamide) nucleic acids (PNAs) and of drugs, as well as their ability to cleave DNA at abasic sites and to inhibit the functioning of connexin hemichannels. Finally, we discuss some aspects of structure–activity relationships in order to explain and improve the target selectivity of AAGs.
1. Introduction
Aminoglycosides (AGs), for example, neomycin B (NEO) 1, paromomycin (PARO) 2, kanamycins A and B (KANA, KANB) 3, 4 and tobramycin 5 (Figure 1), constitute a large family of highly potent broad-spectrum antibiotic drugs. These naturally occurring hydrophilic pseudo-oligosaccharides, which are polycationic species at physiological pH, target ribosomal RNA and disrupt protein synthesis [1]. Unfortunately, the widespread clinical use of AGs has strongly reduced their clinical efficacy through the selection of resistant bacteria [2]. In the search for new antibacterial agents targeting bacterial membranes, our team and others have synthesized and identified antibacterial amphiphilic aminoglycosides (antibacterial AAGs), which constitute a new class of polycationic antibiotic agents [3,4,5,6,7,8]. AAGs can be defined as substances made of a hydrophilic AG core on which lipophilic/hydrophobic group(s) are grafted, to produce, at physiological pH, cationic amphiphilic species.
Originally, AAGs were developed in the search for new AGs that are less susceptible to resistance. In this approach, Hanessian, Westhof and coworkers reported the first example of in vivo antibacterial lipophilic ether-modified derivatives of PARO targeting ribosomal RNA with a novel mode of binding, which are able to inhibit AG-deactivating enzymes [9,10].
In the never-ceasing fight against pathogenic multidrug-resistant bacteria, the identification of new targets, and of the corresponding drugs, is imperative [11], and an interest in membrane-targeting drug is emerging [12]. The expected major advantages of such drugs are (i) their activity against bacterial persisters and antibiotic-tolerant bacterial populations [13], characterized by a low metabolic activity [14,15]; (ii) their low tendency for the development of bacterial resistance [16]; and (iii) the fact that they do not need to cross the bacterial outer membrane (OM) of Gram-negative bacteria.
Indeed, this mode of action should limit the emergence of resistance through drug modifications by intracellular resistance-causing enzymes and efflux pumps expressed by multidrug-resistant (MDR) bacteria. Moreover, interaction with key membrane components, present in multiple copies in the bacterial membranes, can cause many antibacterial destructuring effects. The bacteria that are resistant to these effects have to perform biochemical modifications of multiple membrane components [17]. The corresponding metabolic modifications have a high energetic cost for the selected resistant bacteria, resulting in slow growth, rapid reversibility of the emerging resistance and high sensitivity to antibiotic drugs of other classes.
In such an approach, the antibacterial potential of cationic amphiphilic compounds that target bacterial membranes is attractive from a drug development perspective. The repurposing of the old antibacterial cationic cyclopeptide polymyxin E, colistin (COL, Figure 2), acting on the bacterial outer membrane (OM), as a last-line antibiotic to treat multidrug-resistant (MDR) Gram-negative bacterial infections, especially those caused by antibiotic-resistant Pseudomonas aeruginosa strains, illustrate well the interesting nature of cationic amphiphilic antibiotic drugs [18]. Despite several studies on COL repurposing, many issues related to emerging bacterial resistances, toxicity and pharmacokinetics still need to be elucidated [18,19].
In the search for membrane-targeting antibiotics, we have focused our work on Gram-negative bacteria, such as P. aeruginosa, an opportunistic pathogen that causes a wide range of severe opportunistic infections in patients with serious underlying medical conditions, such as those with burns, surgical wounds or people with cystic fibrosis [20].
The interaction of cationic species with bacterial membrane components such as anionic phospholipids can produce membrane disruption and depolarization. The cell envelope of Gram-negative bacteria contains two membranes, the inner membrane (IM) and the OM, separated by the periplasm. The OM of Gram-negative bacteria has a unique architecture that acts as a potent permeability barrier against antibiotics. The OM is composed of lipopolysaccharide (LPS), phospholipids, outer membrane β-barrel proteins (OMPs) and lipoproteins. These components are synthesized in the cytoplasm or in the IM, and are then selectively transported to the OM by specific transport machines. Recent reviews on the transport and assembly systems of OM components have been published with the aim of developing inhibitors targeting these systems [21,22,23,24].
In the search for new antibacterial agents, we identified broad-spectrum antibacterial AAGs carrying two or three lipophilic groups and, for the first time, we revealed their effects on bacterial membranes [5,25]. There was a corresponding strong increase in the AG lipophilicity resulting in the bacterial target shift from rRNA to membranes and a significant improvement in activity against bacterial strains resistant to the parent AG drugs and to antibiotic drugs of other classes (penicillins, fluoroquinolones, macrolides, etc.). The identification of antibacterial AAGs acting on the bacterial membranes against AG-resistant bacteria and MDR bacteria offered a promising direction for the development of novel antibiotics. Most of the antibacterial AAGs identified were mainly active against susceptible and MDR Gram-positive bacteria and a few appeared to be active against susceptible and resistant Gram-positive and Gram-negative bacteria.
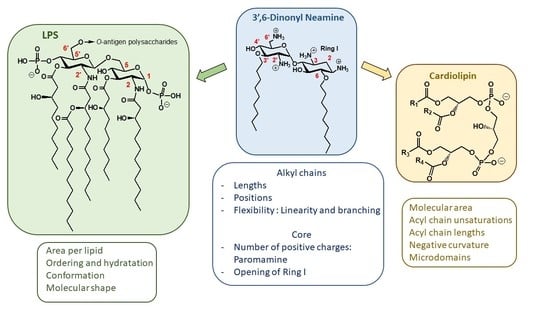
In the first part of this review article, we summarize the results that we obtained in the development of broad-spectrum antibacterial amphiphilic neamine (NEA) 6, paromamine (PARA) 7 and 6-amino-6-deoxy-1-methylglucosamine (1-methyl neosamine, corresponding to ring II in NEO) derivatives (Figure 1), including some recent unpublished results, with a focus on the most active NEA derivatives. Structure–activity and structure–eukaryotic cytotoxicity relationships, as well as specificities and particularities in their mode of action and barriers that need to be crossed in their development as medicinal agents, are delineated. Since the integrity of the biophysical properties of bacterial membranes are required for maintaining their permeability functions, as well as the right environment for proteins embedded within, we explored the effects of AAGs on two major lipids, LPS and cardiolipin (CL), that are involved in one of the most critical biophysical characteristics of the OMs of Gram-negative bacteria, their asymmetry. LPS is located at the outer leaflet of the OM and CL is located mostly within the IM, and also within the OM [26]. Some selectivity results from the binding of AAGs to these lipids in bacterial membranes in comparison to mammalian ones, since bacterial membranes (i) are more negatively charged than eukaryotic membranes; (ii) contain a higher proportion of negative intrinsic curvature lipids, where proteins involved in the formation of the division plane are located [27,28]; and (iii) exhibit a dilational modulus of elasticity that is much lower than the one found in mammalian membranes [29].
At the end of the first part of this review article, other advances in the field of antibacterial AAGs, published since 2016 after the appearance of several review articles [7,8,30,31,32], are reviewed with an emphasis on AAGs made of an AG core conjugated to an adjuvant or an antibiotic drug of another class (antibiotic hybrids), as recently developed by Schweizer, Zhanel and coworkers.
Numerous other biological and biochemical effects of AAGs have been reported, and, in the second part of this article, we illustrate briefly some of these effects, i.e., their antifungal activity; their use as delivery vehicles of nucleic acids, of short peptide nucleic acids and of drugs; their ability to cleave DNA at abasic sites and their ability to inhibit the functioning of connexin hemichannels. Structure–activity relationships are finally discussed, in order to explain and improve their target selectivity.
2. Antibacterial Amphiphilic Aminoglycosides (Antibacterial AAGs)
The chemical strategies used for the preparation of new AGs and AAGs and their biological activities were recently reviewed [1]. The recent progress in AG conjugation for RNA targeting was also summarized in 2020 [33]. AAGs were synthesized by modification of the AG’s primary amine or hydroxyl functions. Lipophilic groups were conjugated to the AG’s core of NEO 1 (NEO), NEA 6, PARO 2, PARA 7, KANA 3 and KANB 4, TOB 5 and nebramine 8 (NEB) (Figure 1) using several strategies [3,4,5,6,7,8,30,31,32,33,34,35,36,37,38]. The amine functions of the selected AG were converted to alkyl- or aryl-amide(s) or to carbamates, leading to a decrease in the number of positive charges present at physiological pH. In another approach, the hydroxyl groups were converted to ether and thioether groups. Such modification(s) of the amine or hydroxyl function(s) of the AG cores should produce AAGs that are not a substrate of AG-deactivating enzymes (AG nucleotidyltransferases (ANTs), AG phosphotransferases (APHs) and AG acetyltransferases (AACs) [2], and therefore produces AAGs which are active against bacteria expressing resistance-causing enzyme(s).
The AAGs identified to be most active against Gram-positive and Gram-negative bacteria are mainly dialkyl and/or trialkyl derivatives of NEA 6 (Figure 1), [5,34,35], of 1-methyl neosamine [36] and of NEB 8, which were identified more recently and are briefly described [37,38]. In this part of the review, we summarize the main results obtained in the development of broad-spectrum antibacterial amphiphilic NEA 6, PARA 7 and 1-methyl neosamine derivatives. Some physicochemical properties of amphiphilic NEA derivatives and a method developed for their dosage are also reported (unpublished results).
2.1. Broad-Spectrum Antibacterial AAGs: Antibacterial NEA, PARA and 6-Amino-6-Deoxy-1-Methylglucosamine Derivatives, Structure–Activity and Structure–Cytotoxicity Relationships, Modes of Action
2.1.1. Synthesis of NEA and PARA Derivatives
Our first works in the field of AGs were developed in the search for anti-HIV agents targeting viral RNA [39]. The AG core of NEA 6, carrying four amine and four hydroxyl functions, has been selected for modification in order to reduce the number of cationic groups present in the synthesized AAGs and, as a consequence, to limit the unspecific binding of the resulting species to biological polyanionic components.
Methods of protection and deprotection of amine and hydroxyl functions of NEA 6, obtained by methanolysis of NEO 1, were developed. From N-tetratritylated NEA 9, the 3′,6-O-di-para-methoxybenzyl (diPMB) and 3′,4′,6-O-triPMB derivatives 10 and 11 (Figure 1) were prepared for selective O-alkylation and deprotection under acidic conditions to give 4′-,5-O-monoalkyl and 4′,5-O-dialkyl derivatives [39,40]. The O-alkylation of N-tetratritylated NEA 9 in DMF using sodium hydride (NaH) as a base allowed the preparation, in one step, of 3′,6-O-dialkyl and 3′,4′,6-O-trialkyl NEA derivatives [5]. Using phase transfer conditions, it was possible to prepare, in high yields, the 6-PMB tetratrityl NEA derivative 12, which was alkylated to selectively produce 3′,4′-O-dialkyl derivatives [36,41]. Methanolysis of PARO 2 gave PARA 7, which was tetratritylated, leading to 13, which was used to prepare 3′,6-O-dialkyl and 3′,4′,6-O-trialkyl PARA derivatives [34]. Major increases in the selectivity and/or reactivity of AAGs in O-mono- and O,O′-di-alkylation were observed under phase-transfer conditions in the presence of tetrabutylammonium fluoride (TBAF) or iodide (TBAI) [41] in comparison to alkylation in DMF solution using NaH [5]. The use of TBAI under phase transfer conditions allowed the rapid and efficient preparation of 3′,6-dialkyl and 3′,6-diarylalkyl NEA derivatives, avoiding the O-trialkylation observed with NaH in DMF [5,33,34].
2.1.2. Structure–Activity Relationships
First Identified Broad-Spectrum Antibacterial Amphiphilic NEA Derivatives
In the search for new antibacterial agents, first, we introduced onto the hydroxyl functions of NEA lipophilic benzyl, 2-naphthylmethylene (2NM) (Figure 3), 2-pyridinylmethylene and 2-quinolinylmethylene groups from the corresponding halides [5]. The resulting 3′,6-di-; 3′,4′-di- and 3′,4′,6-triarylalkyl NEA derivatives were isolated as tetratrifluoroacetate salts, after a final deprotection step in TFA, and were evaluated for their antibacterial activity against susceptible and resistant Gram-positive Staphylococcus aureus (for instance, methicillin-resistant S. aureus (MRSA) and vancomycin-resistant S. aureus (VRSA)) and Gram-negative bacteria (Acinetobacter baumannii, Escherichia coli, Klebsiella pneumonia, P. aeruginosa, etc.).
The 3′,6-di-(14); 3′,4′-di-(15) and 3′,4′,6-tri-(16) 2-naphthylmethylene (2NM) derivatives (Figure 3) showed good activity against susceptible and resistant S. aureus strains expressing resistance pumps (NorA or MsrA), against AG-inactivating enzymes and against MRSA and VRSA strains, which are resistant to methicillin and vancomycin, respectively (minimum inhibitory concentrations (MICs) 4–16 µg/mL), whereas the 3′-, 4′-, 5′-, 6-mono-2NM derivatives were inactive (MICs > 128 µg/mL). The tri2NM derivative 16 showed better antibacterial activity against the selected Gram-positive strains (MICs 2–4 µg/mL). It also exhibited good activity (MICs 4–16 µg/mL) against sensitive and resistant Gram-negative bacteria, both on Enterobacteriaceae and non-Enterobacteriacea, including P. aeruginosa, expressing efflux pumps or AG-deactivating enzymes, or rRNA methylases [2] (Citrobacter amalonaticus arm 06AB0010, E. coli 06AB003arm, Enterobacter aerogenes 06AB008 arm). Derivatives 14 and 16 revealed weak and aspecific binding to a model bacterial 16S rRNA as compared to NEO 1. Derivative 16 also showed a low ability to decrease 3H leucine incorporation into proteins in P. aeruginosa, suggesting that 16 acts against Gram-negative bacteria with a mechanism different from inhibition of protein synthesis, probably by membrane destabilization [5]. For the first time, AAGs (14 and 16) were shown to target the membranes of P. aeruginosa with induction of depolarization [25].
Comparison of the Antibacterial Activities of NEA and PARA Derivatives
In the PARA core, the 6′-amino group of NEA is replaced by a hydroxyl group and, as a consequence, the corresponding AAGs bear, at physiological pH, one less positive charge in comparison to their NEA homologs [34]. The comparison of the antibacterial activities of the previously prepared 2NM derivatives (14 and 16) with those of 1-naphthylmethylene (1NM, 17 and 18) NEA derivatives and of the corresponding PARA derivatives 19–22 (Figure 3), revealed the better good broad-spectrum activity of the 3′,4′,6-triNM NEA derivatives, with the MIC mainly decreased by four times. Therefore, the NEA core was selected in the search for more active compounds than 14–18 [34]. The presence of an amino group at position 6′ that is protonated at physiological pH of the aminoglycoside increases the antibacterial effect, without being essential to the antibacterial effect.
In order to compare the antibacterial activity of 3′,6-dialkyl and 3′,4′,6-trialkyl NEA derivatives and the effect of the alkyl chain lipophilicity on their activity, NEA derivatives carrying two or three linear alkyl groups, butyl (Bu), hexyl (Hx), nonyl (Nn) and octadecyl (Ocd) groups, as well as arylalkyl groups, benzyl and 2-naphthylalkyl with alkyl, methyl, n-propyl, n-butyl and n-hexyl (2NM, 2NP, 2NB and 2NH, respectively), were synthesized and evaluated [34]. The 3′- and 6-mono-Ocd derivatives were also prepared and found to be inactive against Gram-positive and Gram-negative bacteria. The 3′,4′,6-tri2NM (16), -triHx (23) (Figure 3) and the 3′,6-diNn (24), -di2NP (26) and -di2NB (27) NEA derivatives (Figure 4) showed good activity against susceptible and resistant Gram-positive and Gram-negative bacteria, with the 3′,6-dinonyl derivative 24 being the most active, and the di2NP 26 and di2NB 27 also being the most active against Gram-positive bacteria. The main result of this study was the delineation of windows of critical lipophilicity values for the active 3′,6-dialkyl and 3′,4′,6-trialkylNEA derivatives from the plot of 1/MIC versus clogP (calculated octanol/water partition coefficient; see the paragraph below on the fine-tuning of the structure–activity relationships). NEA AAGs 24, 26 and 27 were selected for further study as the most active and the less cytotoxic derivatives at 10 μM to murine J774 macrophages, in comparison to the trihexyl derivative 23 that has shown slightly lower broad-spectrum activity [34]. These three AAGs were shown to be more active against COL-resistant P. aeruginosa (PA272, PA307, PA343 and PA2938; MIC = 2–8 µg/mL) than the 3,’4′,6-tri2NM NEA derivative 16 (MIC = 4–32 µg/mL) [42].
AG antibiotics are effective against biofilms and recently approaches and possible mechanisms for the application of AGs to treat biofilm-associated infections were briefly reviewed [43].
The 3′,6-dinonyl NEA derivative 24 was shown to be bactericidal against P. aeruginosa at its MIC and to inhibit the P. aeruginosa biofilm formation at a two-fold MIC [42].
Comparison of the Antibacterial Activities of 3′,6- and 3′,4′-Dialkyl NEA Derivatives
In order to investigate the role of the attachment position of the second alkyl chain in antibacterial activities and cytotoxicities, the 3′,4′-diNn and -di2NP analogues 25 and 28 of the corresponding 3′,6-dialkyl derivatives 24 and 26 (Figure 4), were synthesized from 6-(para-methoxy)benzyl-N-tetratrityl NEA 12, obtained selectively under phase transfer conditions of alkylation [36,41]. Against susceptible and resistant Gram-positive bacteria, the 3′,6-di2NP and 3′,4′-di2NP derivatives 26 and 28 showed similar good activity. Against Gram-negative bacteria, the activity of 28 was better. The 3′,6-dinonyl derivative 24 appeared to be more active than its 3′,4-dinonyl isomer 25 and was found to be the most active synthesized NEA derivative against susceptible and resistant Gram-positive and Gram-negative bacteria [36].
6-Amino-6-Deoxy-1-Methylglucosamine (1-Methyl Neosamine) Derivatives, Analogues of 3′,4′-Dialkyl NEA Derivatives
In order to evaluate the importance, in the antibacterial activity of 3′,4′-dinonyl derivative 25, of the integrity of the 2-deoxystreptamine ring I (Figure 5), amphiphilic 3,4-dialkyl derivatives 29–36 of 6-amino-6-deoxyglucosamine, named neosamine, corresponding to NEA ring II, were synthesized from the 1-allyl intermediate derivative 37, prepared from N-acetylglucosamine (in 8 or 9 steps) (Figure 5) [36]. The most hydrophilic derivatives 30–36 mainly showed similar good activity against susceptible and resistant Gram-positive and Gram-negative bacteria (MIC = 1–2 and 4–8 µg/mL, respectively), slightly better than the corresponding activity of the 3,4′-dinonyl NEA derivative 25. The azido derivative 29 revealed an activity against Gram-positive bacteria similar to 30–36 but was mainly inactive against Gram-negative bacteria (MIC = 32–>128 µg/mL). The major difference observed in comparison to the 3′,6- and 3′,4′-dinonyl NEA derivatives, 24 and 25, was a better activity against the resistant Acinetobacter lwoffi strain Al88-483 of 30–32 and 34–36 (MIC = 4–8 µg/mL), the only selected bacteria against which 24 revealed an MIC value higher than 2–4 µg/mL (32 µg/mL). These results showed that the integrity of ring I is not necessary to have good broad-spectrum antibacterial activity. They suggest that, at least, one amine function, protonated at physiological pH, should be present in the flexible chain replacing ring I, since the azido derivative 29 is mainly inactive and the hydroxyl derivative 32 is less active than 31 and 33–36 against susceptible and resistant Gram-negative bacteria, especially against P. aeruginosa strains (MICs = 64 and 4–8 µg/mL, respectively).
Fine-Tuning of the Structure–Activity Relationships (SAR)
In the previous studies, the 3′,6-dinonyl NEA derivative 24 has been found to be the most active AAG against susceptible and resistant Gram-negative bacteria with the exception of resistant A. lwoffi (Al88–483). In the search for the best antibacterial amphiphilic NEA derivatives to develop further, different 3′,6- and 3′,4′-dialkyl NEAs having lipophilicity values close to that of 24 were synthesized and studied [35].
3′,6-homodialkyl NEA derivatives carrying n-heptyl (Hp, C7); n-octyl (Oc, C8); n-decyl (De, C10) and n-undecyl (Ud, C11) groups, 38–41, respectively, were prepared (Figure 6). Two 3′,6-heterodialkyl derivatives, 3′-n-heptyl-6′n-undecyl (Ud, C11) 42 and 3′-n-undecyl -6′-n-heptyl (Ud, C11) 43, and two dialkyl derivatives bearing the same branched chain, 3′,6-di(3,7-(dimethyl)octyl (diDiMOc) NEA 46 and its 3′,4′-isomer 47, were also synthesized (Figure 6). Two 3′,6-heterodialkyl NEA derivatives, 42 and 43, having lipophilicities close to that of 24 (3′- or 6-heptyl and 6- or 3′-undecyl) were synthesized in order to evaluate the effects of the presence of dissymmetric alkyl chains on the antibacterial effects and the eukaryotic cytotoxicity. Two fluorescent 3′,6-dialkyl NEA derivatives, 44 and 45, carrying one n-heptyl group and one (1-pyrenyl)butyl group, and also having lipophilicities close to those of 24, were also prepared as probes for mechanistic studies (Figure 6).
Two windows of lipophilicities have been related to the antibacterial activities of the 3′,6-dialkyl and -dialkylaryl derivatives and to the 3′,4′,6-trialkyl and –trialkylaryl derivatives, respectively, through the plot of 1/MIC versus clogP (calculated octanol/water partition coefficient) [34]. The fine-tuning of the delineated critical window of lipophilicity related to the antibacterial activity of the 3′,6-dialkyl and -dialkylaryl derivatives was possible with the new 3′,6-dialkyl derivatives synthesized [35]. It revealed differences between the series of dinaphthylalkyl NEAs and the series of homodialkyl NEAs (Table 1, Figure 7) [35]. Against P. aeruginosa, the dinaphthylpropyl derivative 26 is slightly less active than the alkyl derivatives 24 and 39, despite close lipophilicities. Against Gram-positive bacteria, the activity is similar (MICs 1–2 µg/mL). The dinaphthylalkyl derivative 26 is less cytotoxic (Table 2) than the dialkyl derivatives 24 and 39 at 30 µM. Fridman and coworkers reported that an increased degree of unsaturation in the lipophilic chain of antifungal AAGs derived from TOB 5 significantly reduced the immediate toxicity to mammalian cells in comparison to saturated derivatives [44].
In [35], four new broad-spectrum antibacterial derivatives of similar lipophilicities were identified, two 3′,6-dialkyl derivatives carrying two linear octyl and decyl chains 39 and 40, and, 3′,6- and 3′,4′-dialkyl derivatives bearing the same branched 3,7-(dimethyl)octyl (DiMOc) chains 46 and 47. In comparison to 24 and 25, this study revealed that the grafting of branched alkyl chains of similar lipophilicities (NEAs 46 and 47) preserves or increases the antibacterial activity and increases the eukaryotic cytotoxicity. 3′,6-heterodialkyl NEAs 42–43, of lipophilicities close to those of 24 and 25, showed good broad-spectrum antibacterial activity, and it was shown that the dissymmetry of their chains increases the cytotoxicity. The fluorescent pyrenyl derivatives 44 and 45, having good antibacterial activity, are under study for related mechanistic studies (unpublished results).
The previously identified 3′,6-dinonyl NEA 24 and the 3′,6-dioctyl NEA 39, identified in this study, appeared to be the most active amphiphilic NEAs against susceptible and resistant Gram-positive and Gram-negative bacteria (for example, Table 1) and the least cytotoxic broad spectrum antibacterial 3′,6-dialkyl derivatives (without aryl groups) to mammalian cells (Table 2). These derivatives and the 3′,6-diarylalkyl NEA derivative 26, which showed a better activity against Gram-positive than against Gram-negative bacteria and is the less active cytotoxic derivative at 30 µM (Table 1 and Table 2), merit further interest in an antibacterial development.
Emergence of Resistance to Amphiphilic NEA Derivatives: MIC Changes against P. aeruginosa upon a Long Exposures to AAGs
We studied, in comparison to ciprofloxacin (CIP), the MIC changes against susceptible P. aeruginosa ATCC 27,853 upon several days’ exposure to a half-MIC of 3′,6-di2NP NEA 26 and its 3′,4′-isomer 28 [36], and to the 3′,6-diNn NEA derivative 24 [35].
Exposure of P. aeruginosa to subinhibitory concentrations of CIP or di2NP NEAs caused a decrease in susceptibility that appeared later for the di2NP derivatives. The exposure of susceptible P. aeruginosa to a half-MIC of 24 at different times, over more than one month, also demonstrated the expected high difficulty of resistance emergence to AAGs, with a much weaker and slower increase of MIC in comparison to CIP. Under these conditions, the MIC increased slightly from 1 to 4 µg/mL at day 15, 30 and 38 as compared to the MIC values of CIP, which increased faster, from 0.5 to 16 µg/mL at days 6, 21 and 24 [35].
Solubility of AAGs at High Concentration in Aqueous Solutions and Dosage of 24 for Studies In Vivo (Unpublished Results)
The synthesized AAGs were isolated and evaluated mainly as tetratrifluoroacetates of high Mw (24: 1031 g/mol; 26: 1115 g/mol) isolated after lyophilization as white foams. They are hygroscopic and highly soluble in water, at more than 50 mM for 24 and 26, for example. The solubility S decreases strongly in the presence of glucose (S < 5 mM), of 0.9% aqueous NaCl (S < 3 mM) and of 1× phosphate-buffered saline (PBS) aqueous solution (S < 1 mM). This behavior corresponds to a difficulty in the in vivo antibacterial evaluation of 24 and 26, especially through intravenous injections at high concentration.
The dinonyl derivative 24 does not incorporate a chromophore absorbing in the near and medium UV useful for dosage. Therefore, a spectrofluorimetric method of dosage was optimized by derivatization of 24 with fluorescamine from a method described to quantify the concentration of AGs such as NEO 1 [45,46], KAN and TOB 5 [46,47]. In the reported method, acetone has been used as a solvent of fluorescamine. It was replaced by less volatile acetonitrile, resulting in an increased fluorescence intensity that was found to be linearly correlated to the concentration of the dinonyl derivative 24, in the range of 1 to 30 µM.
2.1.3. Targets and Modes of Action against Gram-Negative Bacteria
In the search for new antibacterials and for the fine-tuning of the most promising AAGs, the understanding of the molecular mechanisms involved in the antibacterial activity is critical. The OM of Gram-negative bacteria has a unique architecture that acts as a potent permeability barrier against antibiotics. Since the biophysical characteristics of bacterial membranes, and especially OM asymmetry, are required to maintain their permeability functions as well as the right environment for proteins embedded within, we focused our studies on the effect of AAGs on the two lipids involved in membrane asymmetry, LPS and cardiolipin.
Anionic LPS
LPS is an essential component of the OM of many Gram-negative bacteria. LPS is a complex macromolecule, comprising the endotoxic lipid A (Figure 8), covalently linked to a polyphosphorylated oligosaccharide core carrying an O-polysaccharide chain (also referred as O-antigen) [48]. The lipid A core serves not only as a hydrophobic anchor of LPS in order to form the bilayer structure, but also as an activator of the innate immune system [48]. This core is made of the diphosphorylated 1,6-N-acetylglucosamine dimer O- and N-acylated with 4–7 fatty acids (Figure 8).
Lipid A structural variations (mainly in acylation patterns) depend upon bacterial species [49]. As an example, in P. aeruginosa, LPS (LpxA) incorporates C10 lipophilic chains grafted on UDP-N-acetylglucosamine, whereas C14 and C12 chains are incorporated in E. coli and Neisseria meningitidis, respectively [50].
In the continuous search for membrane-targeting antibiotics, compounds that interfere in LPS biogenesis or regulatory pathways offer opportunities for development as antibiotics that may be useful against pathogenic bacteria [22,23,51,52].
The electrostatic attraction between the amphiphilic NEA derivatives and the negatively charged lipid or LPS headgroups (i.e., core oligosaccharide and Lipid A phosphates in LPS) is likely the dominant driving force of the interaction [53,54]. However, other parameters governing the interactions between LPS and amphiphilic NEA derivatives, including (i) the area per lipid, (ii) the ordering and the hydration behavior, (iii) the spatial conformation and (iv) the molecular shape of the lipid A moiety, also play a critical role and are discussed hereunder.
First, the area per lipid of the lipid A molecule is critical for characterizing the LPS/lipid A packing. The acyl chains in lipid A occupy a smaller volume than in most of the phospholipids and could be associated with a better packing [55]. Especially, in the P. aeruginosa membrane, the tail volume of the penta-acyl lipid A is smaller as compared to that found in other bacteria having hexa- or hepta-acyl chains in their membranes, such as Salmonella minnesota [55,56,57].
Amphiphilic NEA derivatives were inserted into LPS monolayer, as indicated after spreading them in the subphase and measuring the compression isotherms of rough mutant. A shift to molecular areas higher than those for the pure buffer subphase was observed [42]. This results in a higher packing. Addition of 3′,6-dinonyl NEA 24 in the presence of Ca2+ gave rise to an effect on the molecular area that was weaker than the effect obtained in the absence of Ca2+, suggesting that CaCl2 reduced the insertion of the 3′,6-dinonyl NEA derivative [42].
As mentioned, and demonstrated by displacement assays, it is not only electrostatic interactions that are involved in the binding of amphiphilic NEA derivatives to LPS—the length and branching of acyl chains in LPS are also critical. Different 3′,6-dialkyl NEA derivatives were found to bind to LPS with a clear ranking, depending upon the length of the linear alkyl chain (3′,6-diheptyl NEA 38 < 3′,6-dioctyl NEA 39 < 3′,6-dinonyl NEA 24 < 3′,6-didecyl NEA 40 < 3′,6-undedecyl NEA 41). The presence of a branched alkyl chain also plays a critical role (3′,6-dinonyl NEA 24 < 3′,6-di(dimethyloctyl) NEA 46 (Swain et al., unpublished results).
Second, closely interconnected with LPS/lipid A packing, the order and lipid phase might also be key clues for the interaction between LPS/lipid A and amphiphilic aminoglycoside antibiotics. The order parameter of lipid A from P. aeruginosa can be determined using Fourier-transform infrared (FT-IR) spectroscopy [58]. The antibacterial amphiphilic NEA derivatives make the LPS film state more liquid-like, as suggested by the decrease in the excitation generalized polarization (GPex) of a fluorescent laurdan probe (6-dodecanoyl-2-dimethylamine-naphthalene), which is sensitive to lipid hydration, inserted into LPS micelles [59,60]. This indicates an increasing liquid-like state in the lipid A region of LPS. The effect was dependent upon the presence of aryl groups (3′,6-di2NP 26 < 3′,6-diNB 27 < 3′,4′,6-tri2NM 16). The effect induced by 3′,6-dinonyl NEA 24 was not significantly different from that observed with 3′,6-diNP 26, of similar lipophilicity. In contrast, COL, NEA and the 3′,6-di2NM derivative 14 had no influence on the packing of lipid A in micelles, as indicated by unchanged GPex values compared to the value for the control. The appearance of fluidic material extends previous data related to the formation of a supramolecular network between multi-cationic and multi-anionic substances [61,62]. The liquid-like state in the lipid A region of LPS was likely more predominant than observed with glycerophospholipid. Divalent calcium ions could form salt bridges due to the close proximity of lipid A phosphate groups [63]. This binding is tight enough that the charge density in the solvent can approach zero [55]. All kinks in the water density lines, which indicate the enhanced water penetration, are located around z = 8 nm in the vicinity of the peaks of both lipid A phosphates and calcium ions [55]. The presence of divalent cations explains why the water molecules are able to penetrate deeper into the lipid A leaflet than the phospholipid leaflet.
The conformation of lipid A could be the third critical parameter for designing new potential antibiotics. The spatial conformation of lipid A is an indicator of lipid A bioactivity [58,64,65]. The backbone inclination angle of asymmetric penta-acyl lipid A influences the position and orientation of the phosphate groups in LPS. Other inclination angles also provide information to further determine the lipid A conformation, such as the angles between the membrane surface and the vectors from 4′-carbon to 4′-phosphorus, 1-carbon to 1-phosphorus and 4′-carbon to 1-phosphorus atoms [55]. This might help us to design new positions for substitution or new branching.
Lastly, the molecular shape of LPS/lipid A could play a critical role in its interaction with amphiphilic NEA derivatives. The penta-acyl LPS has a multilamellar structure due to its cylindrical molecular shape. To be biologically appropriate (for interaction with Toll-like receptors, for example), this structure has to be converted into a physiologically active conformation [58]. The transition from a unilamellar into a cubic inverted structure suggests that a subsequent intercalation into the hydrophobic moiety takes place, resulting in a change of the molecular shape of the LPS/lipid A molecules with a more conical conformation [66].
Assuming the volume of the hydrophobic part increases from the linear dialkyl (3′,6-dinonyl NEA 24) to the dinaphthylalkyl (3′,6-di2NM NEA 14, 3′,6-di2NP NEA 26, 3′,6-di2NB NEA 27) and the bulky 3′,4′,6-tri2NM NEA 16 moieties, the maximal binding would be inversely proportional to the volume of the hydrophobic moiety. The 3′,6-dinonyl NEA 24 shows a molecular shape of an inverted cone, with a large hydrophilic part and a small hydrophobic one. This could allow a close interaction between this derivative and the lipid A moiety of the LPS unit, characterized by a conical complementary shape (with a small hydrophilic part and a large hydrophobic part). Very interestingly, LPS acylation can be modified through the activity of the palmitoyl lipid A transferase PagP, leading to new modulation of LPS activity [67].
In summary, the small area per lipid and large order parameter of lipid A indicate a leaflet with low fluidity and highly organized molecules. Calcium ions form salt bridges with phosphates in lipid A and stabilize the corresponding structure. The overall hydrated lipid A leaflet displays solvation differences due to the tilted lipid A backbone. This inclination causes the 1-phosphate in lipid A to project toward the outer aqueous environment and the 4′-phosphate to become buried between the acyl chains of lipid A [55]. Studies have highlighted the formation of a fluidic cross-linked supramolecular network between LPS and amphiphilic NEA derivatives, in parallel with the length and the logP values of the corresponding derivatives. Both hydrophobic and electrostatic interactions are required.
The molecular shape of the 3′,6-dialkyl NEA derivatives induced by the nature of the grafted hydrophobic moieties (naphthylalkyl instead of alkyl) and the flexibility of the hydrophobic moiety are critical for their fluidifying effect and their ability to displace cations bridging LPS. These parameters could be exploited for the development of new amphiphilic NEA derivatives [42].
How these characteristics in the interactions between LPS and amphiphilic NEA derivatives result in antibacterial activity is still unclear. Activity was associated with the self-promoted insertion of the molecule within the lipid A region of the OM outer leaflet in P. aeruginosa, leading to an increase in OM permeability. The fluorescent probe 1-N-phenylnapthylamine (NPN) was used to probe the permeability of the OM, since NPN is a small molecule that cannot effectively cross the OM. It is weakly fluorescent in aqueous solution but fluoresces strongly when it binds to phospholipids [68].
An ascending time dependence of the Ca2+ effect in NPN uptake experiments was observed from 3′,6-di2NM NEA 14 to 3′,6-di2NP NEA 26 and 3′,6-diNB NEA 27 [42]. For linear dialkyl NEA derivatives, no clear effect corresponding to the increase of side chain lengths was observed. Regarding the effect of branched alkyl groups present in the 3′,6-di(dimethyloctyl) derivative 46, the dose required to obtain 50% of the maximal effect is lower for the branched derivative as compared to the unbranched analogues, and even the maximal effect was equal for both compounds (Swain et al., unpublished results).
Cardiolipin (CL)
CL (Figure 8) is a tetra-acylated diphosphatidylglycerol derivative found in plants and animals, but also in bacteria. This structurally unusual phospholipid carries two negative charges due to its dimeric structure, consisting of two phosphatidyl residues connected by a glycerol bridge and four associated fatty acyl chains (made of various alkyl chains R1, R2, R3 and R4) [69]. Due to variability in fatty acyl chains (length, degree of unsaturation, etc.), CL is characterized by numerous molecular species, leading to a high potential for adaptability or responses to stress. CL is known for its sensitivity to stressors, such as the addition of organic solvents or high salt content or the presence of quaternary ammonium compounds [70,71]. Changes in the molecular species suggest CL restructuring. Indeed, CL is a critical phospholipid in maintaining the function and morphology of bacteria. CL was one of the main components of the IM, along with phosphatidylethanolamine (PE) and phosphatidylglycerol (PG) [72,73]. Very interestingly, CL is also located in the OM of most Gram-negative bacteria [26,74].
Like LPS, CL is characterized by its own membrane parameters, including (i) molecular area, (ii) degree of unsaturation and length of acyl chains and (iii) the ability to induce negative curvature and form microdomains, parameters which are critical in the biophysical properties of membranes in which CL is inserted.
With the aim of characterizing the effect of the interactions between amphiphilic NEA derivatives with CL on the molecular area, we determined the compression isotherms corresponding to the lipid monolayers for CL spread on a subphase containing 3′,6-dinonyl NEA 24 (as compared to those of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine (POPE) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG)). The isotherms were shifted to higher molecular areas than the pure buffer subphase, although to a much lower extent for POPE, compared with POPG and CL. At 30 mNewtons/m, a value close to the estimated surface pressure of biological membranes in vivo, the mean molecular area for POPE was not significantly modified by the presence of 3′,6-dinonyl NEA 24 in the subphase. With POPG and CL, the mean molecular area increased by 1.8 and 1.6, respectively. This discrepancy indicates a much higher adsorption of 24 into POPG or CL than into POPE. The shift of the mean molecular area was not significantly different between POPG and CL.
Furthermore, order and interdigitation could be affected by AAGs. Bacterial CL structure is characterized by a high degree of symmetry and unsaturation [71,75,76], as well as by long fatty acyl chains. Both the degree of unsaturation and the length of acyl chains can modulate order and crosstalk between leaflets [77,78,79]. CL might be a reservoir for unsaturated fatty acid chains, as suggested by the content of unsaturated fatty acid in cls/cls2-mutant cells, which is approximatively 40% lower than in wild-type cells [71]. The relative concentrations of C16 and C18 for the barotolerant Pseudomonas sp. BT1 were approximately 60% and 40%, respectively [72]. PE and CL have a phase transition well above that of PG when the fatty acid content is identical for both lipids. In general, lipids with long fatty acyl chains undergo phase transition at higher temperature than lipids with short chains [80]. Long chains explain why CL stimulates changes in the physical properties of the membrane and why it decreases the lateral interaction within the monolayer leaflet, which favors the creation of membrane folds [81].
Another striking characteristic of CL is the comparatively small cross-section of its headgroup relative to the cross-section of its four large tail groups. This discrepancy results in a molecular shape with a large intrinsic negative curvature [82,83,84,85] and a non-bilayer anionic phospholipid also described previously for PE [86] that facilitates the insertion of membrane proteins. The cross-sectional size difference further explains the location of CL-enriched regions at the pole and/or the division septum [87], which can in turn be related to the polar localization of many proteins, including those involved in cell division and osmosensing [88]. The intrinsic negative curvature of CL can result in the formation of microdomains (clusters).
The presence and the role of these microdomains can be affected by membrane-acting antibiotics. Using membrane models mimicking P. aeruginosa plasma membrane composition (POPE:POPG:CL), we demonstrated the ability of 3′,6-dinonyl NEA 24 to induce in a CL dependent manner, to increase membrane permeability through reduced hydration and decreased ability of the membrane to mix and fuse, as shown by monitoring calcein release, GPex of laurdan and fluorescence dequenching of octadecyl rhodamine B, respectively [89]. After incubation with 3′,6-dinonyl NEA 24, CL was co-localized with PE in small clusters, sections with increased membrane curvature, and fusion points in the joined giant unilamellar vesicles (GUV) population. Moreover, a clear overall lipid reorganization was observed [89].
CL-interacting proteins and functions regulated by CL are affected by the amphiphilic AG, as we demonstrated an inhibition of the respiratory chain and changes in bacterial shape. The latter effect was characterized by the loss of the bacterial rod shape through a decrease in length and an increase in curvature. It resulted from the effect on MreB, a CL-dependent cytoskeleton protein, as well as a direct effect of 3′,6-dinonyl NEA 24 on CL [90]. In E. coli, CL is known to enhance the activity of the glycosyltransferase MurG involved in peptidoglycan biosynthesis [91]. Some correlation (phosphatidic acid > CL > PG) with the activity of dynamin-related Protein 1 (Drp1) has been reported [92]. In addition, CL content plays a critical role since low levels of CL in the membrane of Pseudomonas putida allow the tetradecyltrimethylammonium cation (TTAB) to cross the membrane, reach its site of action and kill the cell, as bacteria cannot counteract the fluidizing effect of the detergent [71]. On the contrary, the addition of CL to the culture medium delayed the growth of P. aeruginosa, favored asymmetrical growth and enhanced the efficiency of 3′,6-dinonyl NEA 24 [93].
These results shed light on how targeting CL microdomains may be of great interest for developing new antibacterial therapies. Some recent evidence also highlights the role of CL for outer membrane vesicle (OMV) formation. These vesicles could be important as inter-kingdom players. Experimental evidence that bacterial OMVs, by sequestering of cationic peptides, may protect pathogenic yeast against the combined action of antifungal drugs has been reported [94].
2.1.4. Targets and Modes of Action against Gram-Positive Bacteria
We also investigated the mechanism of action against Gram-positive bacteria, S. aureus and Bacillus subtilis [95]. Time-killing experiments were performed to demonstrate the bactericidal effect induced by 3′,6-dinonyl NEA 24 against S. aureus MSSA and MRSA (methicillin susceptible and resistant S. aureus, respectively). The displacement of the BODIPY™-TR cadaverine probe bound to lipoteichoic acids (LTA) showed that 24 interacts with the bacterial surface components. The ability of 24 to enhance membrane depolarization and induce membrane permeability was highlighted, using fluorescent probes, DiSC3C(5) and propidium iodide, respectively. These effects were observed for both MSSA and MRSA, as well as for B. subtilis. The disruption of membrane integrity of the bacterial cell wall was revealed by electronic microscopy, and changes in the localization of lipids from the enriched-septum region and the impairment of the formation of the septum were observed by fluorescence microscopy. This study revealed that 24 interferes with multiple targets, suggesting a low ability of Gram-positive bacteria to acquire resistance to this antibacterial agent.
2.2. Recent Reports on AAGs in the Field of Antibacterial Agents
Here, the main reports in the field of antibacterial AAGs, published after the appearance of several review articles since 2016 [7,8,30,31,32], are summarized with an emphasis on AAGs made of an AG core conjugated to an adjuvant or an antibiotic drug of another class (antibiotic hybrids), as recently developed by Schweizer, Zhanel and coworkers.
The grafting of a metal binding site on antimicrobial peptides (AMPs) improves their antibacterial efficiency [96,97,98]. In the search for antibacterial AAGs bearing a metal binding motif, 3′,4′,6-tri2NM NEA (16) [22] derivatives functionalized at position 5 through a short spacer by a Zn(II) or Cu(II) chelating group, tris(2-pyridylmethyl)amine (TPA), di(picolyl)amine (DPA) and tetraazacyclotetradecane (Cyclam) were synthesized [99]. NEA-cyclam and Zn(NEA-TPA) derivatives were found to be the most efficient compounds active against clinical MDR strain isolate Enterobacter aerogenes EA289, with MICs in the range of 16–4 and 4 μM, respectively, whereas usual antibiotics such as β-lactams and phenicols were inactive and ciprofloxacin was weakly active. NEA-Cyclam and Zn(NEA-TPA) were shown to target and permeabilize the OM of EA289. All NEA conjugates were able to block the efflux of 1,2′-dinaphthylamine in EA289 by acting on the efflux transporter located in the inner membrane [99].
A series of pyrene-NEO (PYR-NEO) conjugates were synthesized and their binding affinity to A-site RNA targets, resistance to AG-deactivating enzymes and antibacterial activity against a wide variety of bacterial strains of clinical relevance were studied [100]. As observed previously with NEA conjugates [35], the conjugation significantly alters the affinities of NEO for bacterial A-site targets. PYR-NEO conjugates exhibited broad-spectrum activity towards Gram-positive bacteria, including improved activity against NEO-resistant methicillin-resistant S. aureus (MRSA) strains. The conjugation significantly increased the resistance of NEO to AG-deactivating enzyme modification.
New antibacterial properties of amphiphilic AG conjugates were also recently highlighted. Antimicrobial hybrids that are AAGs have emerged in a promising strategy to combat bacterial resistance as drugs alone and as adjuvants in combination with existing antibiotics. The concepts, advances and challenges of antibiotic hybrids, including AAGs linked to an established antibiotic agent, were reviewed recently [101,102]. Such AAG hybrids can promote the influx of antibiotics through the OM of Gram-negative bacteria via the self-promoted uptake mechanism by displacement of the divalent cations (Ca2+ or Mg2+) that stabilize LPS [103].
Lysine-NEO conjugates have been synthesized and exhibited mainly decreased antibacterial activities [104]. Amphiphilic tobramycin (TOB) conjugates to lysine and anthracenyl groups 48 (Figure 9) appeared to be able to sensitize MDR Gram-negative bacteria to antibiotic drugs [105,106].
Combination studies indicate that these amphiphilic TOB-lysine conjugates 48 sensitize Gram-negative bacteria to antibiotics. The most potent derivative (n = 11, Figure 9) synergizes rifampicin and minocycline against MDR and extensively drug resistant (XDR) P. aeruginosa isolates and enhances efficacy of both antibiotics in the Galleria mellonella larvae in vivo infection model [105]. Mode of action studies indicate that the amphiphilic TOB-lysine adjuvants enhance OM cell penetration and affect the proton motive force, which energizes efflux pumps. The TOB-lysine conjugate 48 (n = 11) (Figure 9) also potentiates the antibacterial efficacy of eight clinically used antibiotics against wild-type, multidrug-resistant and extensively drug-resistant P. aeruginosa isolates from Canadian hospitals [106]. Antibiotics that are synergistic with 48 (n = 11) included moxifloxacin (MOX), ciprofloxacin (CIP), erythromycin, chloramphenicol, trimethoprim, novobiocin, linezolid, and fosfomycin. Novobiocin showed the highest synergy.
TOB conjugates to the efflux pump inhibitors (EPIs) 1-(1′-naphthylmethyl)piperazine (NMP) (49), paroxetine (PAR) (50), and dibasic naphthyl peptide (DBP) (51) (Figure 9) also overcome resistance against MDR P. aeruginosa isolates by enhancing bacterial OM penetration and reducing efflux [107]. These AAGs enhance the synergy and efficacy of EPIs in combination with tetracycline antibiotics against MDR Gram-negative bacteria, including P. aeruginosa. In addition to potentiating tetracycline antibiotics, TOB-EPI conjugates can also suppress resistance development to the tetracycline antibiotic minocycline, thereby providing a strategy to develop more effective adjuvants to rescue tetracycline antibiotics from resistance in MDR Gram-negative bacteria. A strong synergy with MOX, ciprofloxacin (CIP), rifampicin and fosfomycin was also observed against MDR/XDR P. aeruginosa [108].
AAG hybrids incorporating a fluoroquinolone were synthesized through conjugation of the MOX and CIP to the TOB core via an aliphatic alkyl linker. Among the TOB-MOX AAGs 52–54 (Figure 10) synthesized, the hybrid 52 was shown to enhance OM permeability and reduce efflux by dissipating the proton motive force of the cytoplasmic membrane in P. aeruginosa [109]. It protects Galleria mellonella larvae from the lethal effects of MDR P. aeruginosa. Attempts to select for resistance over a period of 25 days resulted in a two-fold increase in the MIC for the hybrid, whereas MOX or TOB resulted in a 16- and 512-fold increase in MIC. TOB-ciprofloxacin hybrid (TOB-CIP) adjuvants, in which CIP is attached at the TOB 5- (hybrids 55, Figure 10), 2″- or 6″-position, were shown to rescue the activity of fluoroquinolone antibiotics against MDR and XDR P. aeruginosa isolates in vitro and enhance fluoroquinolone efficacy in the Galleria mellonella in vivo infection model [110]. Structure–activity studies revealed that the presence of both TOB and CIP, which are separated by a C12 tether, is critical for the function of the adjuvant. Mechanistic studies indicate that the antibacterial modes of CIP are retained, whereas the role of TOB is limited to destabilization of the OM in the hybrid.
In vitro screening of six anticancer drugs for their potential use in antimicrobial therapy suggested the feasibility of repurposing the anticancer drug mitomycin C against MDR Gram-negative bacteria [111]. In combination with the TOB-CIP hybrid 55 (n = 12, Figure 10), the antibacterial activity of mitomycin C was enhanced against MDR clinical isolates of P. aeruginosa, A. baumannii, E. coli, K. pneumoniae and Enterobacter cloacae. Synergy was inherent to TOB-CIP and neither TOB nor CIP individually synergized with mitomycin C. The TOB-based hybrid adjuvants able to potentiate multiple classes of legacy antibiotics against various MDR Gram-negative bacteria were modified by replacing the TOB domain with the smaller pseudo-disaccharide NEB through selective cleavage of the α-D-glucopyranosyl linkage of TOB 8, which is an NEA (7) analogue lacking the 3′-hydroxyl function (Figure 2 and Figure 10) [112]. The hybrid AAGs NEB-moxifloxacin (NEB-MOX) 56, NEB-ciprofloxacin (NEB-CIP) 57 and NEB-1-(1′-naphthylmethyl)piperazine (NEB-NMP) 58 were synthesized (Figure 10). Potent synergism was found for combinations of NEB-based hybrid adjuvants with multiple classes of antibiotics, including fluoroquinolones (MOX and CIP), tetracyclines (minocycline) and rifamycins (rifampicin) against both wild-type and MDR P. aeruginosa clinical isolates. The combination of the optimized NEB-CIP hybrid 56 (Figure 10) and rifampicin protects G. mellonella larvae from the lethal effects of extensively drug-resistant (XDR) P. aeruginosa. Mechanistic evaluation of NEB-based hybrid adjuvants revealed that the hybrids affect the OM and IM of wild-type P. aeruginosa PAO1.
TOB-polymyxin B-3 hybrids showed potent activity against carbapenem-resistant as well as MDR or extensively drug-resistant (XDR) P. aeruginosa clinical isolates [113]. The most potent hybrid incorporating a C12 spacer was able to synergize with currently used antibiotics against wild-type and MDR/XDR P. aeruginosa and Acinetobacter baumannii.
More recently, TOB-rifampicin conjugates were also shown to break the intrinsic resistance in P. aeruginosa, due to chromosomally encoded low OM permeability and constitutively over-expressed efflux pumps, and sensitize MDR and XDR P. aeruginosa to doxycycline and chloramphenicol in vitro and in vivo [114]. Tetracyclines and chloramphenicol are model compounds for bacteriostatic effects, but when combined with TOB-rifampicin adjuvants, their effects became bactericidal at sub MIC levels. Potentiation of tetracyclines was found to be correlated with SAR of this class of drugs and consistent with OM permeabilization and efflux pump inhibition.
Conjugation of TOB and cyclam to give 59 (Figure 11) also abrogates the ribosomal effects of TOB but confers a potent adjuvant property that restores the full antibiotic activity of aztreonam and meropenem against carbapenem-resistant P. aeruginosa [115]. Therapeutic levels of susceptibility were attained in several MDR clinical isolates, and time-kill assays revealed a synergistic dose-dependent pharmacodynamic relationship. A triple combination of the adjuvant with ceftazidime/avibactam (approved), aztreonam/avibactam (phase III) and meropenem/avibactam enhances the efficacies of β-lactam/beta-lactamase inhibitors against recalcitrant strains, suggesting rapid access of the combination to their periplasmic targets. The adjuvants, and their combinations, were non-hemolytic and non-cytotoxic, and preliminary in vivo evaluation of 59 (n = 8) in G. mellonella suggested therapeutic potential for the double and triple combinations. The TOB–cyclam conjugate 59 (n = 8) mitigates the effect of OprD/OprF porin loss in P. aeruginosa and potentiates β-lactam/β-lactamase inhibitors against carbapenem-resistant clinical isolates, highlighting the complexity of resistance to β-lactam antibiotics.
The NEB-cyclam conjugate 60 (Figure 11) was shown to potentiate β-lactam antibiotics, as well as other antibiotic drugs, against P. aeruginosa in vitro [116]. This adjuvant is able to synergize with β-lactams aztreonam and ceftazidime against MDR and extremely drug-resistant clinical isolates through a hypothesized mechanism of OM permeabilization.
AGs were also modified for obtaining antibacterial amphiphilic nanoparticles. The brain neurotransmitter dopamine has been polymerized under aerobic conditions to produce polydopamine (PDA). Recently, self-polymerized PDA nanoparticles were tethered to AGs (gentamicin, KANA and NEO) [117]. These nanoconjugates were evaluated for their antimicrobial potency against various bacterial strains, including resistant ones, and their cytocompatibility. Of the three nanoconjugates (PDA-gentamicin, PDA-KANA and PDA-NEO), the PDA-KANA (PDA-K) nanoconjugate exhibited the highest activity against potent pathogens, the least toxicity to human embryonic kidney (HEK 293) cells and intense toxic effects to human glioblastoma (U87) cells.
2.3. AAG Positioning as Potential Antibacterial Drug Candidates, Toxicity
Dialkyl and trialkyl NEA derivatives have broad-spectrum antibacterial activity against susceptible and resistant Gram-positive and Gram-negative bacteria and their activity is related to favorable lipophilicity windows. Fridman and coworkers identified one amphiphilic 4′,5,6-triheptyl NEB derivative 80 (Figure 12) that is active against susceptible and resistant Gram-positive and Gram-negative bacteria (MRSA, E. coli, K. Pneumonia, P. aeruginosa strains, etc.) [37]. The corresponding trihexyl and trioctyl derivatives were found to be active against Gram-positive bacteria and inactive or weakly active against Gram-negative bacteria. As a consequence, the antibacterial Gram-positive and Gram-negative activity of amphiphilic NEB derivatives corresponds to a lipophilicity window that is much narrower than the windows delineated in the NEA series for trialkyl and dialkyl derivatives. This result is probably related to the higher lipophilicity of the NEB derivatives in comparison to the corresponding 3′,4′,6-trialkyl NEA derivatives [34,35].
A chemical way to increase the antimicrobial activity of the anti-Gram-positive and anti-Gram-negative NEB, TOB and PARO AAGs was reported in 2015 [38]. AAGs acting against a panel of Gram-positive and Gram-negative bacteria were obtained by di-N-methylation of all amine functions of the corresponding AAGs; for example, the 4′,5-dinonyl NEB derivative 81 was methylated to give the tetra-N-dimethyl derivative 82. However, strong hemolytic effects of the N-dimethyl NEB, TOB and PARO derivatives were observed and were related to the strong increase in lipophilicity in comparison to the parent non-N-dimethylated AAGs (an increase in the calculated logP by about a three orders of magnitude). Cetrimonium and gramicidin D, which are both in antibacterial topical clinical use, appeared to be significantly more hemolytic than all studied antibacterial AAGs. These results suggest a possible means of development of potent and broad-spectrum antibacterial membrane-disrupting AAGs for the treatment of persistent topical infections [38].
In order to evaluate the immediate toxicity of antibacterial AAGs, we performed a toxicity study in SKH1 male mice with AAGs 24 and 26 delivered in aqueous solution at concentrations higher than 5 µM, intravenously, intraperitoneally and subcutaneously in 100 µL injections twice a day. This study revealed immediate toxic effects (tail necrosis or swelling in the area of the injections and high mean body weight decrease; unpublished results obtained in collaboration with the preclinical Contract Research Organization (CRO), VOXCAN, Marcy l’Étoile, France). Such effects could be related, at least in part, to cellular membrane effects and to a low solubility in saline and buffered aqueous solutions. These results show that antibacterial AAGs have to be formulated to be delivered in vivo at high concentrations, for example in liposomes like the anionic and amphiphilic antifungal drug amphothericin B, which self-aggregates and is highly toxic [118]. Such formulations should allow the reduction of the concentration-dependent local toxic effects and the release of the AAGs at an efficient concentration.
The antibacterial potential of AAGs in antibiotherapy as adjuvants to antibiotic drugs also suggests their potential use at lower concentrations, rather than alone as antibiotic drugs. Recently, other adjuvants that are membrane-affecting small molecules were shown to be able to suppress COL resistance by abolishing or reducing the extent of lipid A modification induced by the mcr-1 gene and pmrAB system in E. coli [119,120]. AAGs 24, 26 and 27 are active against clinical COL-resistant P. aeruginosa strains (MICs = 28 µg/mL) and they cooperatively bind to LPS, increasing the OM permeability [42]. The much weaker and slower increase of MIC, in comparison to the fluoroquinolone CIP, of the 3′,6-dinonyl (24) and 3′,6-di2NP (26) NEA derivatives observed against P. aeruginosa illustrates the difficulty of resistance emergence to AAGs. These results suggest that 24 and/or 26 could be used at low concentrations as adjuvants, to reduce resistance to COL.
Another application of antibacterial AAGs could be found in their development as materials useful for antibacterial and anti-biofilm coating of surfaces, for example of catheters. Since trialkyl NEA derivatives are broad-spectrum antibacterial agents, their covalent attachment by one of the three alkyl groups to insoluble crosslinked polymers could produce interesting antibacterial materials.
3. Other Biological Activities of AAGs
Since the antibacterial activity of AAGs is related to narrow windows of lipophilicity and their molecular shape, it should be possible to discriminate different targets in adjusting, for example, the AAG lipophilicity and/or the corresponding hydrophilic/hydrophobic balance for different biological and medicinal applications.
3.1. Recent Advances in the Field of Antifungal AAGs
Recent years saw the expansion of AAGs in the search for new antifungal agents and probes for related mechanistic studies. The corresponding advances were reviewed in 2020 [121]. AAGs were shown to disrupt fungal cell membranes. As an example, the first antifungal non-antibacterial AAGs identified were KAN derivatives. Their development, through the delineation of structure–activity relationships, led to the novel antifungal agent K20 (Figure 13), which is capable of inhibiting many fungal species such as Fusarium graminearum, the causal agent in wheat Fusarium head blight (FHB) [122,123,124,125,126,127,128]. Plasma membrane permeabilization is probably the principal antifungal mechanism of action of AAGs, leading to the suggestion that they mechanistically and structurally comprise a novel class of antifungal agents [121,124,127].
Numerous antifungal AAG derivatives of KANA and KANB, NEB, trehalose, TOB and NEO were identified as antifungal agents or fluorogenic derivatives used as probes for related mechanistic studies. Recently, in a collaboration with D. Aldebert and M. Cornet (TIMC-IMAG/University Grenoble Alpes), we also identified some interesting antifungal antibacterial amphiphilic NEA and neosamine derivatives (unpublished results).
3.2. AAG Vehicles for Nucleic Acids, Effects on DNA or RNA
3.2.1. Gene and siRNA Delivery
AAGs have also received interest in relation to different medicinal applications, especially for intracellular delivery of active agents, for example, nucleic acids or analogues, non-covalently or covalently bound to AAGs or AGs. The synthesis and development of gene delivery vehicles was recently reviewed [129]. Here, we mainly illustrate the precursor works of J.M. Lehn, P. Lehn and coworkers.
Amphiphilic TOB, KANA, PARO, NEO and NEA derivatives were demonstrated to be efficient intracellular delivery vehicles for biologically relevant molecules such as genes [130,131,132,133,134] and small interfering RNA (siRNA) [135]. The transfection potential of KANA and of its tri-guanidinylated derivative, conjugated to cholesterol through the formation of a carbamate group, was explored [130,131,132]. The KANA conjugate 61 (Figure 14) was highly efficient for gene transfection into a variety of mammalian cell lines when used either alone or as a liposomal formulation with the neutral phospholipid dioleoylphosphatidylethanolamine (DOPE). The polyguanidinylated derivative mediated in vitro gene transfection. In addition, the colloidally stable KANA-cholesterol conjugate/DOPE lipoplexes were found to be efficient for in vivo gene transfection into mouse airways. AAGs consisting of cholesteryl or dioleyl moieties linked via various spacers to PARO or NEO headgroups are also efficient for gene transfection both in vitro and into mouse airways in vivo, and the physico-chemical properties of their DNA lipocomplexes were investigated in order to delineate structure–activity relationships [130].
NEA derivatives 62–65, bearing long dialkyl chains, one or two NEA headgroups and four to ten protonatable amine functions, were also prepared for gene transfection through the selective alkylation of the 4′- or 5-hydroxyl function in ring I and ring II of NEA, respectively (Figure 14) [134]. The transfection activity of the twelve derivatives synthesized was investigated in vitro in gene transfection experiments, using several mammalian cell lines. The most efficient derivative, 63 (Figure 15), carries two octadecyl chains linked to a diaminoethyl spacer attached at the 4′-position of the NEA core. Both the presence of that spacer and such an attachment position, i.e., at an extremity of the NEA core, appear to play a role in the increase of the gene transfection efficiency.
Chemically synthesized small interfering RNAs (siRNAs) can be used to induce specific and reversible gene expression silencing in mammalian cells. Cationic lipids commonly used for DNA transfection have also been used for siRNA transfection. To deliver siRNA into cells, two dioleyl chains were grafted through a succinyl spacer to the TOB, KANA, PARO and NEO cores to give AAGs 66, 67, 68 and 69, respectively (Figure 16) [135]. The most active AAG/siRNA complexes for gene silencing were the PARO and NEO derivatives 68 and 69, which were characterized as small particles exhibiting lamellar microdomains, corresponding to siRNA sandwiched between the lipid bilayers.
More recently, in the search for gene delivery vehicles with inherent antibacterial properties, NEO AAGs 1, PARO 2 and NEA 6 were conjugated to the tetramino-tetrahexyloxycalix [4]arene scaffold [136]. The three synthesized conjugates exhibited greater DNA binding ability than the gold standard transfectant 25 kDa bPEI, as well as a striking DNA packing ability. An antibacterial effect of the NEO and PARO AAGs and their DNA lipoplexes was observed against E. coli.
3.2.2. Peptide (Polyamide) Nucleic Acid (PNA)-AG Conjugates to Target RNA
In the antisense approach developed to target, for example, messager RNA sequences by hybridization and to inhibit the corresponding translation, conjugates of AGs (NEO, NEA, PARA, ribostamycin and methyl neobiosamine) to oligo-2′-deoxyribonucleotides (ODNs) were synthesized in order to allow the ODN cellular uptake [137,138,139,140,141,142]. However, in the resulting conjugates, the strong binding of AGs to the ODN through intramolecular charge–charge interaction between the protonated AG core and the phosphodiester backbone can disturb the selective binding to the RNA target.
Peptide nucleic acid oligomers (PNAs) are a class of antisense DNA analogues devoid of charges under physiological conditions, first synthesized by Buchardt, Nielsen and coworkers in 1991 [143]. Their design, based upon a repeating N-(2-aminoethyl)glycine polyamide backbone to which nucleobases are attached through a methylene carbonyl linkage to the α-amino group, allows the perfect H-bonding to the RNA and DNA complementary sequences. PNAs are chemically stable and show superior resistance to nucleases and proteases than nucleic acids. However, their low solubility in water and their poor cellular uptake hampered their development in the antisense approach. PNAs can be used in an antibacterial approach, consisting in specifically targeting any single pathogen. For example, potent antibacterial antisense peptide–peptide nucleic acid conjugates were developed against P. aeruginosa [144]. A recent review highlighted the potential of gene-specific oligonucleotides and PNAs as antibacterial agents in order to broaden the range of potential targets to any gene with a known sequence in any bacterium [145].
AGs are attractive cationic vehicles to conjugate to PNAs. The resulting AAG conjugates should be more soluble in water than the PNAs, and the conjugation could allow an efficient cellular uptake of the PNAs. Therefore, chemical methods for the conjugation of PNAs to AGs have been developed [142,146,147,148,149,150]. We conjugated the NEA core with a 16-mer PNA targeting HIV-1 TAR RNA (anti-TAR PNA) [146]. In the resulting conjugate 73 (Figure 16), the presence of the NEA core not only strongly increased the solubility in water and enhanced the cellular uptake of PNA, it also conferred a unique metal ion-independent, target-specific RNA cleavage property to the conjugate [146,148].
We also conjugated the 6-amino-6-deoxy-1-methylglucosamine (1-methyl neosamine) core corresponding to ring II of NEA, to a 16-mer PNA targeting HIV-1 TAR RNA [151]. The resulting conjugate 74 (Figure 17) was stable under acidic conditions and displayed very high target specificity in vitro and strongly inhibited Tat-mediated transactivation of HIV-1 LTR transcription in a cell culture system. The fluorescent derivative 75 was efficiently taken up by the human cells and well distributed in both the cytosol and nucleus without endosomal entrapment because co-treatment with endosome-disrupting agent had no effect on its cellular distribution.
A strategy consisting of recognizing and selectively targeting RNA duplex sequences (ds) with chemically modified dsRNA-binding PNAs (dbPNAs), incorporating thiopseudoisocytosine (L) and guanidine-modified 5-methyl cytosine (Q) residues at physiologically relevant conditions, was developed to facilitate the sequence-specific recognition of Watson−Crick G-C and C-G pairs [152]. A short 10-mer dbPNA targeting a highly conserved panhandle duplex structure of influenza A virion, not accessible to traditional antisense DNA or RNA with a similar length, was conjugated to the NEA core to enhance the cellular uptake [153]. It was shown to inhibit innate immune receptor RIG-I binding to panhandle structure and thus RIG-I ATPase activity. These findings provide the foundation for developing novel dbPNAs for the detection of influenza viral RNAs and therapeutics with optimal antiviral and immunomodulatory activities. An antisense PNA was also conjugated to the NEA core to enter cells and was used to target and probe the tau pre-mRNA exon 10 5′-splice hairpin structure through strand invasion [154].
3.2.3. Some Particular Effects of AAGs on DNA and RNA
The study of 6″-substituted variants of TOB, derivatives with C-12 or C-14 linear alkyl substituents, revealed a potent inhibition of reverse transcription in vitro, which was related to nucleic acid binding with high affinity and the formation of high-molecular weight complexes [155]. Stable complex formation was observed with DNA or RNA in single- or double-stranded form, suggesting that the formation of micelles and/or vesicles with surface-bound nucleic acids may be a useful tool to localize nucleic acids to surfaces or deliver nucleic acids to cells or organelles.
Abasic sites are probably the most common lesions in DNA, resulting from the hydrolytic cleavage of glycosidic bonds that can occur spontaneously and through DNA alkylation, especially at N-7 of purine [156]. The Nobel Prize in Chemistry for 2015 was awarded to T. Lindahl, P. Modrich and A. Sancar for mapping at a molecular level how cells repair damaged DNA in order to preserve genetic information. Compounds able to specifically bind and react at abasic sites have attracted much attention for therapeutic and diagnostic purposes. We studied the efficient cleavage activity of AG antibiotic drugs and of some AAGs at abasic sites, introduced either by depurination in a plasmidic DNA or site-specifically in a synthetic oligonucleotide [157]. NEO was found to be the most efficient cleaving AG drug, followed by the AAG 76 (Figure 18) resulting from the conjugation of the NEA core to the nucleic base adenine (EC50 = 0.09 and 0.15 µM, respectively, and EC50(NEA) = 0.9 µM).
3.3. AAGs as Intracellular Delivery Vectors of Drugs
The cationic amphiphilic stimuli-responsive azobenzene-NEO conjugate 77 (Figure 18) was synthesized and its self-assembly in aqueous solutions into nanostructures, in which eosin and aspirin were successfully encapsulated, was demonstrated [158]. The ability of the resulting nanostructures to act as drug carriers, in a UV- and visible light-mediated release pattern or through azoreductase-mediated cleavage of the azo moiety, was also investigated. Stimuli responsiveness of nanostructures and their on/off-switch-like behavior appeared to be of interest, in relation to their use as controlled drug delivery systems and in other biomedical applications, such as colon-specific delivery and gene delivery. Amphiphilic PARO-derived nanoparticles were also prepared for drug delivery from mPEG-PAE (methoxy-terminated poly(ethylene glycol)-poly(aminoether)) nanoparticles [159]. They were able to carry significant amounts of the anticancer drug doxorubicin, and cell-based studies indicated that the nanoparticles loaded with doxorubicin were able to induce a significant loss in the viability of cancer cells.
3.4. A New Target of AAGs
Connexin hemichannels (HCs) from adjacent cells form gap junctional channels to mediate cell-to-cell communication. Abnormal opening of “free” undocked HCs can produce cell damage and participate in the mechanism of disorders such as cardiac infarction, stroke, deafness, skin diseases and cataracts. Therefore, inhibitors of connexin HCs have great pharmacological potential. AGs have been recently identified as connexin HC inhibitors, but their antibiotic effect is an issue for the treatment of disorders where infections do not play a role [160]. Several AAGs without antibiotic effect were synthesized and tested for their inhibition against connexin HCs, using a newly developed cell-based bacterial growth complementation assay. Several leads, 79 (Figure 19), that are non-bactericidal and non-toxic or moderately toxic to mammalian HeLa cells, with superior potency than the parent compound, KANA, were identified.
4. Discussion and Conclusions
AAGs have been found to have many potential applications of biological and medicinal interest. Several interesting key targets were identified for antibacterial AAGs—ribosomal RNA (a target of AAGs carrying small lipophilic groups) [9,10], as well as LPS [25,42,89] and cardiolipin [89,90,93] in membranes of Gram-negative bacteria and lipoteichoic acids in membranes of Gram-positive bacteria [95]. The presence of these different bacterial targets allows for broad-spectrum antibacterial activity and limits the emergence of resistance to antibiotic AAGs. It could be a limitation in the bacterial/mammalian cell selectivity leading to toxic effects, since cardiolipin is present in mammalian cell membranes. The selectivity of AAGs for bacterial membranes should be improved through mechanistic studies and the careful design of the active species, for example, in order to selectively target LPS or cardiolipin clusters, especially LPS, which is a unique component of bacterial membranes.
We showed that the broad-spectrum antibacterial activity of AAGs is dependent on their lipophilicity, corresponding to narrow windows of clogP (or clogD) [34,35]. Several types of AAGs may be defined, for example, from the composition of their lipophilic part. A first type corresponds to antibacterial and/or antifungal AAGs carrying one or more lipophilic groups (alkyl, arylalkyl) including only carbon and hydrogen atoms, like broad-spectrum antibacterial NEA and PARA derivatives [5,34,35,36]. Such AAGs target bacterial and/or fungal membranes. The identification of non-antibacterial antifungal AAGs targeting fungal membranes demonstrates that the main targets of AAGs can be discriminated [121,122,123,124,125,126,127]. This selectivity could be found in the difference between monoalkylated and di- or tri-alkylated derivatives of similar lipophilicities.
A second type of AAGs corresponds to AG conjugates in which the lipophilic part incorporates heteroatoms (mainly O and N). In a promising strategy to combat bacterial resistance, antibacterial AAGs have also emerged as adjuvants in combination with existing antibiotics. AG conjugates to an efflux pump inhibitor or to an antibiotic drug of another class (antibiotic hybrids) can promote, as adjuvants, the uptake of antibiotics through the OM of Gram-negative bacteria via the self-promoted uptake mechanism by displacement of the divalent cations (Ca2+ or Mg2+), which stabilize LPS [101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116]. The high affinity of strongly lipophilic AAGs of the first and second types for nucleic acids was used to develop efficient non-antibacterial vehicles for nucleic acid uptake in mammalian cells [129,130,131,132,133,134,135]. AAGs have also been employed for drug delivery [158,159] and their cationic characters should be exploited to deliver anionic drugs. More recently, weakly lipophilic monoalkyl AAGs were demonstrated to be non-antibacterial inhibitors of connexin hemichannels [160].
Altogether, these results suggest that it should be possible to discriminate different anionic targets through a fine adjustment of the lipophilicity, of the number of positive charges present at physiological conditions, of the hydrophilicity/hydrophobicity balance and the corresponding molecular shape of AAGs. In our opinion, the number of charges should be limited in order to increase the selectivity, as well as the lipophilicity, which increased the cytotoxicity in the NEA series. The detailed understanding of the mechanisms of the antibacterial and antifungal effects, as well as the internalization in mammalian cells, of AAGs having different lipophilicities, and of their lipoplexes, should contribute strongly to a structural design that can improve their activity and their selectivity. There is no doubt that they will be used as efficient tools in many future biological, pharmacological and medicinal studies.
Funding
This research received no external funding.
Acknowledgments
Many thanks to all who contributed to the works reported here and to the financial agencies that supported them, the Belgian Funds for Scientific Research (Fonds National de la Recherche Scientifique (grants no. 3.4.588.10, 3.4.578.12, T.1003.14, J.0205.16, and T.175.20), French National Research Agency (ANR; “Physique et Chimie du Vivant” 1998 and ANR-07-PCVI-0030), Indo−French Centre for the Promotion of Advanced Research (IFCPAR, project no. 3405-1), Région Rhône-Alpes (ARC 1, grant no. 12-00887201), LABEX ARCANE (ANR-11-LABX-003 and CBH-EUR-GS (ANR-17-EURE-0003)), SATT Linksium Grenoble Alpes, (project ANTIGRAM), «Fondation pour la Recherche Médicale» (FRM, post-doctoral grant for CD and JK, DBF20161136768).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AAG | Amphiphilic aminoglycoside |
| AG | Aminoglycoside |
| Bu | n-butyl |
| CIP | Ciprofloxacin |
| CL | Cardiolipin |
| clogP | Calculated log(octanol/water partition coefficient) |
| COL | Colistin |
| DiMOc | 3,7-(dimethyl)octyl |
| DBP | Dibasic naphthyl peptide |
| De | Decyl |
| EPI | Efflux pump inhibitor |
| Hx | n-hexyl |
| Hp | n-heptyl |
| IM | Inner membrane |
| KANA | Kanamycin A 3 |
| KANB | Kanamycin B 4 |
| LPS | Lipopolysaccharides |
| MDR | Multidrug-resistant |
| MIC | Minimum inhibitory concentration |
| MOX | Moxifloxacin |
| MRSA | Methicillin resistant S. aureus |
| 2NB | 2-naphthylbutyl |
| NEA | Neamine 6 |
| NEB | Nebramine 8 |
| NEO | Neomycin B 1 |
| 2NH | 2-naphthylhexyl |
| 2NM | 2-naphthylmethyl |
| NMP | 1-(1′-naphthylmethyl)piperazine |
| Nn | n-nonyl |
| 2NP | 2-naphthylpropyl |
| Oc | n-octyl |
| Ocd | n-octadecyl (C18) |
| OM | Outer membrane |
| PAR | Paroxetine |
| PARA | Paromamine 7 |
| PARO | Paromomycin 2 |
| PDA | Polydopamine |
| PE | Phosphatidylethanolamine |
| PG | Phosphatidylglycerol |
| PMB | para-methoxybenzyl |
| POPE | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine |
| POPG | 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) |
| PNA | Peptide (polyamide) nucleic acid |
| SAR | Structure-activity relationships |
| TOB | Tobramycin 5 |
| Tr | Trityl |
| Ud | n-undecyl (C11) |
| XDR | Extensively drug-resistant |
References
- Chandrika, N.T.; Garneau-Tsodikova, S. Comprehensive review of chemical strategies for the preparation of new aminoglycosides and their biological activities. Chem. Soc. Rev. 2018, 47, 1189–1249. [Google Scholar]
- Garneau-Tsodikova, S.; Labby, K.J. Mechanisms of resistance to aminoglycoside antibiotics: Overview and perspectives. MedChemComm 2016, 7, 11–27. [Google Scholar]
- Zhang, J.; Chiang, F.I.; Takemoto, J.Y.; Bensaci, M.; Litke, A.; Czyryca, P.G.; Chang, C.W.T. Surprising alteration of antibacterial activity of 5′’-modified neomycin against resistant bacteria. J. Med. Chem. 2008, 51, 7563–7573. [Google Scholar] [PubMed] [Green Version]
- Bera, S.; Zhanel, G.G.; Schweizer, F. Design, synthesis and antibacterial activities of neomycin-lipid conjugates: Polycationic lipids with potent gram-positive activity. J. Med. Chem. 2008, 51, 6160–6164. [Google Scholar]
- Baussanne, I.; Bussière, A.; Halder, S.; Ganem-Elbaz, C.; Ouberai, M.; Riou, M.; Paris, J.M.; Ennifar, E.; Mingeot-Leclercq, M.P.; Décout, J.L.; et al. Synthesis and antimicrobial evaluation of amphiphilic neamine derivatives. J. Med. Chem. 2010, 53, 119–127. [Google Scholar]
- Herzog, I.M.; Green, K.D.; Berkov-Zrihen, Y.; Feldman, M.; Vidavski, R.R.; Eldar-Boock, A.; Satchi-Fainaro, R.; Eldar, A.; Garneau-Tsodikova, S.; Fridman, M.; et al. 6″-Thioether tobramycin analogues: Towards selective targeting of bacterial membranes. Angew. Chem. Int. Ed. Engl. 2012, 51, 1–6. [Google Scholar]
- Gorityala, B.K.; Guchhait, G.; Schweizer, F. Carbohydrates in drug design and discovery. In Chapter Amphiphilic Aminoglycoside Antimicrobials in Antibacterial Discovery; Jimenez-Barbero, J., Canada, F.J., Martin-Santamaria, S., Eds.; Royal Society of Chemistry: London, UK, 2015; pp. 255–285. [Google Scholar]
- Mingeot-Leclercq, M.P.; Décout, J.L. Bacterial lipid membranes as promising targets to fight antimicrobial resistance, molecular foundations and illustration through the renewal of aminoglycoside antibiotics and emergence of amphiphilic aminoglycosides. MedChemComm 2016, 7, 586–611. [Google Scholar]
- François, B.; Szychowski, J.; Adhikari, S.S.; Pachamuthu, K.; Swayze, E.E.; Griffey, R.H.; Migawa, M.T.; Westhof, E.; Hanessian, S. Antibacterial aminoglycosides with a modified mode of binding to the ribosomal-RNA decoding site. Angew. Chem. Int. Ed. 2004, 43, 6735–6738. [Google Scholar]
- Szychowski, J.; Kondo, J.; Zahr, O.; Auclair, K.; Westhof, E.; Hanessian, S.; Keillor, J.W. Inhibition of aminoglycoside-deactivating enzymes APH(3′)-IIIa and AAC(6′)-II by amphiphilic paromomycin O2″-ether analogues. ChemMedChem 2011, 6, 1961–1966. [Google Scholar]
- Baral, B.; Mozafari, M.R. Strategic moves of “superbugs” against available chemical scaffolds: Signaling, regulation, and challenges. ACS Pharmacol. Transl. Sci. 2020, 3, 373–400. [Google Scholar]
- MacNair, C.R.; Tsai, C.N.; Brown, E.D. Creative targeting of the gram-negative outer membrane in antibiotic discovery. Ann. N. Y. Acad. Sci. 2020, 1459, 69–85. [Google Scholar] [PubMed]
- Schmidt, N.W.; Deshayes, S.; Hawker, S.; Blacker, A.; Kasko, A.M.; Wong, G.C.L. Engineering persister-specific antibiotics with synergistic antimicrobial functions. ACS Nano 2014, 8, 8786–8793. [Google Scholar] [PubMed] [Green Version]
- Allison, K.R.; Brynildsen, M.P.; Collins, J.J. Heterogeneous bacterial persisters and engineering approaches to eliminate them. Curr. Opin. Microbiol. 2011, 14, 593–598. [Google Scholar] [PubMed] [Green Version]
- Hurdle, J.G.; O’Neill, A.J.; Chopra, I.; Lee, R.E. Targeting bacterial membrane function: An underexploited mechanism for treating persistent infections. Nat. Rev. Microbiol. 2011, 9, 62–75. [Google Scholar]
- Ganewatta, M.S.; Tang, C. Controlling macromolecular structures towards effective antimicrobial polymers. Polymers 2015, 63, A1–A29. [Google Scholar]
- Steinbuch, K.B.; Fridman, M. Mechanisms of resistance to membrane-disrupting antibiotics in gram-positive and gram-negative bacteria. MedChemComm 2016, 7, 86–102. [Google Scholar]
- Yin, J.; Meng, Q.; Cheng, D.; Fu, J.; Luo, Q.; Liu, Y.; Yu, Z. Mechanisms of bactericidal action and resistance of polymyxins for Gram-positive bacteria. Appl. Microbiol. Biotechnol. 2020, 104, 3771–3780. [Google Scholar]
- Moubareck, C.A. Polymyxins and bacterial membranes: A review of antibacterial activity and mechanisms of resistance. Membranes 2020, 10, 30. [Google Scholar]
- Gellatly, S.L.; Hancock, R.E. Pseudomonas aeruginosa: New insights into pathogenesis and host defenses. Pathog. Dis. 2013, 67, 159–173. [Google Scholar]
- Airoldi, C.; Sommaruga, S.; Merlo, S.; Sperandeo, P.; Cipolla, L.; Polissi, A.; Nicotra, F. Targeting bacterial membranes: Identification of Pseudomonas aeruginosa D-arabinose-5P isomerase and NMR characterisation of its substrate recognition and binding properties. Chembiochem 2011, 12, 719–727. [Google Scholar]
- Putker, F.; Bos, M.P.; Tommassen, J. Transport of lipopolysaccharide to the Gram-negative bacterial cell surface. FEMS Microbiol. Rev. 2015, 39, 985–1002. [Google Scholar] [PubMed] [Green Version]
- Sperandeo, P.; Martorana, A.M.; Polissi, A. Lipopolysaccharide biogenesis and transport at the outer membrane of Gram-negative bacteria. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2017, 1862, 1451–1460. [Google Scholar] [PubMed]
- Choi, U.; Lee, C.R. Antimicrobial agents that inhibit the outer membrane assembly machines of Gram-negative bacteria. J. Microbiol. Biotechnol. 2019, 29, 1–10. [Google Scholar] [PubMed]
- Ouberai, M.; El Garch, F.; Bussière, A.; Riou, M.; Alsteens, D.; Lins, L.; Baussanne, I.; Dufrêne, Y.F.; Brasseur, R.; Décout, J.L.; et al. The Pseudomonas aeruginosa membranes: A target for a new amphiphilic aminoglycoside derivative? Biochem. Biophys. Acta Biomembr. 2011, 1808, 1716–1727. [Google Scholar]
- Dong, H.; Zhang, Z.; Tang, X.; Huang, S.; Li, H.; Peng, B.; Dong, C. Structural insights into cardiolipin transfer from the inner membrane to the outer membrane by PbgA in gram-negative bacteria. Sci. Rep. 2016, 6, 10. [Google Scholar]
- Bramkamp, M.; van Baarle, S. Division site selection in rod-shaped bacteria. Curr. Opin. Microbiol. 2009, 12, 683–688. [Google Scholar]
- Renner, L.D.; Weibel, D.B. MinD and MinE interact with anionic phospholipids and regulate division plane formation in Escherichia Coli. J. Biol. Chem. 2012, 287, 38835–38844. [Google Scholar]
- Evans, E.A.; Waugh, R. Osmotic correction to elastic area compressibility measurements on red cell membrane. Biophys. J. 1977, 20, 307–313. [Google Scholar]
- Fosso, Y.; Li, Y.; Garneau-Tsodikova, S. New trends in the use of aminoglycosides. MedChemComm 2014, 5, 1075–1091. [Google Scholar]
- Chandrika, N.T.; Garneau-Tsodikova, S.A. Review of patents (2011–2015) towards combating resistance to and toxicity of aminoglycosides. MedChemComm 2016, 7, 50–68. [Google Scholar]
- Bera, S.; Mondal, D.; Palit, S.; Schweizer, F. Structural modifications of the neomycin class of aminoglycosides. MedChemComm 2016, 7, 1499–1534. [Google Scholar] [CrossRef]
- Aradi, K.; di Giorgio, A.; Duca, M. Recent progresses in aminoglycoside conjugation for RNA targeting: Antimicrobials and beyond. Chem. Eur. J. 2020, 26. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Bussière, A.; Ouberai, M.; Baussanne, I.; Jolivalt, C.; Mingeot-Leclercq, M.P.; Décout, J.L. Tuning the antibacterial activity of amphiphilic neamine derivatives and comparison to paromamine homologues. J. Med. Chem. 2013, 56, 7691–7705. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Kempf, J.; Briée, F.; Swain, J.; Mingeot-Leclercq, M.P.; Décout, J.L. Broad-spectrum antibacterial amphiphilic aminoglycosides: A new focus on the structure of the lipophilic groups extends the series of active dialkyl NEAs. Eur. J. Med. Chem. 2018, 157, 1512–1525. [Google Scholar] [CrossRef]
- Zimmermann, L.; Das, I.; Désiré, J.; Sautrey, G.; Barros, R.S.V.; El Khoury, M.; Mingeot-Leclercq, M.P.; Décout, J.L. New broad-spectrum antibacterial amphiphilic aminoglycosides active against resistant bacteria: From neamine derivatives to smaller neosamine analogues. J. Med. Chem. 2016, 59, 9350–9369. [Google Scholar] [CrossRef]
- Berkov-Zrihen, Y.; Herzog, I.M.; Benhamou, R.I.; Feldman, M.; Steinbuch, K.B.; Shaul, P.; Lerer, S.; Eldar, A.; Fridman, M. Tobramycin and Nebramine as pseudo-oligosaccharide scaffolds for the development of antimicrobial cationic amphiphiles. Chem. Eur. J. 2015, 21, 4340–4349. [Google Scholar] [CrossRef]
- Benhamou, R.I.; Shaul, P.; Herzog, I.M.; Fridman, M. Di-N-methylation of anti-Gram-positive aminoglycoside-derived membrane disruptors improves antimicrobial potency and broadens spectrum to Gram-negative bacteria. Angew. Chem. Int. Ed. Engl. 2015, 54, 13617–13621. [Google Scholar] [CrossRef]
- Riguet, E.; Désiré, J.; Bailly, C.; Décout, J.L. A route for preparing new NEA derivatives targeting HIV-1 TAR RNA. Tetrahedron 2004, 60, 8053–8064. [Google Scholar] [CrossRef]
- Riguet, E.; Désiré, J.; Boden, O.; Ludwig, V.; Göbel, M.; Bailly, C.; Décout, J.L. NEA dimers targeting the HIV-1 TAR RNA. Bioorg. Med. Chem. Lett. 2005, 15, 4651–4655. [Google Scholar] [CrossRef]
- Jackowski, O.; Bussière, A.; Vanhaverbeke, C.; Baussanne, I.; Peyrin, E.; Mingeot-Leclercq, M.P.; Décout, J.L. Major increases of the reactivity and selectivity in aminoglycoside O-alkylation due to the presence of fluoride ions. Tetrahedron 2012, 68, 737–746. [Google Scholar] [CrossRef]
- Sautrey, G.; Zimmermann, L.; Deleu, M.; Delbar, A.; Souza, M.L.; Jeannot, K.; Van, B.; Buyck, J.M.; Décout, J.L.; Mingeot-Leclercq, M.P. New amphiphilic NEA derivatives active against resistant Pseudomonas aeruginosa and their interactions with lipopolysaccharides. Antimicrob. Agents Chemother. 2014, 58, 4420–4430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, F.; Nguyen Pham, D.T.; Kim, Y.M. Alternative strategies for the application of aminoglycoside antibiotics against the biofilm-forming human pathogenic bacteria. Appl. Microbiol. Biotechn. 2020, 104, 1955–1976. [Google Scholar] [CrossRef] [PubMed]
- Steinbuch, K.B.; Benhamou, R.I.; Levin, L.; Stein, R.; Fridman, M. Increased degree of unsaturation in the lipid of antifungal cationic amphiphiles facilitates selective fungal cell disruption. ACS Infect. Dis. 2018, 4, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Kusnlr, J.; Barna, K. Fluorimetric determination of some basic antibiotics at very low concentrations. Z. Anal. Chem. 1974, 271, 288. [Google Scholar] [CrossRef]
- El-Shabrawy, Y. Fluorimetric determination of aminoglycoside antibiotics in pharmaceutical preparations and biological fluids. Spectrosc. Lett. 2002, 35, 99–109. [Google Scholar] [CrossRef]
- Tekkeli, S.E.K.; Önal, A.; Sagirli, A.O. Spectrofluorimetric determination of tobramycin in human serum and pharmaceutical preparations by derivatization with fluorescamine. Luminescence 2012, 29, 87–91. [Google Scholar] [CrossRef]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide Endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef] [Green Version]
- Raetz, C.R.H.; Reynolds, C.M.; Trent, M.S.; Bishop Russell, E. Lipid A modification systems in gram-negative bacteria. Annu. Rev. Biochem. 2007, 76, 295–329. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Quinn, P.J. Lipopolysaccharide: Biosynthetic pathway and structure modification. Prog. Lipid Res. 2010, 49, 97–107. [Google Scholar] [CrossRef]
- Bohl, T.E.; Aihara, H. Current progress in the structural and biochemical characterization of proteins involved in the assembly of lipopolysaccharide. Int. J. Microbiol. 2018, 2018, 5319146. [Google Scholar] [CrossRef] [Green Version]
- Qiao, S.; Luo, Q.; Zhao, Y.; Zhang, X.C.; Huang, Y. Structural basis for lipopolysaccharide insertion in the bacterial outer membrane. Nature 2014, 511, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Lam, N.H.; Ha, B.Y. Surface-lattice model describes electrostatic interactions of ions and polycations with bacterial lipopolysaccharides: Ion valence and polycation’s excluded area. Langmuir 2014, 30, 13631–13640. [Google Scholar] [CrossRef] [PubMed]
- Neville, F.; Hodges, C.S.; Liu, C.; Konovalov, O.; Gidalevitz, D. In situ characterization of lipid A interaction with antimicrobial peptides using surface X-ray scattering. Biochim. Biophys. Act. 2006, 1758, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Schertzer, J.W.; Yong, X. Molecular dynamics modeling of pseudomonas aeruginosa outer membranes. Phys. Chem. Chem. Phys. 2018, 20, 23635–23648. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, K.; Funari, S.S.; Koch, M.H.; Seydel, U. Investigation into the acyl chain packing of endotoxins and phospholipids under near physiological conditions by WAXS and FTIR spectroscopy. J. Struct. Biol. 1999, 128, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Dias, R.P.; Lin, L.; Soares, T.A.; Alexov, E. Modeling the electrostatic potential of asymmetric lipopolysaccharide membranes: The MEMPOT algorithm Implemented in DelPhi. J. Comput. Chem. 2014, 35, 1418–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandenburg, K.; Andra, J.; Muller, M.; Koch, M.H.; Garidel, P. physicochemical properties of bacterial glycopolymers in relation to bioactivity. Carbohydr. Res. 2003, 338, 2477–2489. [Google Scholar] [CrossRef] [PubMed]
- Parasassi, T.; Gratton, E.; Yu, W.M.; Wilson, P.; Levi, M. Two-photon fluorescence microscopy of Laurdan generalized polarization domains in model and natural membranes. Biophys. J. 1997, 72, 2413–2429. [Google Scholar] [CrossRef] [Green Version]
- Kathi Scheinpflug, K.; Krylova, O.; Strahl, H. Measurement of cell membrane fluidity by laurdan GP: Fluorescence spectroscopy and microscopy. Methods Mol. Biol. 2017, 1520, 159–174. [Google Scholar]
- Pristovsek, P.; Kidric, J. Solution structure of polymyxins B and E and effect of binding to lipopolysaccharide: An NMR and molecular modeling study. J. Med. Chem. 1999, 42, 4604–4613. [Google Scholar] [CrossRef]
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure-activity relationships of polymyxin antibiotics. J. Med. Chem. 2010, 53, 1898–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beveridge, T.J. Structures of gram-negative cell walls and their derived membrane vesicles. J. Bacteriol. 1999, 181, 4725–4733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seydel, U.; Oikawa, M.; Fukase, K.; Kusumoto, S.; Brandenburg, K. Intrinsic conformation of lipid A is responsible for agonistic and antagonistic activity. Eur. J. Biochem. 2000, 267, 3032–3039. [Google Scholar] [CrossRef] [PubMed]
- Mashburn-Warren, L.; Howe, J.; Garidel, P.; Richter, W.; Steiniger, F.; Roessle, M.; Brandenburg, K.; Whiteley, M. Interaction of quorum signals with outer membrane lipids: Insights into prokaryotic membrane vesicle formation. Mol. Microbiol. 2008, 69, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Howe, J.; Hammer, M.; Alexander, C.; Rossle, M.; Fournier, K.; Mach, J.P.; Waelli, T.; Gorczynski, R.M.; Ulmer, A.J.; Zahringer, U.; et al. Biophysical characterization of the interaction of endotoxins with hemoglobins. Med. Chem. 2007, 3, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalebroux, Z.D.; Matamouros, S.; Whittington, D.; Bishop, R.E.; Miller, S.I. PhoPQ Regulates acidic glycerophospholipid content of the Salmonella typhimurium outer membrane. Proc. Natl. Acad. Sci. USA 2014, 111, 1963–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helander, I.M.; Mattila-Sandholm, T. Fluorometric assessment of Gram-negative bacterial permeabilization. J. Appl. Microbiol. 2000, 88, 213–219. [Google Scholar] [CrossRef]
- Olofsson, G.; Sparr, E. Ionization constants pKa of cardiolipin. PLoS ONE 2013, 8, e73040. [Google Scholar] [CrossRef] [Green Version]
- Boeris, P.S.; Domenech, C.E.; Lucchesi, G.I. Modification of phospholipid composition in pseudomonas putida a ATCC 12633 induced by contact with tetradecyltrimethylammonium. J. Appl. Microbiol. 2007, 103, 1048–1054. [Google Scholar] [CrossRef]
- Lopez, G.A.; Heredia, R.M.; Boeris, P.S.; Lucchesi, G.I. Content of cardiolipin of the membrane and sensitivity to cationic surfactants in Pseudomonas putida. J. Appl. Microbiol. 2016, 121, 1004–1014. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, H.; Takami, H.; Inoue, A.; Horikoshi, K. Effects of hydrostatic pressure and temperature on growth and lipid composition of the inner membrane of Barotolerant Pseudomonas Sp. BT1 isolated from the deep-sea. Biosci. Biotechnol. Biochem. 2000, 64, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broniatowski, M.; Mastalerz, P.; Flasinski, M. Studies of the interactions of ursane-type bioactive terpenes with the model of escherichia coli inner membrane-langmuir monolayer approach. Biochim. Biophys. Acta 2015, 1848, 469–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalebroux, Z.D.; Edrozo, M.B.; Pfuetzner, R.A.; Ressl, S.; Kulasekara, B.R.; Blanc, M.P.; Miller, S.I. Delivery of cardiolipins to the Salmonella outer membrane is necessary for survival within host tissues and virulence. Cell. Host Microbe 2015, 17, 441–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghorbal, S.K.; Chatti, A.; Sethom, M.M.; Maalej, L.; Mihoub, M.; Kefacha, S.; Feki, M.; Landoulsi, A.; Hassen, A. Changes in membrane fatty acid composition of Pseudomonas aeruginosa in response to UV-C radiations. Curr. Microbiol. 2013, 67, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Pizzuto, M.; Lonez, C.; Baroja-Mazo, A.; Martinez-Banaclocha, H.; Tourlomousis, P.; Gangloff, M.; Pelegrin, P.; Ruysschaert, J.M.; Gay, N.J.; Bryant, C.E. Saturation of acyl chains converts cardiolipin from an antagonist to an activator of toll-like receptor-4. Cell. Mol. Life Sci. 2019, 76, 3679–3680. [Google Scholar] [CrossRef] [Green Version]
- Garrett, T.A.; O’Neill, A.C.; Hopson, M.L. Quantification of cardiolipin molecular species in escherichia coli lipid extracts using liquid chromatography/electrospray ionization mass spectrometry. Rapid. Commun. Mass Spectrom. 2012, 26, 2267–2274. [Google Scholar] [CrossRef]
- Oemer, G.; Lackner, K.; Muigg, K.; Krumschnabel, G.; Watschinger, K.; Sailer, S.; Lindner, H.; Gnaiger, E.; Wortmann, S.B.; Werner, E.R.; et al. Molecular structural diversity of mitochondrial cardiolipins. Proc. Natl. Acad. Sci. USA 2018, 115, 4158–4163. [Google Scholar] [CrossRef] [Green Version]
- Prossnigg, F.; Hickel, A.; Pabst, G.; Lohner, K. Packing behaviour of two predominant anionic phospholipids of bacterial cytoplasmic membranes. Biophys. Chem. 2010, 150, 129–135. [Google Scholar] [CrossRef]
- Pluschke, G.; Overath, P. Function of phospholipids in Escherichia coli. Influence of changes in polar head group composition on the lipid phase transition and characterization of a mutant containing only saturated phospholipid acyl chains. J. Biol. Chem. 1981, 256, 3207–3212. [Google Scholar]
- Nichols-Smith, S.; Teh, S.Y.; Kuhl, T.L. Thermodynamic and mechanical properties of model mitochondrial membranes. Biochim. Biophys. Acta 2004, 1663, 82–88. [Google Scholar] [CrossRef]
- Dowhan, W. Molecular basis for membrane phospholipid diversity: Why are there so many lipids? Annu. Rev. Biochem. 1997, 66, 199–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlovsky, Y.; Chernomordik, L.V.; Kozlov, M.M. Lipid intermediates in membrane fusion: Formation, structure, and decay of hemifusion diaphragm. Biophys. J. 2002, 83, 2634–2651. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.F.; Martinou, J.C.; Fornallaz-Mulhauser, M.; Hughes, D.W.; Epand, R.M. The apoptotic protein tBid promotes leakage by altering membrane curvature. J. Biol. Chem. 2002, 277, 32632–32639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beales, P.A.; Bergstrom, C.L.; Geerts, N.; Groves, J.T.; Vanderlick, T.K. Single vesicle observations of the cardiolipin-cytochrome C interaction: Induction of membrane morphology changes. Langmuir 2011, 27, 6107–6115. [Google Scholar] [CrossRef] [Green Version]
- Rogasevskaia, T.P.; Coorssen, J.R. A new approach to the molecular analysis of docking, priming, and regulated membrane fusion. J. Chem. Biol. 2011, 4, 117–136. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.M.; Epand, R.F. Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochim. Biophys. Acta 2009, 1788, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Romantsov, T.; Gonzalez, K.; Sahtout, N.; Culham, D.E.; Coumoundouros, C.; Garner, J.; Kerr, C.H.; Chang, L.; Turner, R.J.; Wood, J.M. Cardiolipin synthase A colocalizes with cardiolipin and osmosensing transporter ProP at the poles of Escherichia coli cells. Mol. Microbiol. 2018, 107, 623–638. [Google Scholar] [CrossRef] [Green Version]
- Sautrey, G.; El Koury, M.; Dos Santos, A.G.; Zimmermann, L.; Deleu, M.; Lins, L.; Décout, J.L.; Mingeot-Leclercq, M.P. Negatively charged lipids as a potential target for new amphiphilic aminoglycoside antibiotics: A biophysical study. J. Biol. Chem. 2016, 291, 13864–13874. [Google Scholar] [CrossRef] [Green Version]
- El Khoury, M.; Swain, J.; Sautrey, G.; Zimmermann, L.; Van der Smissen, P.; Décout, J.L.; Mingeot-Leclercq, M.P. Targeting bacterial cardiolipin enriched microdomains: An antimicrobial strategy used by amphiphilic aminoglycoside antibiotics. Sci. Rep. 2017, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Van den Brink-van der Laan, E.; Boots, J.W.P.; Spelbrink, R.E.J.; Kool, G.M.; Breukink, E.; Killian, J.A.; de Kruijff, B. Membrane interaction of the glycosyltransferase MurG: A special role for cardiolipin. J. Bacteriol. 2003, 185, 3773–3779. [Google Scholar] [CrossRef] [Green Version]
- Ugarte-Uribe, B.; Müller, H.M.; Otsuki, M.; Nickel, W.; García-Sáez, A.J. Dynamin-related protein 1 (Drp1) promotes structural intermediates of membrane division. J. Biol. Chem. 2014, 289, 30645–30656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swain, J.; El Khoury, M.; Kempf, J.; Briée, F.; van der Smissen, P.; Décout, J.L.; Mingeot-Leclercq, M.P. Effect of cardiolipin on the antimicrobial activity of new amphiphilic aminoglycoside derivative on P. aeruginosa. PLoS ONE 2018, 13, e0201752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roszkowiak, J.; Jajor, P.; Guła, G.; Gubernator, J.; Żak, A.; Drulis-Kawa, Z.; Augustyniak, D. Interspecies outer membrane vesicles (OMVs) modulate the sensitivity of pathogenic bacteria and pathogenic yeasts to cationic peptides and serum complement. Int. J. Mol. Sci. 2019, 20, 5577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swain, J.; El Khoury, M.; Flament, A.; Dezanet, C.; Briée, F.; van der Smissen, P.; Decout, J.L.; Mingeot-Leclercq, M.P. Antimicrobial activity of amphiphilic neamine derivatives: Understanding the mechanism of action on gram-positive bacteria. Biochem. Biophys. Acta Biomembr. 2019, 1861, 10. [Google Scholar] [CrossRef] [PubMed]
- Libardo, M.D.; Cervantes, J.L.; Salazar, J.C.; Angeles-Boza, A.M. Improved bioactivity of antimicrobial peptides by addition of amino-terminal copper and nickel (ATCUN) binding motifs. ChemMedChem. 2014, 9, 1892–1901. [Google Scholar] [CrossRef] [Green Version]
- Libardo, M.D.J.; Nagella, S.; Lugo, A.; Pierce, S.; Angeles-Boza, A.M. Copper binding tripeptide motif increases potency of the antimicrobial peptide anoplin via reactive oxygen species generation. Biochem. Biophys. Res. Commun. 2015, 456, 446–451. [Google Scholar] [CrossRef]
- Joyner, J.C.; Hodnick, W.F.; Cowan, T.D.; Boyd, R.; Cowan, J.A. Antimicrobial metallopeptides with broad nuclease and ribonuclease activity. Chem. Commun. 2013, 49, 2118–2120. [Google Scholar] [CrossRef]
- Allam, A.; Maigre, L.; Alves de Sousa, R.; Dumont, E.; Vergalli, J.; Pages, J.M.; Artaud, I. New amphiphilic neamine conjugates bearing a metal binding motif active against MDR E. aerogenes gram-negative bacteria. Eur. J. Med. Chem. 2017, 12, 748–756. [Google Scholar] [CrossRef]
- Story, S.; Skriba, M.J.; Maiti, K.; Ranjan, N.; Degtyareva, N.N.; Green, K.D.; Khodaverdian, V.; Oyelere, A.K.; Garneau-Tsodikova, S.; Arya, D.P.; et al. Synthesis, antimicrobial activity, attenuation of aminoglycoside resistance in MRSA, and ribosomal A-site binding of pyrene-neomycin conjugates. Eur. J. Med. Chem. 2019, 163, 381–393. [Google Scholar] [CrossRef]
- Domalaon, R.; Idowu, T.; Zhanel, G.G.; Schweizer, F. Antibiotic hybrids: The next generation of agents and adjuvants against gram-negative pathogens? Clin. Microb. Rev. 2018, 31, 45. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; Datta, P. Next-generation strategy for treating drug resistant bacteria: Antibiotic hybrids. Indian J. Med. Res. 2019, 7, 97–106. [Google Scholar]
- Schweizer, F. Enhancing uptake of antibiotics into gram-negative bacteria using nonribosome-targeting aminoglycoside-based adjuvants. Future Med. Chem. 2019, 11, 1519–1522. [Google Scholar] [CrossRef] [PubMed]
- Bera, S.; Zhanel, G.; Schweizer, F. Synthesis and antibacterial activity of amphiphilic lysine-ligated neomycin B conjugates. Carbohydr. Res. 2011, 346, 560–568. [Google Scholar]
- Lyu, Y.; Yang, X.; Goswami, S.; Gorityala, B.K.; Idowu, T.; Domalaon, R.; Zhanel, G.G.; Shan, A.; Schweizer, F. Amphiphilic tobramycin-lysine conjugates sensitize multidrug resistant gram-negative bacteria to rifampicin and minocycline. J. Med. Chem. 2017, 60, 3684–3702. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.; Domalaon, R.; Yang, X.; Schweizer, F. Amphiphilic lysine conjugated to tobramycin synergizes legacy antibiotics against wild-type and multidrug-resistant Pseudomonas aeruginosa. Peptide Sci. 2019, 111, 7. [Google Scholar] [CrossRef]
- Yang, X.; Goswami, S.; Gorityala, B.K.; Domalaon, R.; Lyu, Y.; Kumar, A.; Zhanel, G.G.; Schweizer, F. A Tobramycin vector enhances synergy and efficacy of efflux pump inhibitors against multidrug-resistant Gram-negative bacteria. J. Med. Chem. 2017, 60, 3913–3932. [Google Scholar] [CrossRef]
- Yang, X.; Domalaon, R.; Lyu, Y.; Zhanel, G.G.; Schweizer, F. Tobramycin-linked efflux pump inhibitor conjugates synergize fluoroquinolones, rifampicin and fosfomycin against multidrug-resistant Pseudomonas aeruginosa. J. Clin. Med. 2018, 7, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorityala, B.K.; Guchhait, G.; Goswami, S.; Fernando, D.M.; Kumar, A.; Zhanel, G.G.; Schweizer, F. Hybrid antibiotic overcomes resistance in P. aeruginosa by enhancing outer membrane penetration and reducing efflux. J. Med. Chem. 2016, 59, 8441–8455. [Google Scholar]
- Gorityala, B.K.; Guchhait, G.; Fernando, D.M.; Deo, S.; McKenna, S.A.; Zhanel, G.G.; Kumar, A.; Schweizer, F. Adjuvants based on hybrid antibiotics overcome resistance in Pseudomonas aeruginosa and enhance fluoroquinolone efficacy. Angew. Chem. Int. Ed. Engl. 2016, 55, 555–559. [Google Scholar] [CrossRef]
- Domalaon, R.; Ammeter, D.; Brizuela, M.; Gorityala, B.K.; Zhanel, G.G.; Schweizer, F. Repurposed antimicrobial combination therapy: Tobramycin-Ciprofloxacin hybrid augments activity of the anticancer drug Mitomycin C against Multidrug-Resistant Gram-Negative bacteria. Front. Microbiol. 2019, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Ammeter, D.; Idowu, T.; Domalaon, R.; Brizuela, M.; Okunnu, O.; Bi, L.; Guerrero, Y.A.; Zhanel, G.G.; Kumar, A.; et al. Amphiphilic nebramine-based hybrids rescue legacy antibiotics from intrinsic resistance in multidrug-resistant gram-negative bacilli. Eur. J. Med. Chem. 2019, 175, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Domalaon, R.; Yang, X.; Lyu, Y.; Zhanel, G.G.; Schweizer, F. Polymyxin B-3-tobramycin hybrids with Pseudomonas aeruginosa-selective antibacterial activity and strong potentiation of rifampicin, minocycline, and vancomycin. ACS Infect. Dis. 2017, 3, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Idowu, T.; Arthur, G.; Zhanel, G.G.; Schweizer, F. Heterodimeric rifampicin-tobramycin conjugates break intrinsic resistance of Pseudomonas aeruginosa to doxycycline and chloramphenicol in vitro and in a Galleria mellonella in vivo model. Eur. J. Med. Chem. 2019, 174, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Idowu, T.; Ammeter, D.; Arthur, G.; Zhanel, G.G.; Schweizer, F. Potentiation of β-lactam antibiotics and β -lactam/β-lactamase inhibitor combinations against MDR and XDR Pseudomonas aeruginosa using non-ribosomal Tobramycin-Cyclam conjugates. J. Antimicrob. Chemother. 2019, 74, 2640–2648. [Google Scholar] [CrossRef] [PubMed]
- Ammeter, D.; Idowu, T.; Zhanel, G.G.; Schweizer, F. Development of a nebramine-cyclam conjugate as an antibacterial adjuvant to potentiate beta-lactam antibiotics against multidrug-resistant P. aeruginosa. J. Antibiot. 2019, 72, 816–826. [Google Scholar] [CrossRef]
- Singh, I.; Priyam, A.; Jha, D.; Dhawan, G.; Gautam, H.K.; Kumar, P. Polydopamine-aminoglycoside nanoconjugates: Synthesis, characterization, antimicrobial evaluation and cytocompatibility. Mater. Sci. Eng. C 2020, 107, 11. [Google Scholar] [CrossRef]
- Torrado, J.J.; Espada, R.; Ballesteros, M.P.; Torrado-Santiago, S. Amphotericin B formulations and drug targeting. J. Pharm. Sci. 2008, 97, 2405–2425. [Google Scholar]
- Barker, W.T.; Martin, S.E.; Chandler, C.E.; Nguyen, T.V.; Harris, T.L.; Goodell, C.; Melander, R.J.; Doi, Y.; Ernst, R.K.; Melander, C. Small molecule adjuvants that suppress both chromosomal and mcr-1 encoded colistin-resistance and amplify colistin efficacy in polymyxin-susceptible bacteria. Bioorg. Med. Chem. 2017, 25, 5749–5753. [Google Scholar] [CrossRef]
- Kathayat, D.; Antony, L.; Deblais, L.; Helmy, Y.A.S.; Caria, J.; Rajashekara, G. Small molecule adjuvants potentiate Colistin activity and attenuate resistance development in Escherichia coli by affecting pmrAB system. Infect. Drug Resist. 2020, 13, 2205–2222. [Google Scholar] [CrossRef]
- Howard, K.C.; Dennis, E.K.; Watt, D.S.; Garneau-Tsodikova, S. A comprehensive overview of the medicinal chemistry of antifungal drugs including: Perspectives and promise. Chem. Soc. Rev. 2020, 49, 2426–2480. [Google Scholar] [CrossRef]
- Chang, C.W.T.; Takemoto, J.Y. Antifungal amphiphilic aminoglycosides. Med. Chem. Commun. 2014, 5, 1048–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.W.T.; Fosso, M.; Kawasaki, Y.; Shrestha, S.; Bensaci, M.F.; Wang, J.; Evans, C.K.; Takemoto, J.Y. Divergent synthesis of three classes of antifungal amphiphilic kanamycin derivatives. J. Antibiot. 2010, 63, 667–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemoto, J.Y.; Wegulo, S.N.; Yuen, G.Y.; Stevens, J.A.; Jochum, C.C.; Chang, C.W.T.; Kawasaki, Y.; Miller, G.W. Suppression of wheat Fusarium head blight by novel amphiphilic aminoglycoside fungicide K20. Fungal Biol. 2017, 122, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Subedi, Y.P.; AlFindee, M.N.; Takemoto, J.Y.; Chang, C.W.T. Antifungal amphiphilic kanamycins: New life for an old drug. MedChemComm 2018, 9, 909–919. [Google Scholar] [CrossRef]
- Subedi, Y.P.; Roberts, P.; Grilley, M.; Takemoto, J.Y.; Chang, C.T. Development of fungal selective amphiphilic kanamycin: Cost-effective synthesis and use of fluorescent analogs for mode of action investigation. ACS Infect. Dis. 2019, 5, 473–483. [Google Scholar] [CrossRef]
- Alfindee, M.N.; Subedi, Y.P.; Grilley, M.M.; Takemoto, J.Y.; Chang, C.W.T. Antifungal activities of 4″,6″-disubstituted amphiphilic kanamycins. Molecules 2019, 24, 1882. [Google Scholar] [CrossRef] [Green Version]
- Subedi, Y.P.; Pandey, U.; Alfindee, M.N.; Montgomery, H.; Roberts, P.; Wight, J.; Nichols, G.; Grilley, M.; Takemoto, J.Y.; Chang, C.W.T.; et al. Scalable and cost-effective tosylation-mediated synthesis of antifungal and fungal diagnostic 6″-Modified amphiphilic kanamycins. Eur. J. Med. Chem. 2019, 182, 8. [Google Scholar] [CrossRef]
- Bellucci, M.C.; Volonterio, A. Aminoglycosides: From antibiotics to building blocks for the synthesis and development of gene delivery vehicles. Antibiotics 2020, 9, 504. [Google Scholar] [CrossRef]
- Belmont, P.; Aissaoui, A.; Hauchecorne, M.; Oudrhiri, N.; Petit, L.; Vigneron, J.P.; Lehn, J.M.; Lehn, P. Aminoglycoside-derived cationic lipids as efficient vectors for gene transfection in vitro and in vivo. J. Gene Med. 2002, 4, 517–526. [Google Scholar] [CrossRef]
- Sainlos, M.; Belmont, P.; Vigneron, J.P.; Lehn, P.; Lehn, J.M. Aminoglycoside-derived cationic lipids for gene transfection: Synthesis of kanamycin A derivatives. Eur. J. Org. Chem. 2003, 15, 2764–2774. [Google Scholar] [CrossRef]
- Sainlos, M.; Hauchecorne, M.; Oudrhiri, N.; Zertal-Zidani, S.; Aissaoui, A.; Vigneron, J.P.; Lehn, J.M.; Lehn, P. Kanamycin A-derived cationic lipids as vectors for gene transfection. ChemBioChem 2005, 6, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Mevel, M.; Sainlos, M.; Chatin, B.; Oudrhiri, N.; Hauchecorne, M.; Lambert, O.; Vigneron, J.P.; Lehn, P.; Pitard, B.; Lehn, J.M. Paromomycin and neomycin B derived cationic lipids: Synthesis and transfection studies. J. Control. Release 2012, 158, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, T.; Baussanne, I.; Halder, S.; Carmoy, N.; Montier, T.; Lehn, P.; Décout, J.L. Synthesis and transfection properties of a series of lipidic neamine derivatives. Bioconjugate Chem. 2009, 20, 2032–2046. [Google Scholar] [CrossRef] [PubMed]
- Desigaux, L.; Sainlos, M.; Lambert, O.; Chèvre, R.; Letrou-Bonneval, E.; Vigneron, J.P.; Lehn, P.; Lehn, J.M.; Pitard, B. Self-assembled lamellar complexes of siRNA with lipidic aminoglycoside derivatives promote efficient siRNA delivery and interference. Proc. Natl. Acad. Sci. USA 2007, 104, 16534–16539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bono, N.; Pennetta, C.; Sganappa, A.; Giupponi, E.; Sansone, F.; Volonterio, A.; Candiani, G. Design and synthesis of biologically active cationic amphiphiles built on the calix [4]arene scaffold. Int. J. Pharm. 2018, 549, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Irudayasamy, C.; Xi, H.; Arya, D.P. Sequence specific targeting of RNA with an oligonucleotide-neomycin conjugate. Bioconjugate Chem. 2007, 18, 160–169. [Google Scholar]
- Ketomäki, K.; Virta, P. Synthesis of aminoglycoside conjugates of 2′-O-methyl oligoribonucleotides. Bioconjugate Chem. 2008, 19, 766–777. [Google Scholar] [CrossRef]
- Kiviniemi, A.; Virta, P.; Lönnberg, H. Utilization of intrachain 4′-C-azidomethylthymidine for preparation of oligodeoxyribonucleotide conjugates by click chemistry in solution and on a solid support. Bioconjugate Chem. 2008, 19, 1726–1734. [Google Scholar] [CrossRef]
- Kiviniemi, A.; Virta, P.; Lönnberg, H. Solid supported synthesis and click conjugation of 4′-C-alkyne functionalized oligodeoxyribonucleotides. Bioconjugate Chem. 2010, 21, 1890–1901. [Google Scholar] [CrossRef]
- Kiviniemi, A.; Virta, P. Synthesis of aminoglycoside-3′- conjugates of 2′-O-methyl oligoribonucleotides and their invasion to a 19F labeled HIV-1 TAR model. Bioconjugate Chem. 2011, 22, 1559–1566. [Google Scholar] [CrossRef]
- Mei, H.; Xing, L.; Cai, L.; Hong-Wei, J.; Zhao, P.; Yang, Z.J.; Zhang, L.R.; Zhang, L.H. Studies on the synthesis of neamine−dinucleosides and neamine−PNA conjugates and their interaction with RNA. Bioorg. Med. Chem. Lett. 2008, 18, 5355–5358. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, A.; Nielsen, P. Potent antibacterial antisense peptide–peptide nucleic acid conjugates against Pseudomonas aeruginosa. Nucleic Acid Ther. 2012, 22, 323–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojciechowska, M.; Rownicki, M.; Mieczkowski, A.; Miszkiewicz, J.; Trylska, J. Antibacterial peptide nucleic acids-facts and perspectives. Molecules 2020, 25, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riguet, E.; Tripathi, S.; Chaubey, B.; Désiré, J.; Pandey, V.N.; Décout, J.L. A peptide nucleic acid−neamine conjugate that targets and cleaves HIV-1 TAR RNA inhibits viral replication. J. Med. Chem. 2004, 47, 4806–4809. [Google Scholar] [CrossRef]
- Charles, I.; Arya, D.P. Synthesis of neomycin−DNA/peptide nucleic acid conjugates. J. Carbohydr. Chem. 2005, 24, 145–160. [Google Scholar] [CrossRef]
- Soonsil, H.; Hyun, L.K.; Jaehoon, Y. A strategy for the design of selective RNA binding agents. Preparation and RRE RNA binding affinities of a neomycin−peptide nucleic acid heteroconjugate library. Bioorg. Med. Chem. Lett. 2006, 16, 4757–4759. [Google Scholar]
- Chaubey, B.; Tripathi, S.; Désiré, J.; Baussanne, I.; Décout, J.L.; Pandey, V.N. Mechanism of RNA cleavage catalyzed by sequence specific polyamide nucleic acid−neamine conjugate. Oligonucleotides 2007, 17, 302–313. [Google Scholar] [CrossRef]
- Alguacil, J.; Defaus, S.; Claudio, A.; Trapote, A.; Masides, M.; Robles, J. A straigtforward preparation of aminoglycoside−dinucleotide and−diPNA conjugates via Click ligation assisted by microwaves. Eur. J. Org. Chem. 2010, 20, 3102–3109. [Google Scholar] [CrossRef]
- Das, I.; Désiré, J.; Manvar, D.; Baussanne, I.; Décout, J.L. A peptide nucleic acid-aminosugar conjugate targeting transactivation response element of HIV-1 RNA genome shows a high bioavailability in human cells and strongly inhibits Tat-mediated transactivation of HIV-1 transcription. J. Med. Chem. 2012, 55, 6021–6032. [Google Scholar] [CrossRef]
- Devi, G.; Yuan, Z.; Lu, Y.; Zhao, Y.; Chen, G. Incorporation of thio-pseudoisocytosine into triplex-forming peptide nucleic acids for enhanced recognition of RNA duplexes. Nucleic Acids Res. 2014, 42, 4008–4018. [Google Scholar] [CrossRef] [PubMed]
- Kesy, J.; Patil, K.; Subaschandrabose, R.; Shu, Z.; Yong, H.Y.; Zimmermann, L.; Ong, A.A.L.; Toh, D.F.K.; Krishna, M.S.; Yang, L.; et al. A Short chemically-modified dsRNA-binding PNA (dbPNA) inhibits influenza viral replication by targeting viral RNA panhandle structure. Bioconjugate Chem. 2019, 30, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Ong, A.A.L.; Tan, J.; Bhadra, M.; Dezanet, C.; Patil, K.M.; Chong, M.S.; Kierzek, R.; Decout, J.L.; Roca, X.; Chen, G.; et al. RNA secondary structure-based design of antisense peptide nucleic acids for modulating disease-associated aberrant tau pre-mRNA alternative splicing. Molecules 2019, 24, 3020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, L.; Zhu, H.; Fosso, M.Y.; Garneau-Tsodikova, S.; Fredrick, K. Modified aminoglycosides bind nucleic acids in high-molecular-weight complexes. Antibiotics 2020, 9, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, M.M. The formamidopyrimidines: Purine lesions formed in competition with 8-oxopurines from oxidative stress. Accounts Chem. Res. 2012, 45, 588–597. [Google Scholar] [CrossRef] [Green Version]
- Perigolo, M.; Constant, J.F.; Peuchmaur, M.; Pitta, I.; Décout, J.L. Antibiotic drugs aminoglycosides cleave DNA at abasic sites: Shedding new light on their toxicity? Chem. Res. Tox. 2013, 26, 1710–1719. [Google Scholar] [CrossRef]
- Deka, S.R.; Yadav, S.; Mahato, M.; Sharma, A.K. Azobenzene-aminoglycoside: Self-assembled smart amphiphilic nanostructures for drug delivery. Colloid Surface B 2015, 135, 150–157. [Google Scholar] [CrossRef]
- Miryala, B.; Godeshala, S.; Grandhi, T.S.P.; Christensen, M.D.; Tian, Y.; Rege, K. Aminoglycoside-derived amphiphilic nanoparticles for molecular delivery. Colloid Surface B 2016, 146, 924–937. [Google Scholar] [CrossRef]
- AlFindee, M.N.; Subedi, Y.P.; Fiori, M.C.; Krishnan, S.; Kjellgren, A.; Altenberg, G.A.; Chang, C.W.T. Inhibition of connexin hemichannels by new amphiphilic aminoglycosides without antibiotic activity. ACS Med. Chem. Lett. 2018, 9, 697–701. [Google Scholar] [CrossRef]
Figure 1.
Structures of natural antibiotic aminoglycosides 1–5, of some corresponding constitutive derivatives 6–8 and of synthetic intermediates used to prepare amphiphilic aminoglycosides (AAGs) 9–13 (Tr = trityl group = triphenylmethyl, PMB = para-methoxyphenyl group).
Figure 1.
Structures of natural antibiotic aminoglycosides 1–5, of some corresponding constitutive derivatives 6–8 and of synthetic intermediates used to prepare amphiphilic aminoglycosides (AAGs) 9–13 (Tr = trityl group = triphenylmethyl, PMB = para-methoxyphenyl group).

Figure 2.
Structure of polymyxin E (COL), showing the five amine functions protonated at physiological pH.
Figure 2.
Structure of polymyxin E (COL), showing the five amine functions protonated at physiological pH.

Figure 3.
Structures of the first identified broad-spectrum antibacterial amphiphilic neamine (NEA) (6) and paromamine (PARA) (7) derivatives [5,34].

Figure 4.
Structures of the identified broad-spectrum antibacterial amphiphilic dialkyl (24 and 25) and dialkylnaphthyl (26–28) NEA derivatives [34].
Figure 4.
Structures of the identified broad-spectrum antibacterial amphiphilic dialkyl (24 and 25) and dialkylnaphthyl (26–28) NEA derivatives [34].

Figure 5.
Structures of the broad-spectrum antibacterial 3′,4′-dinonyl NEA derivative and of the corresponding antibacterial analogues synthesized in the 1-methyl neosamine series [36].
Figure 5.
Structures of the broad-spectrum antibacterial 3′,4′-dinonyl NEA derivative and of the corresponding antibacterial analogues synthesized in the 1-methyl neosamine series [36].

Figure 6.
Structures of antibacterial homodialkyl (38–41, 46, 47) and heterodialkyl (42–45) NEA derivatives synthesized [35].
Figure 6.
Structures of antibacterial homodialkyl (38–41, 46, 47) and heterodialkyl (42–45) NEA derivatives synthesized [35].

Figure 7.
Values of 1/(MIC (mL/µg) as a function of clogP values for 3′,6-dinaphthylalkyl NEAs (di2NM 14, di2NP 26, di2NB 27 and di2-naphthylhexyl) and 3′,6-dialkyl NEAs (diC4, diC6, diC7 38, diC8 39, diC9 24, diC10 40, diC11 41 and diC18). (A) Against MRSA; (B) against susceptible P. aeruginosa ATCC 27853. Naphthylalkyl derivatives: red squares; alkyl derivatives: green triangles [35].
Figure 7.
Values of 1/(MIC (mL/µg) as a function of clogP values for 3′,6-dinaphthylalkyl NEAs (di2NM 14, di2NP 26, di2NB 27 and di2-naphthylhexyl) and 3′,6-dialkyl NEAs (diC4, diC6, diC7 38, diC8 39, diC9 24, diC10 40, diC11 41 and diC18). (A) Against MRSA; (B) against susceptible P. aeruginosa ATCC 27853. Naphthylalkyl derivatives: red squares; alkyl derivatives: green triangles [35].

Figure 8.
Comparison of the structures of lipid A and cardiolipin (CL) to the structure of the antibacterial 3′,6-dinonyl NEA derivative 24.
Figure 8.
Comparison of the structures of lipid A and cardiolipin (CL) to the structure of the antibacterial 3′,6-dinonyl NEA derivative 24.

Figure 9.
Structures of the tobramycin (TOB) conjugates to lysine 48 [105,106], and to the efflux pump inhibitors (EPIs), 1-(1′-naphthylmethyl)piperazine (NMP) (49), paroxetine (PAR) (50) and dibasic naphthyl peptide (DBP) (51) [107,108].

Figure 10.
Structures of the TOB (52–54) and nebramine (NEB) (56) conjugates to the fluoroquinolones moxifloxacin (MOX) and ciprofloxacin (CIP) [109,111], respectively 55 and 57, and, of the NEB conjugates to the efflux pump inhibitor 1-(1′-naphthylmethyl)piperazine (NEB-NMP) 58 [112].


Figure 12.
Structures of the synthesized broad-spectrum antibacterial 4′,5,6-tri- and 4′,5-di-alkylated NEB derivatives [37,38].

Figure 13.
Structure of the amphiphilic aminoglycoside K20, capable of inhibiting many fungal species such as Fusarium graminearum, the causal agent wheat Fusarium head blight (FHB) [125,126,127,128].

Figure 14.
Structure of one of the kanamycin (KANA)-cholesterol conjugates, 61, developed for gene transfection [130].
Figure 14.
Structure of one of the kanamycin (KANA)-cholesterol conjugates, 61, developed for gene transfection [130].

Figure 15.
Structures of the most efficient NEA-based vectors for gene transfection [134].
Figure 15.
Structures of the most efficient NEA-based vectors for gene transfection [134].

Figure 16.
Structures of the AAGs developed for small interfering RNA (siRNA) delivery made of the TOB, KANA, PARO and NEO cores, respectively, linked to two dioleyl chains by a succinyl spacer [135].
Figure 16.
Structures of the AAGs developed for small interfering RNA (siRNA) delivery made of the TOB, KANA, PARO and NEO cores, respectively, linked to two dioleyl chains by a succinyl spacer [135].

Figure 17.
Structures of the anti-HIV (anti-TAR RNA) PNA conjugates to NEA [145,149] and to 6-amino-6-deoxy-1-methylglucosamine (1-methyl neosamine) [151].

Figure 18.
Structure of the most efficient DNA-cleaving AAG identified, 76, at abasic sites [157], and of the amphiphilic azobenzene-NEO conjugate 77 forming nanostructures [158].

Figure 19.
Structures of AAGs that are non-bactericidal and non-toxic or moderately toxic to mammalian HeLa cells, which are connexin hemichannel (HC) inhibitors [160].
Figure 19.
Structures of AAGs that are non-bactericidal and non-toxic or moderately toxic to mammalian HeLa cells, which are connexin hemichannel (HC) inhibitors [160].


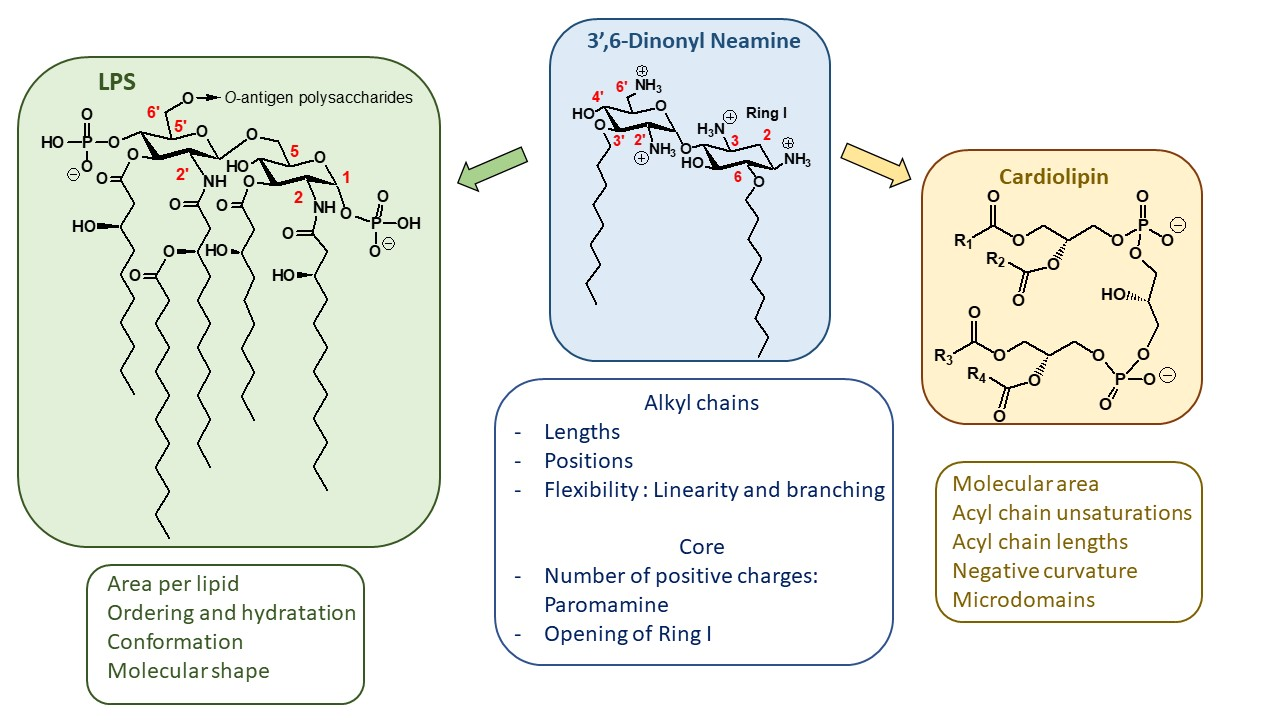{kind=link}
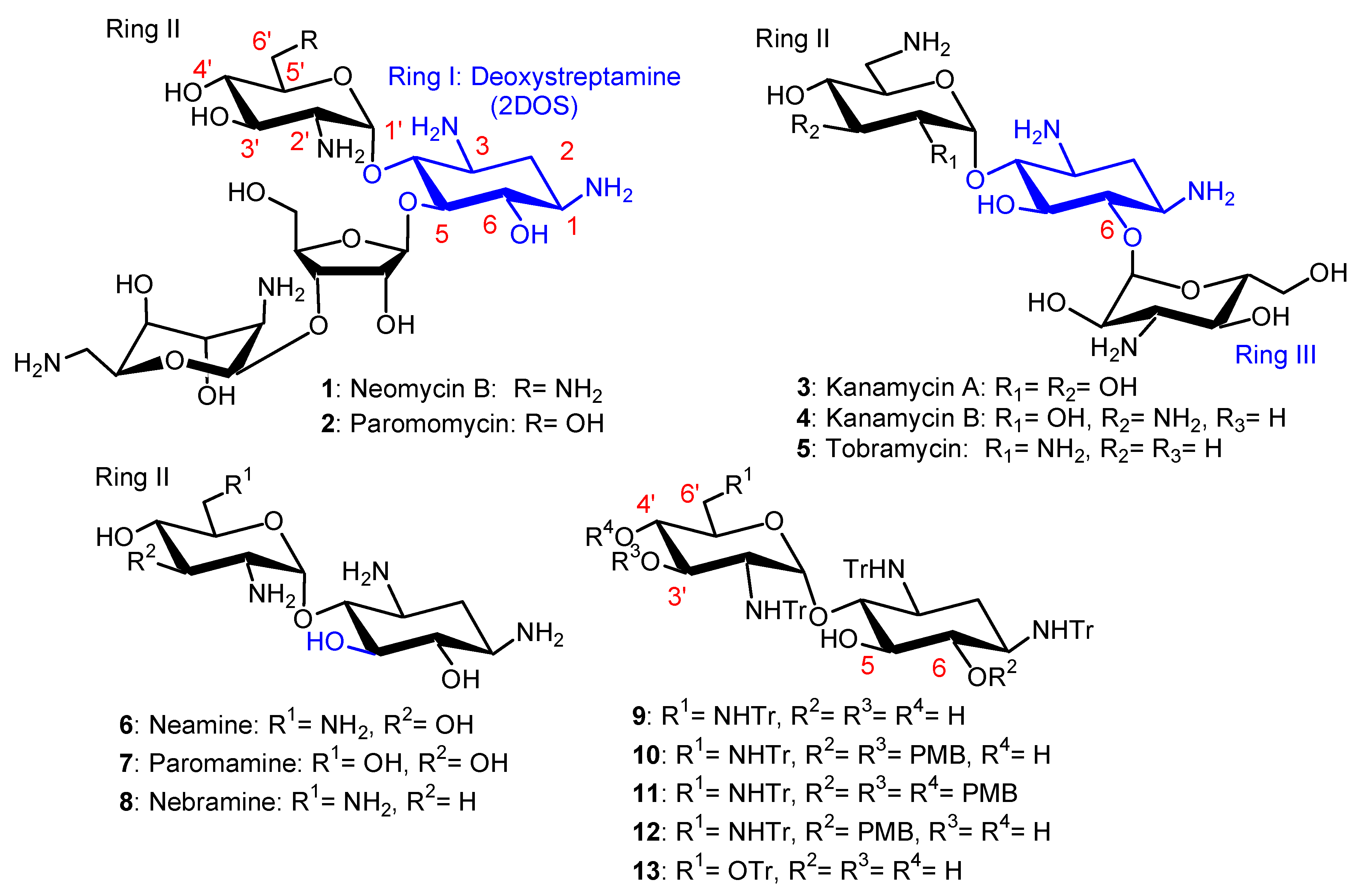{kind=link}
{kind=link}
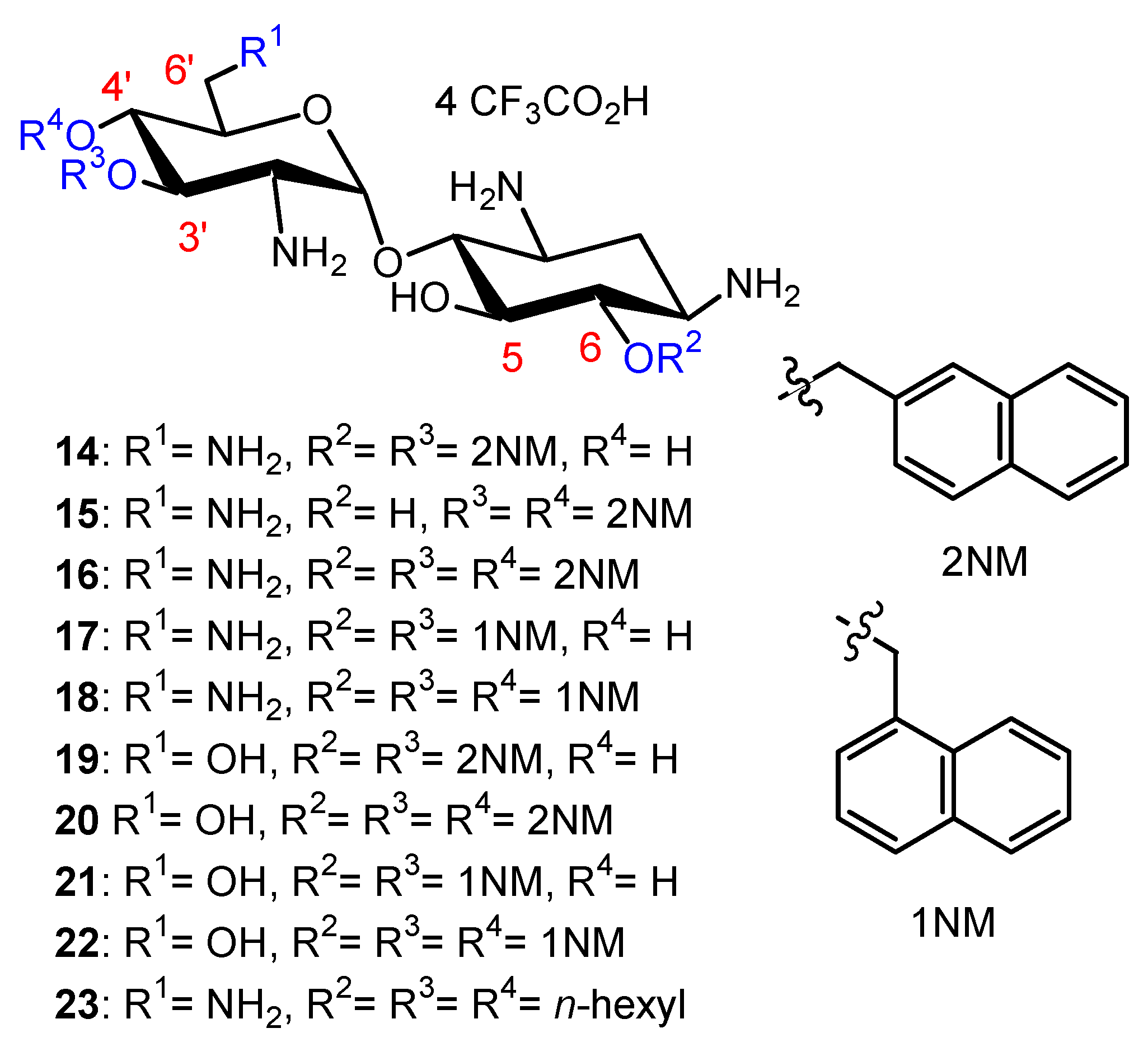{kind=link}
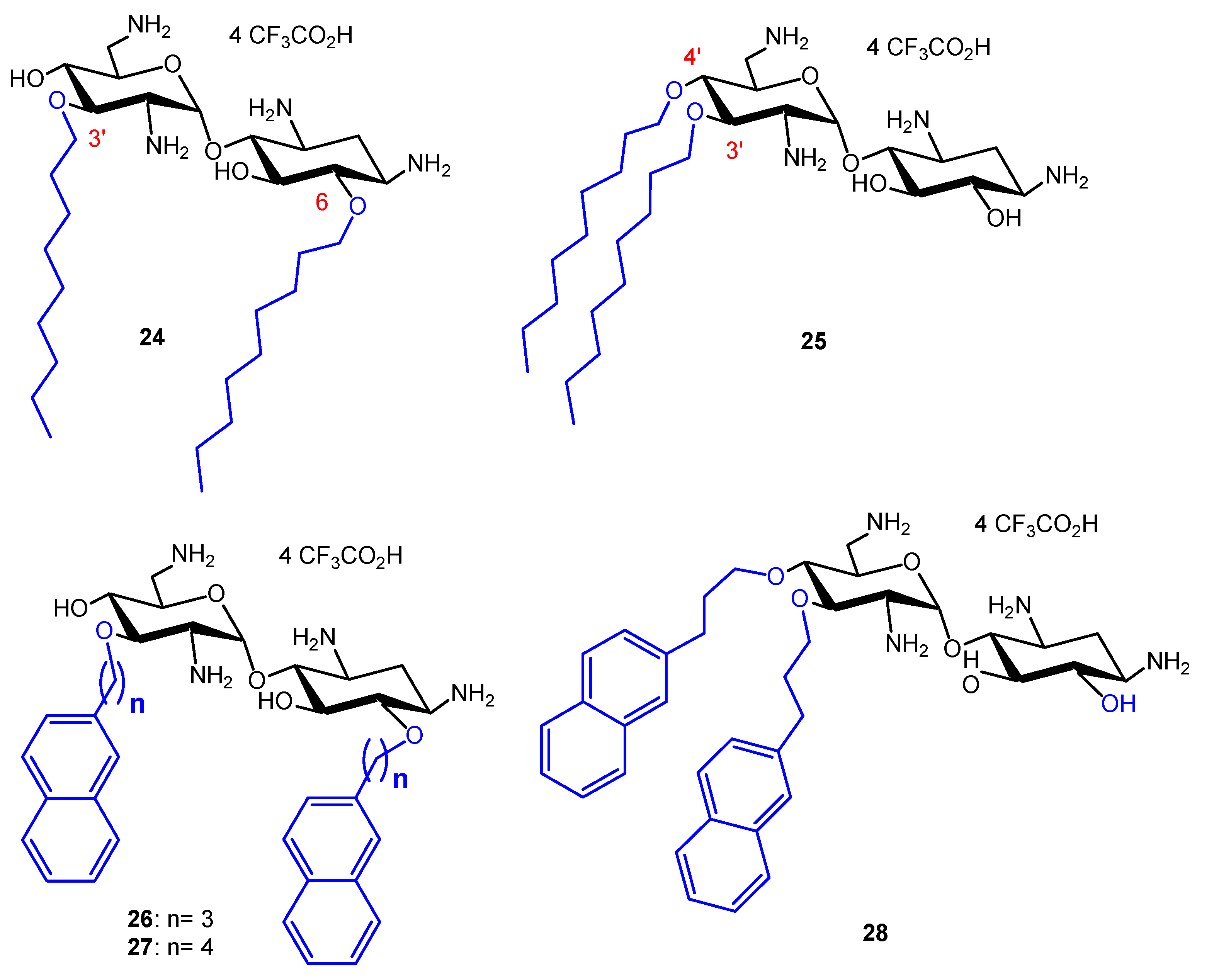{kind=link}
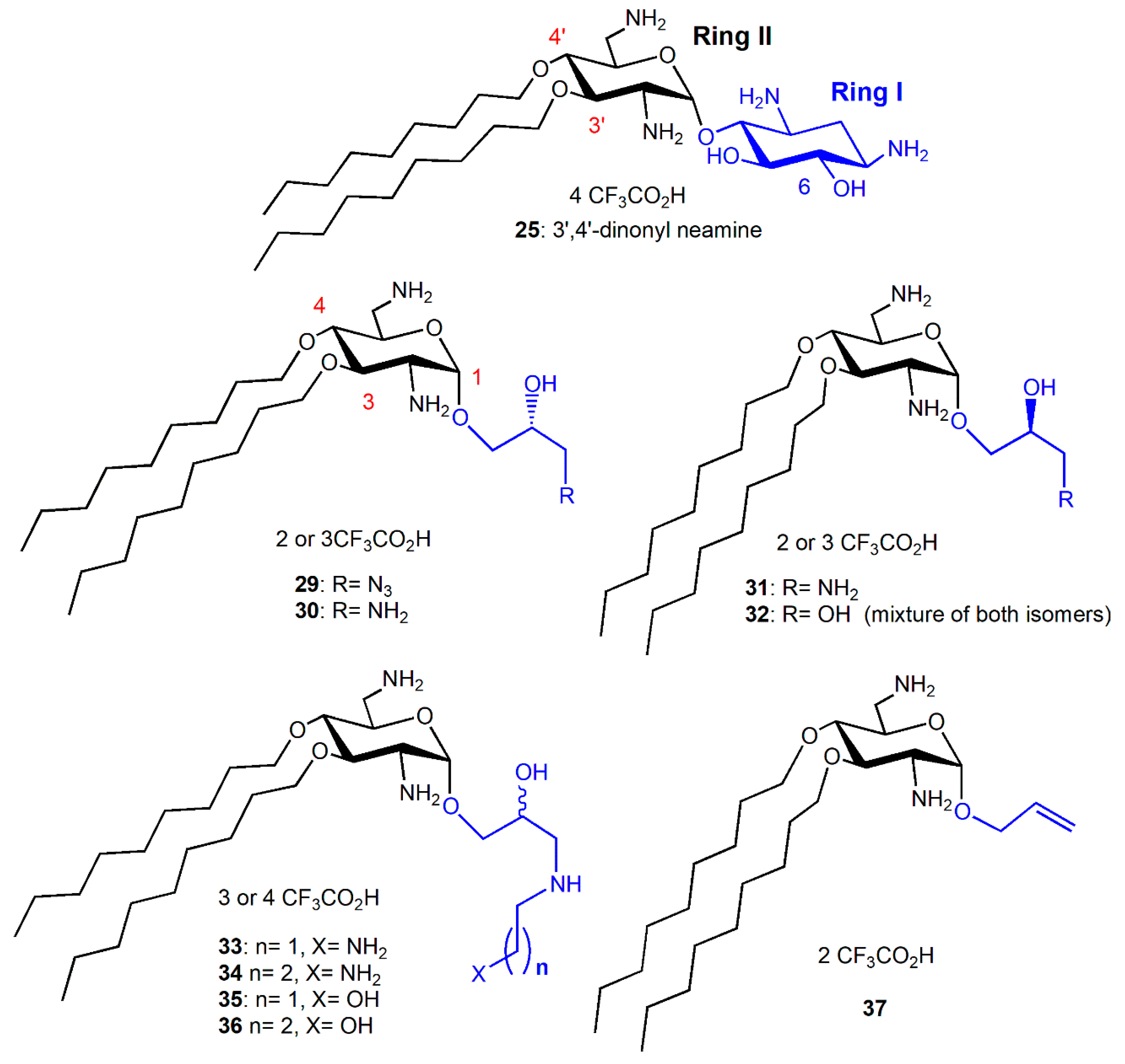{kind=link}
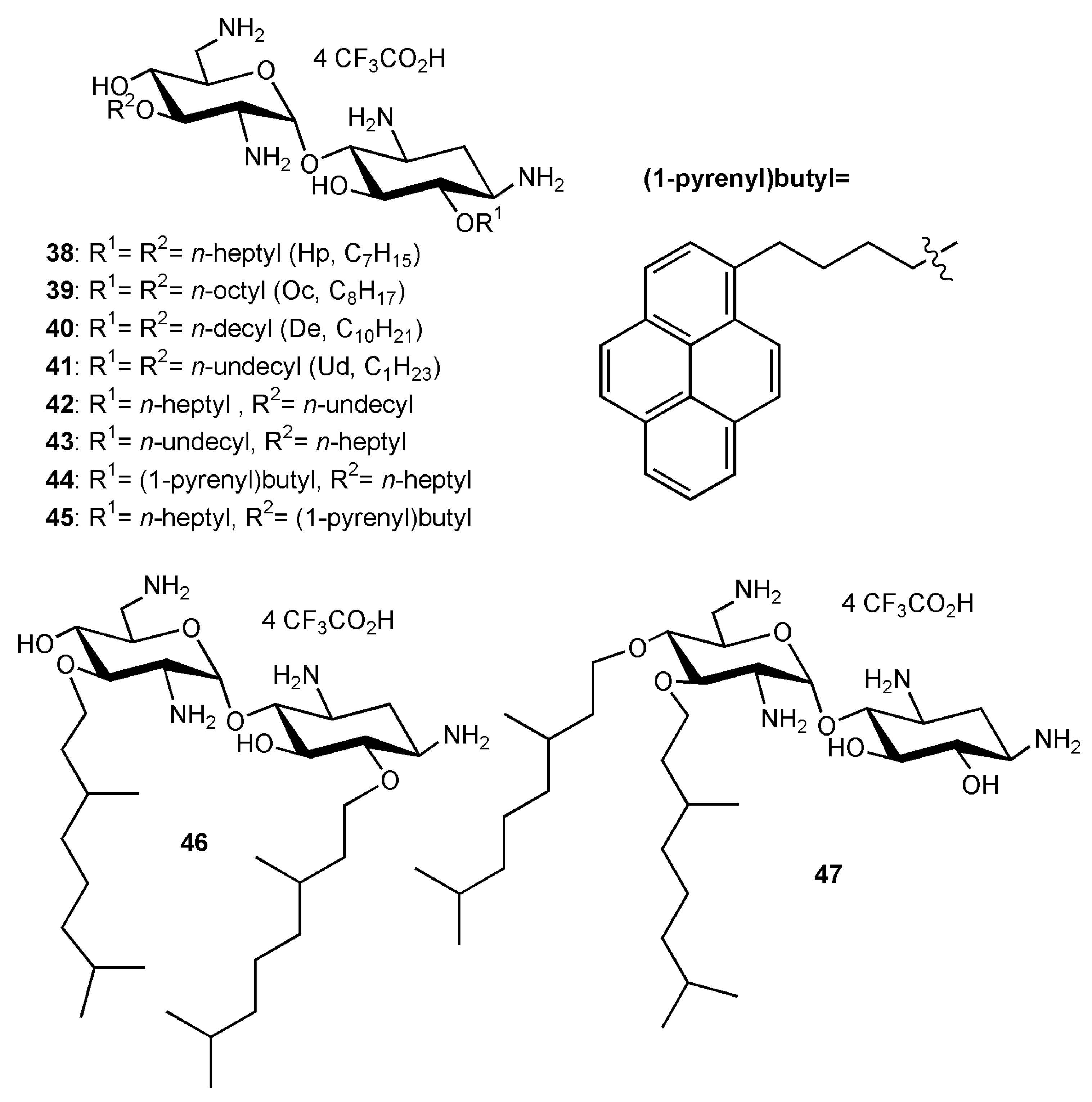{kind=link}
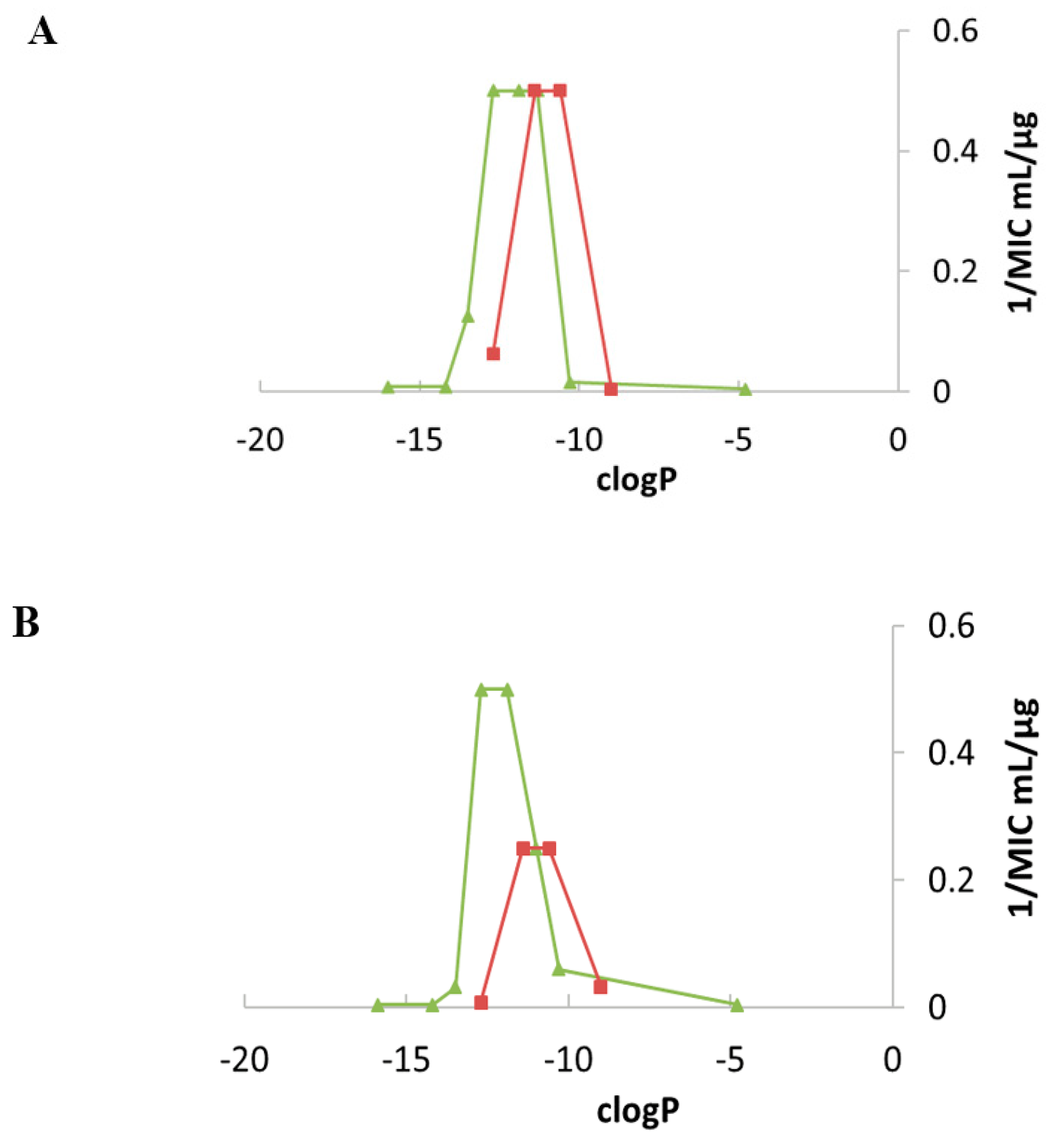{kind=link}
{kind=link}
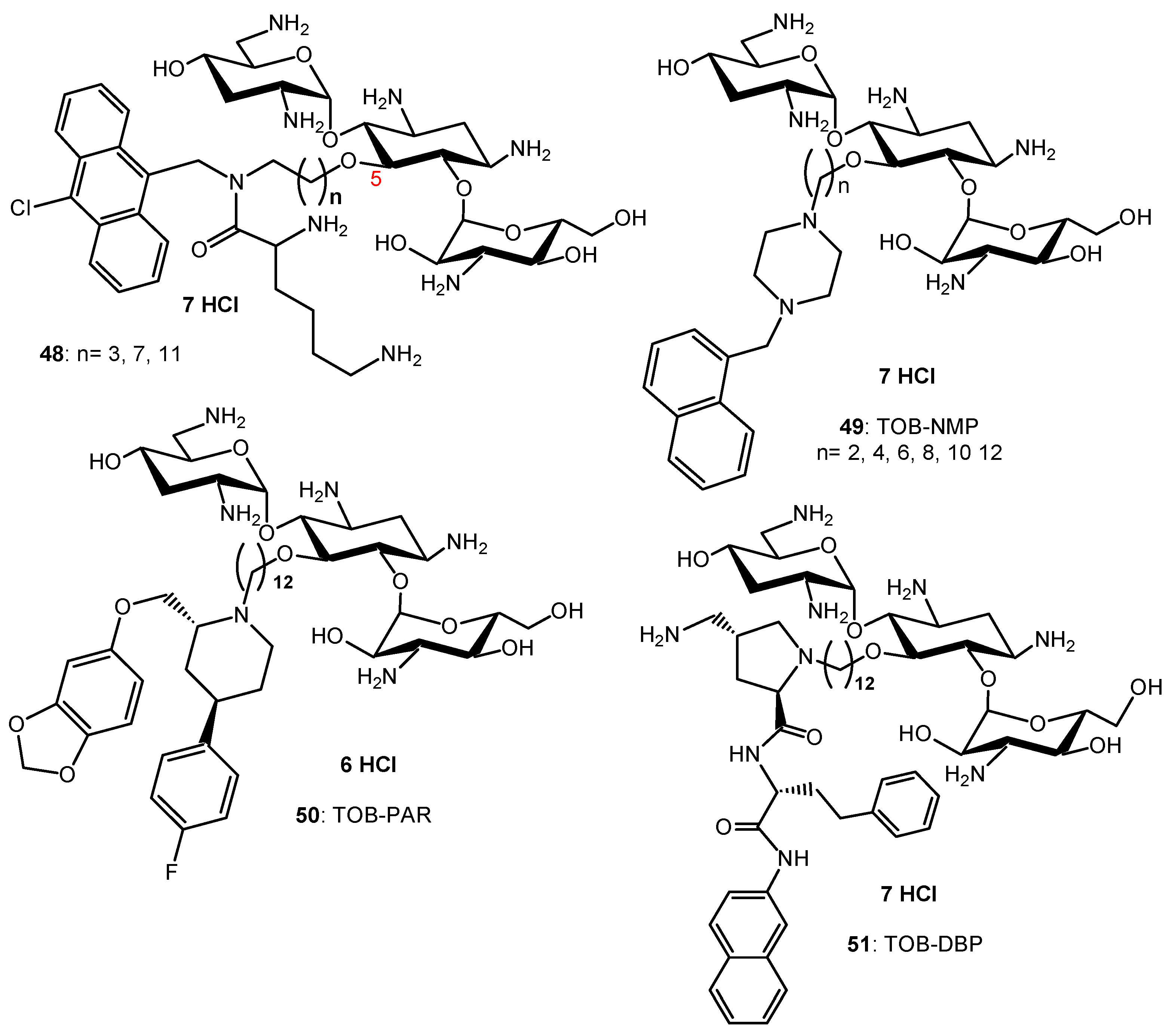{kind=link}
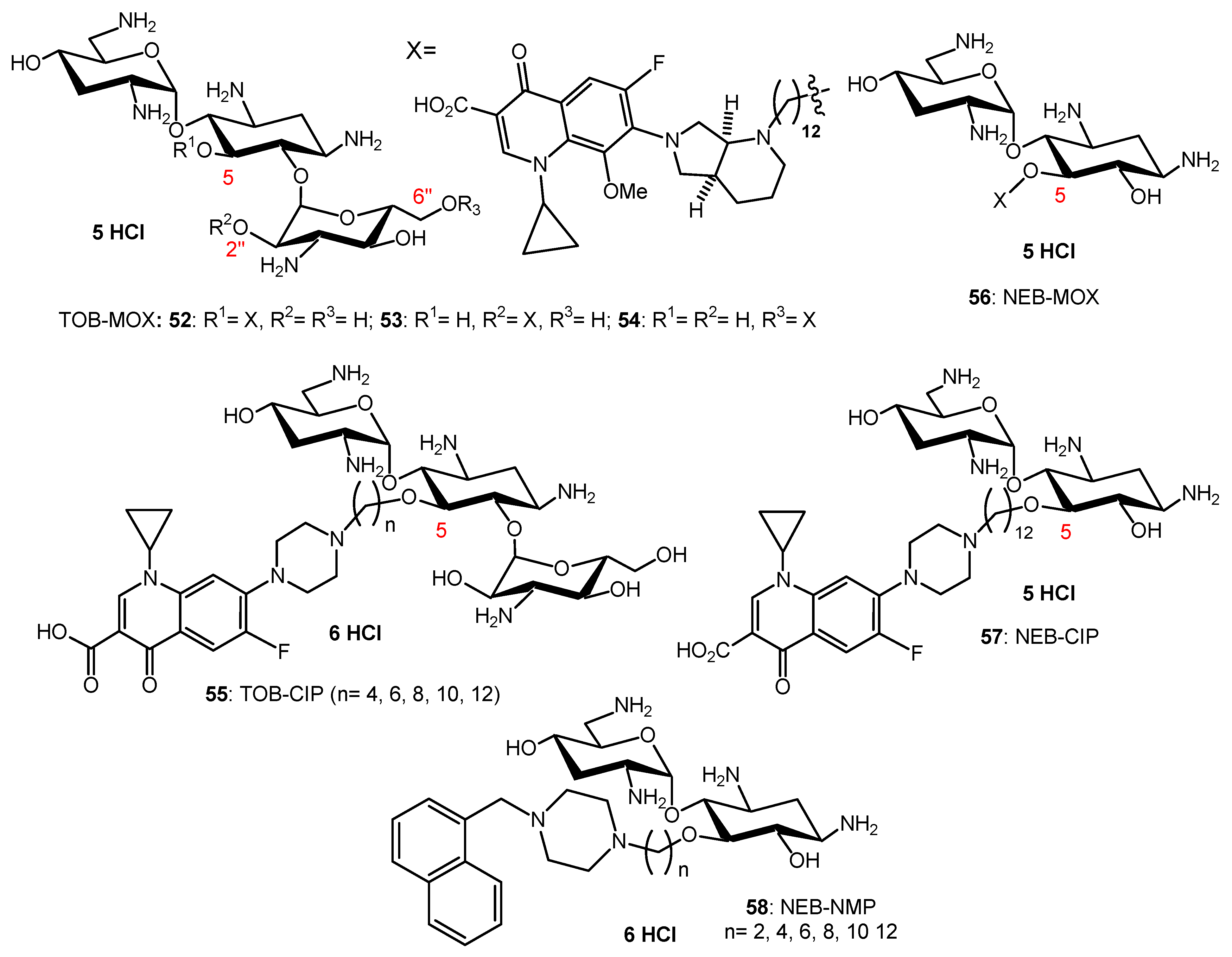{kind=link}
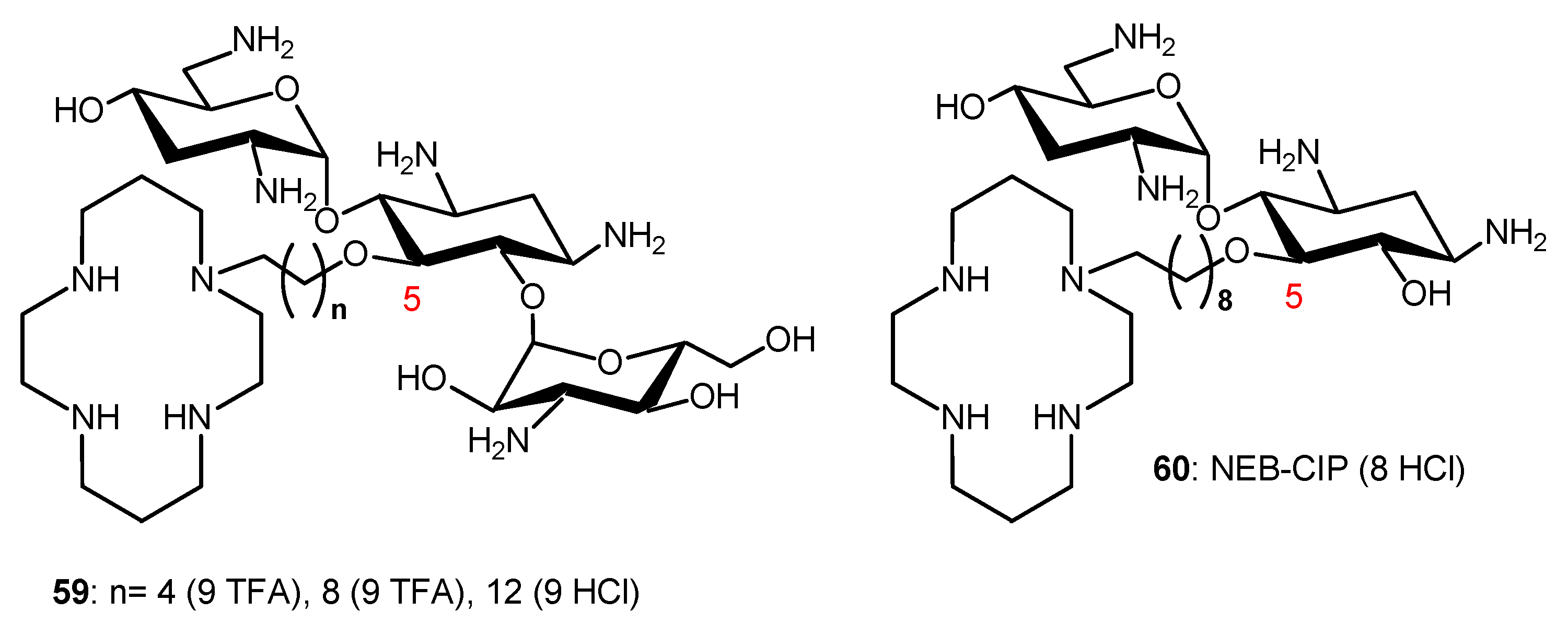{kind=link}
{kind=link}
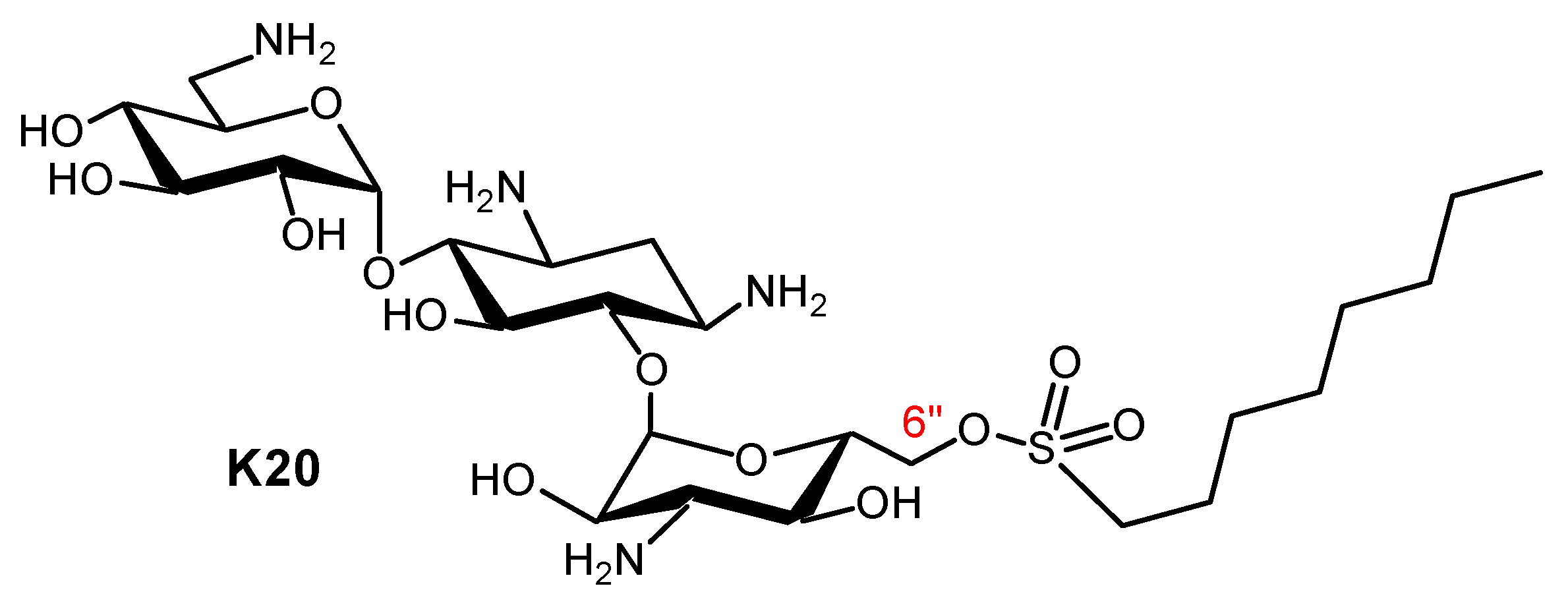{kind=link}
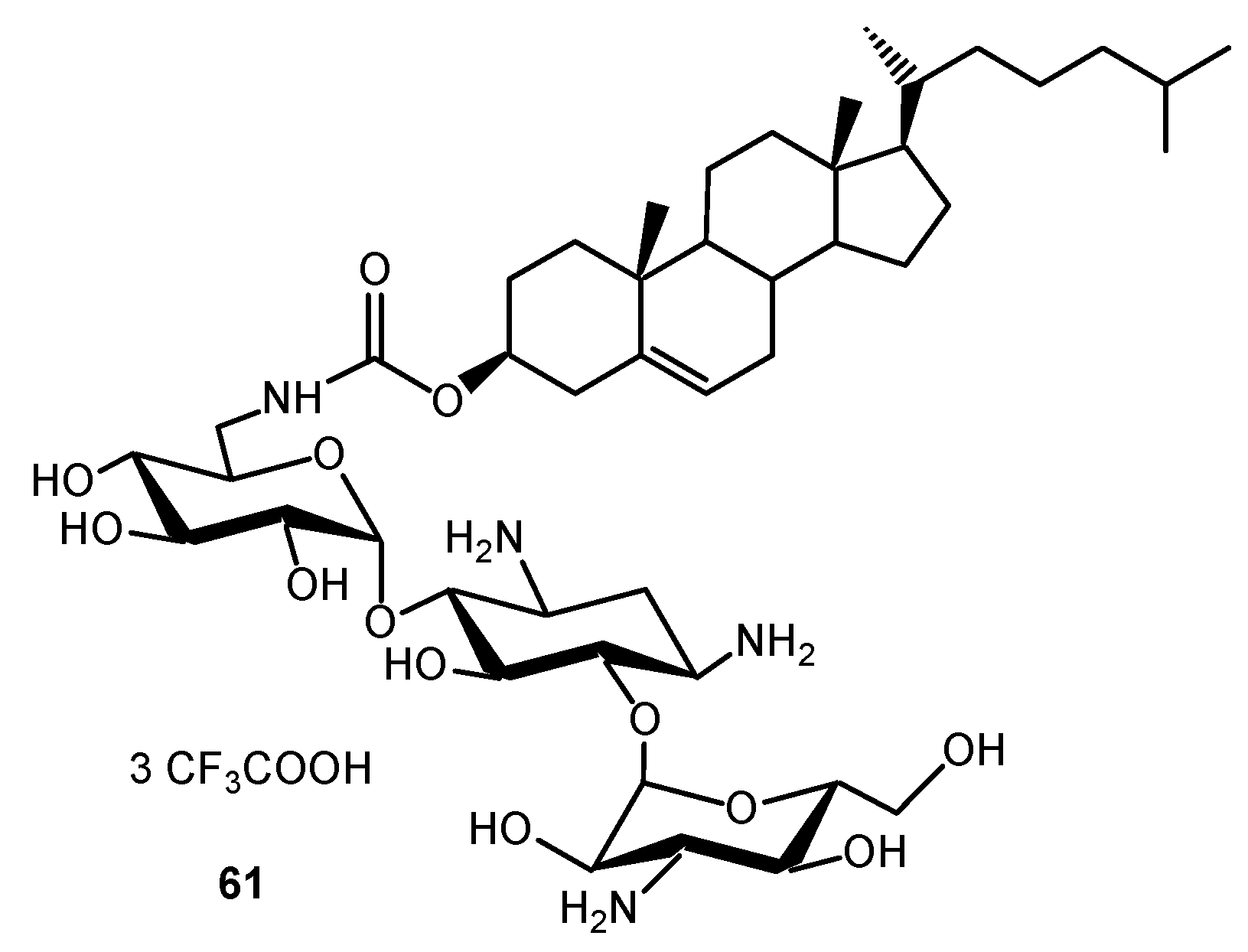{kind=link}
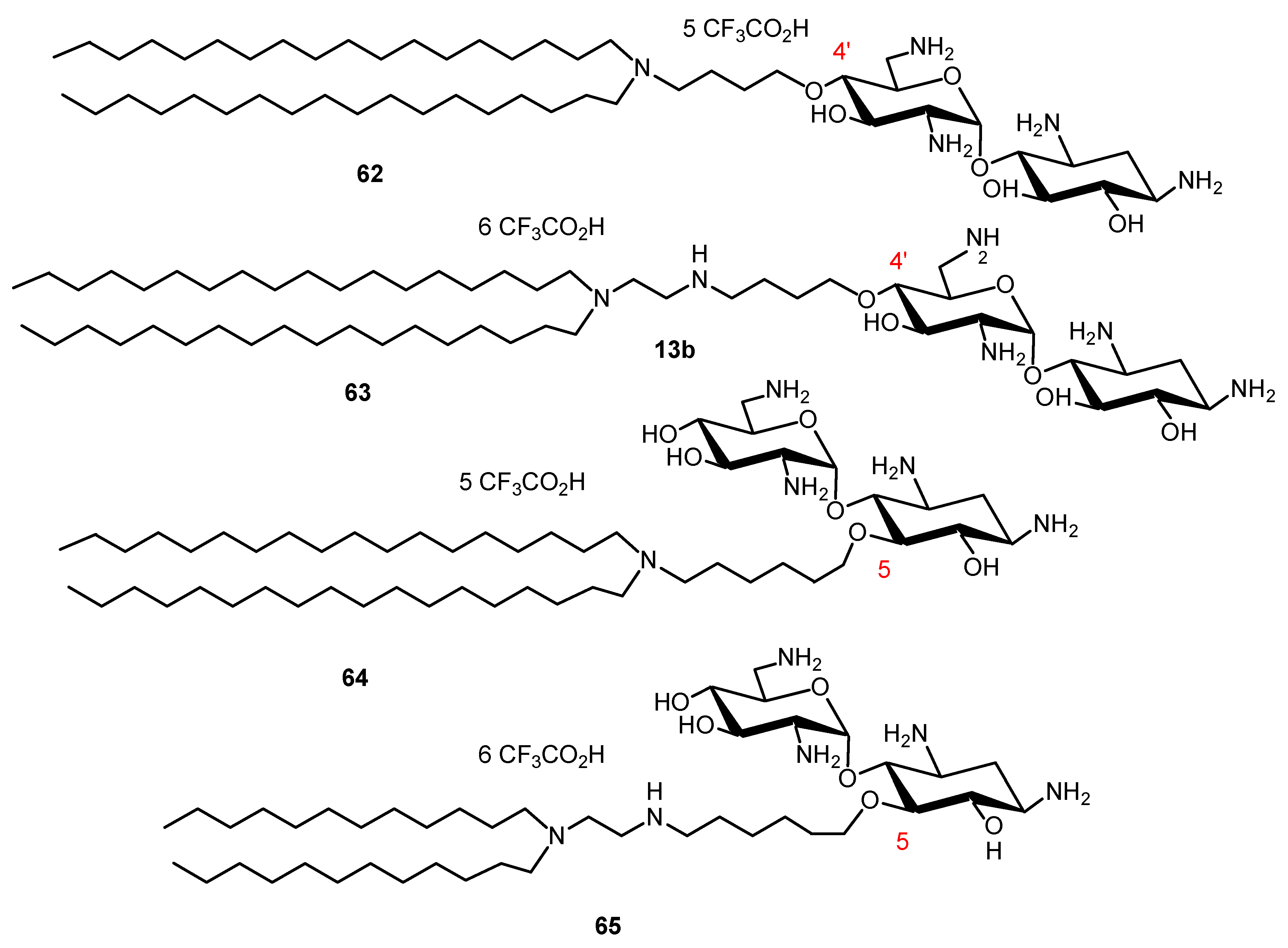{kind=link}
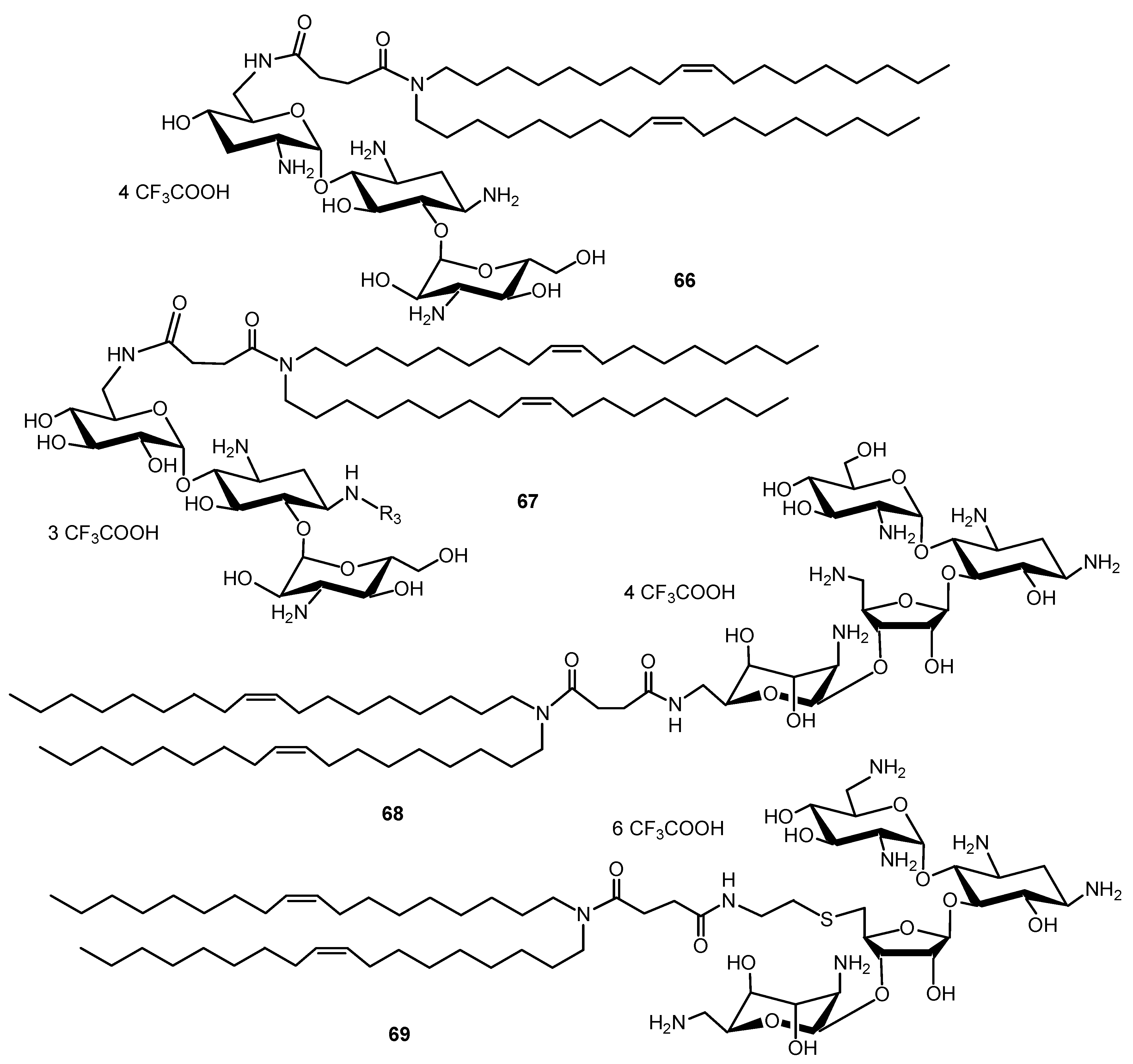{kind=link}
{kind=link}
{kind=link}
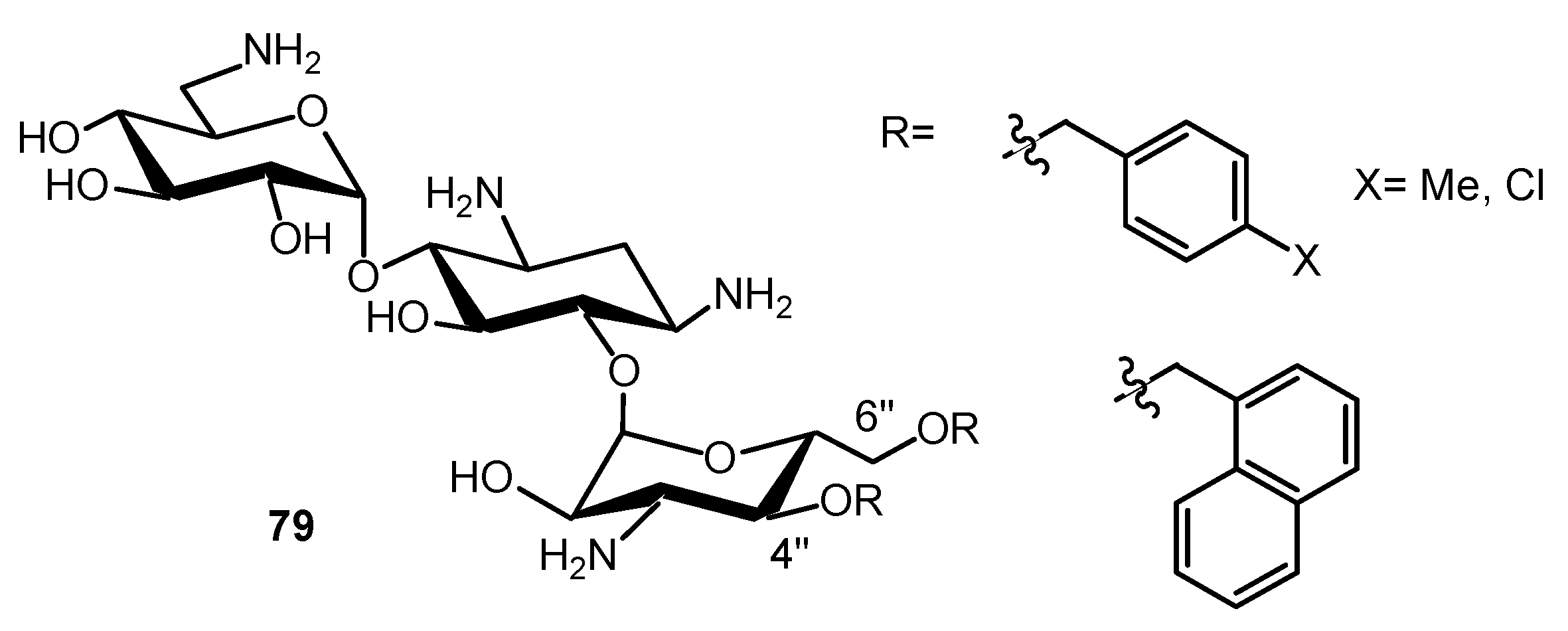{kind=link}
Table 1.
Minimum inhibitory concentrations (MICs) of the NEA derivatives 24, 26, 39 and of NEO 1 and NEA 6, representative aminoglycosides (AGs) against susceptible and resistant Staphylococcus aureus and Pseudomonas aeruginosa strains [35]. MRSA: methicillin-resistant S. aureus.
Table 1.
Minimum inhibitory concentrations (MICs) of the NEA derivatives 24, 26, 39 and of NEO 1 and NEA 6, representative aminoglycosides (AGs) against susceptible and resistant Staphylococcus aureus and Pseudomonas aeruginosa strains [35]. MRSA: methicillin-resistant S. aureus.
| AGs | Lipophilicity Expressed as clogP | MIC µg/mL | ||||||
|---|---|---|---|---|---|---|---|---|
| S. aureus | P. aeruginosa | |||||||
| ATCC 25923 | SA-1 Pump NorA | ATCC 33592 HA-MRSA | ATCC 27853 | Psa. FO3 a | PA22 b | PA406 c | ||
| NEO 1 | −29.9 | 1–2 | 0.5–1 | >128 | 64 | 128 | 32–64 | 2–4 |
| NEA 6 | −19.4 | 16–32 | 8 | >128 | >128 | >128 | >128 | 64 |
| 3′,6-diNn 24 | −11.9 | 1 | 1 | 2–4 | 2–4 | 4–8 | 4 | 2–4 |
| 3′,6-di2NP 26 | −11.4 | 2 | 2 | 2 | 8-16 | 16 | 16 | 2–4 |
| 3′,6-diOc 39 | −12.7 | 1 | 1 | 2 | 2 | 8 | 8 | 2 |
a: Psa.F03 AAC6′-IIA; b: surexp MexXY; c: PAO509.5 ∆triABC.
Table 2.
Viability (%) of murine J774 macrophages determined using the MTT assay in the presence of 10 and 30 µM of the NEA derivatives 24, 26 and 39 in comparison to NEO 1 and NEA 6, representative AGs; the numbers of independent experiments are mentioned after the viability values in brackets [35].
Table 2.
Viability (%) of murine J774 macrophages determined using the MTT assay in the presence of 10 and 30 µM of the NEA derivatives 24, 26 and 39 in comparison to NEO 1 and NEA 6, representative AGs; the numbers of independent experiments are mentioned after the viability values in brackets [35].
| AAG | Lipophilicity Expressed as clogP | Viability % | |
|---|---|---|---|
| 10 µM | 30 µM | ||
| NEO 1 | −29.9 | 87.3 (10) | 69.8 (2) |
| NEA 6 | −19.4 | 94.8 (9) | 84.4 (2) |
| 3′,6-diNn 24 | −11.9 | 86.7 (9) | 67.4 (3) |
| 3′,6-di2NP 26 | −11.4 | 91.1 (13) | 89.5 (2) |
| 3′,6-diOc 39 | −12.7 | 91.3 (4) | 65.1 (3) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dezanet, C.; Kempf, J.; Mingeot-Leclercq, M.-P.; Décout, J.-L. Amphiphilic Aminoglycosides as Medicinal Agents. Int. J. Mol. Sci. 2020, 21, 7411. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197411
AMA Style
Dezanet C, Kempf J, Mingeot-Leclercq M-P, Décout J-L. Amphiphilic Aminoglycosides as Medicinal Agents. International Journal of Molecular Sciences. 2020; 21(19):7411. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197411
Chicago/Turabian StyleDezanet, Clément, Julie Kempf, Marie-Paule Mingeot-Leclercq, and Jean-Luc Décout. 2020. "Amphiphilic Aminoglycosides as Medicinal Agents" International Journal of Molecular Sciences 21, no. 19: 7411. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21197411
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.