Yeast Rpn4 Links the Proteasome and DNA Repair via RAD52 Regulation



Abstract
:
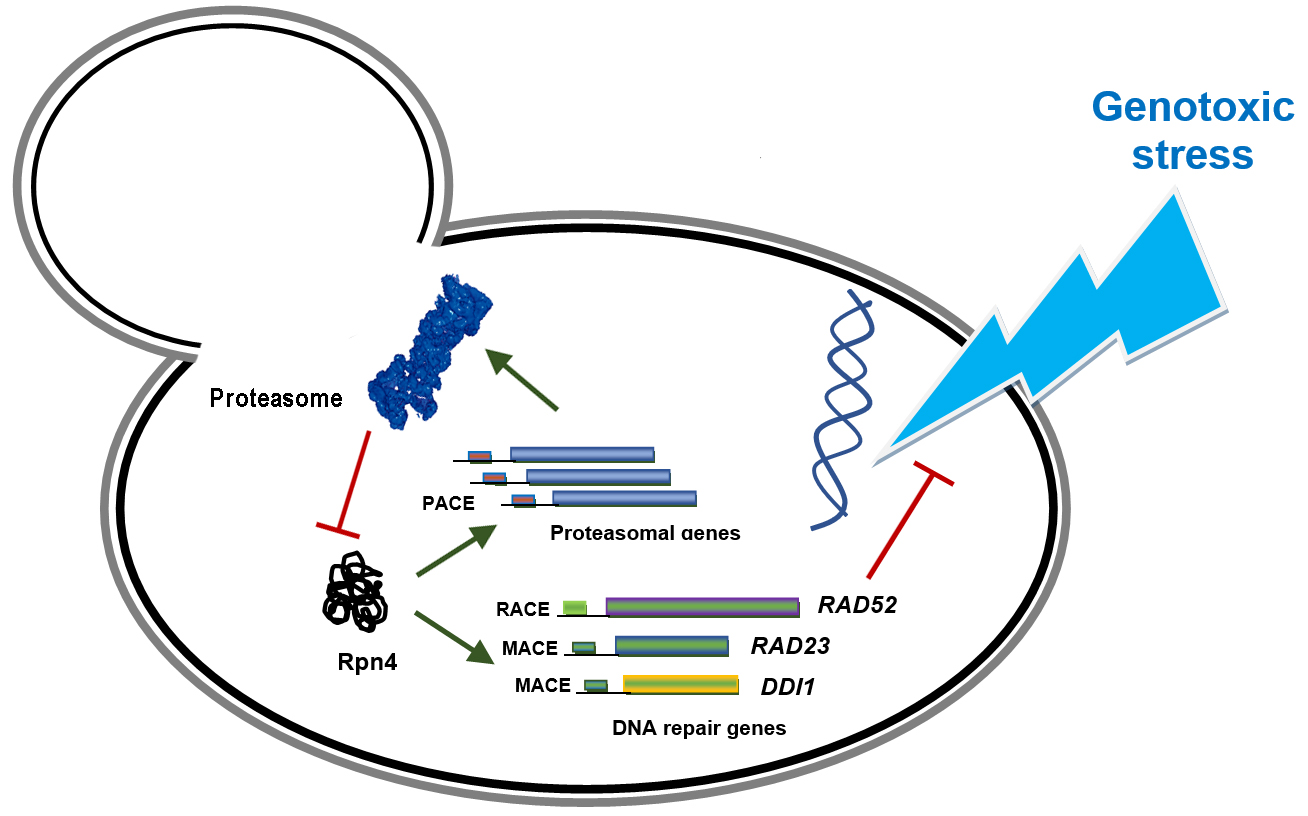{kind=link}
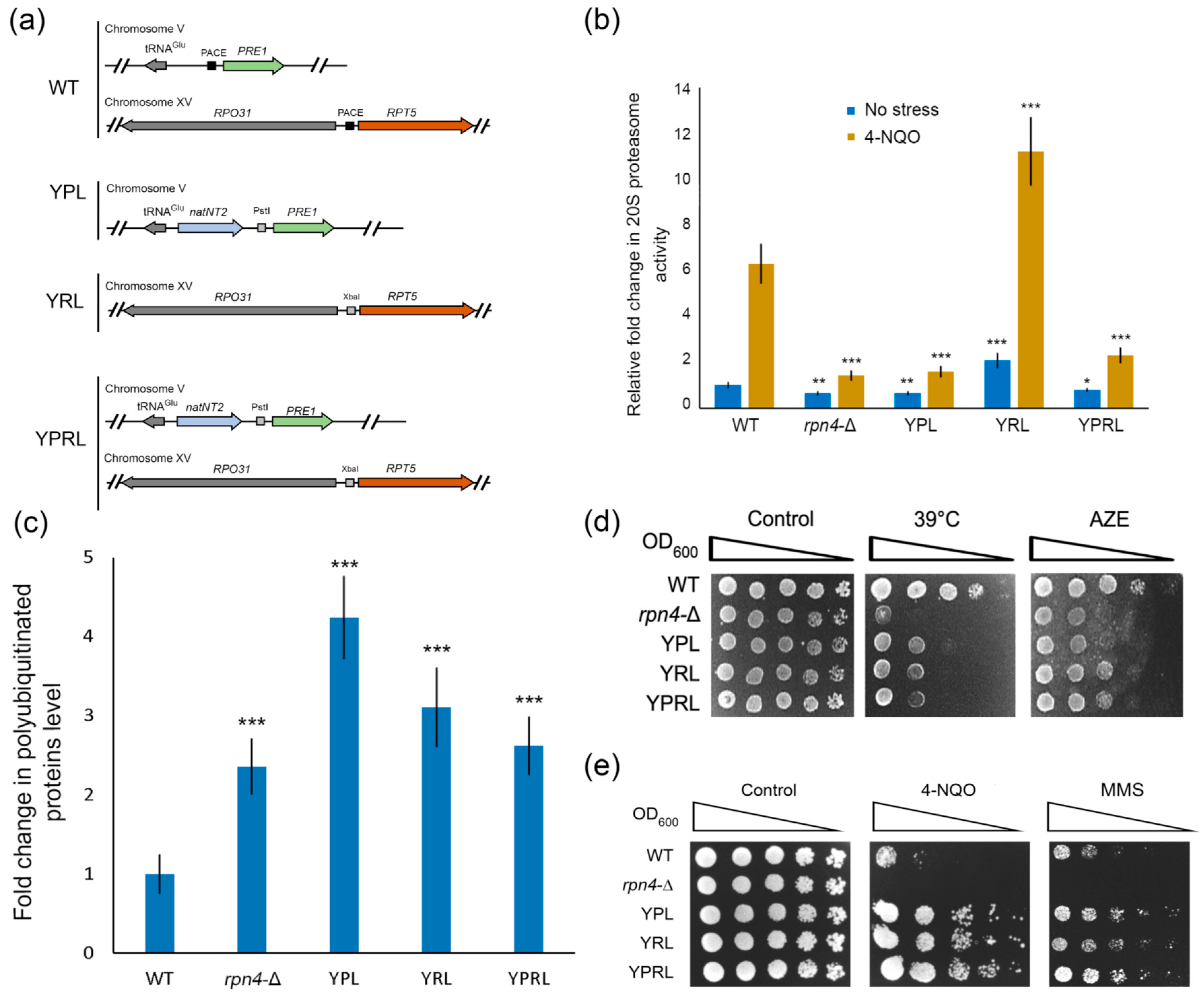{kind=link}
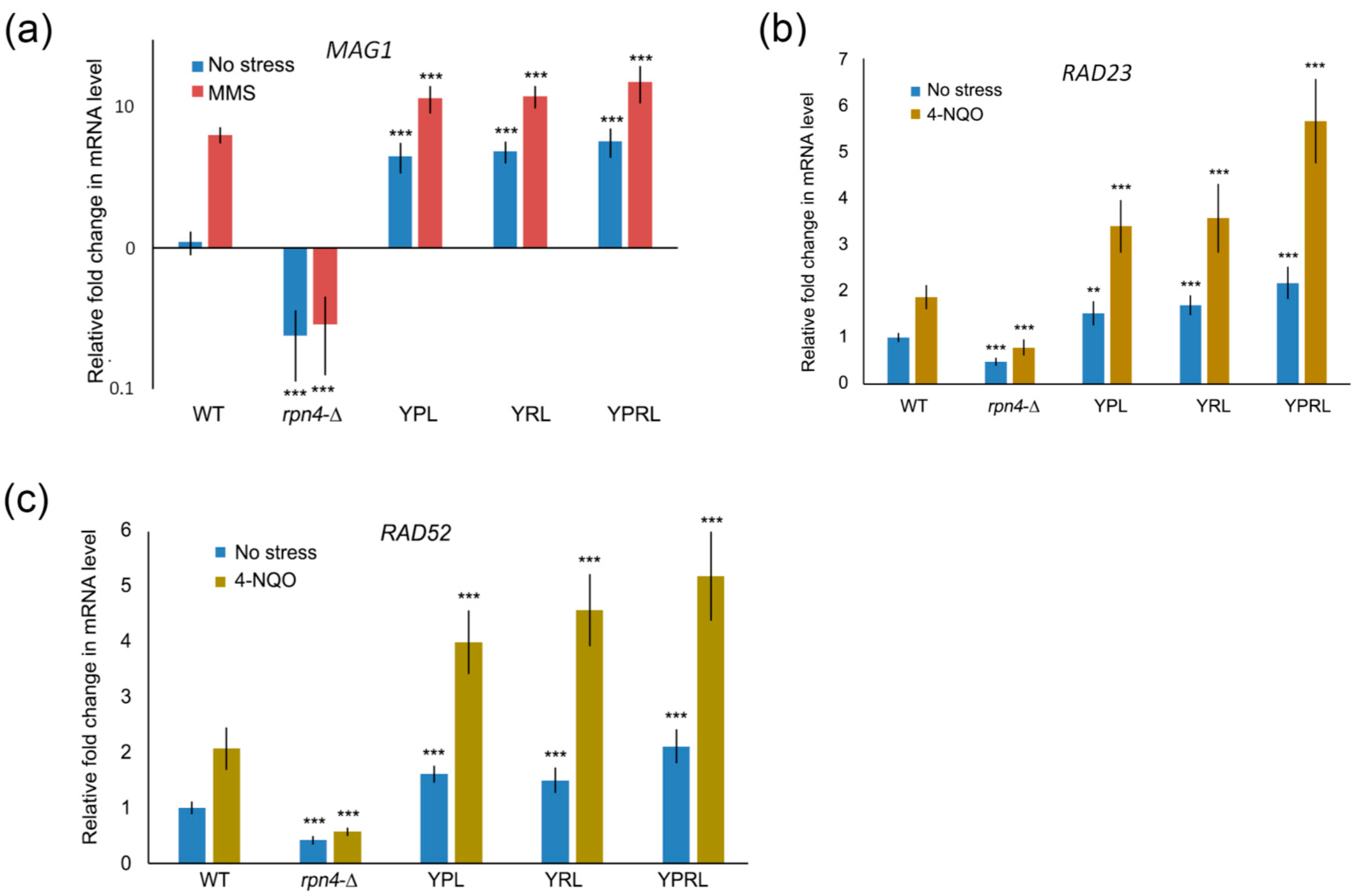{kind=link}
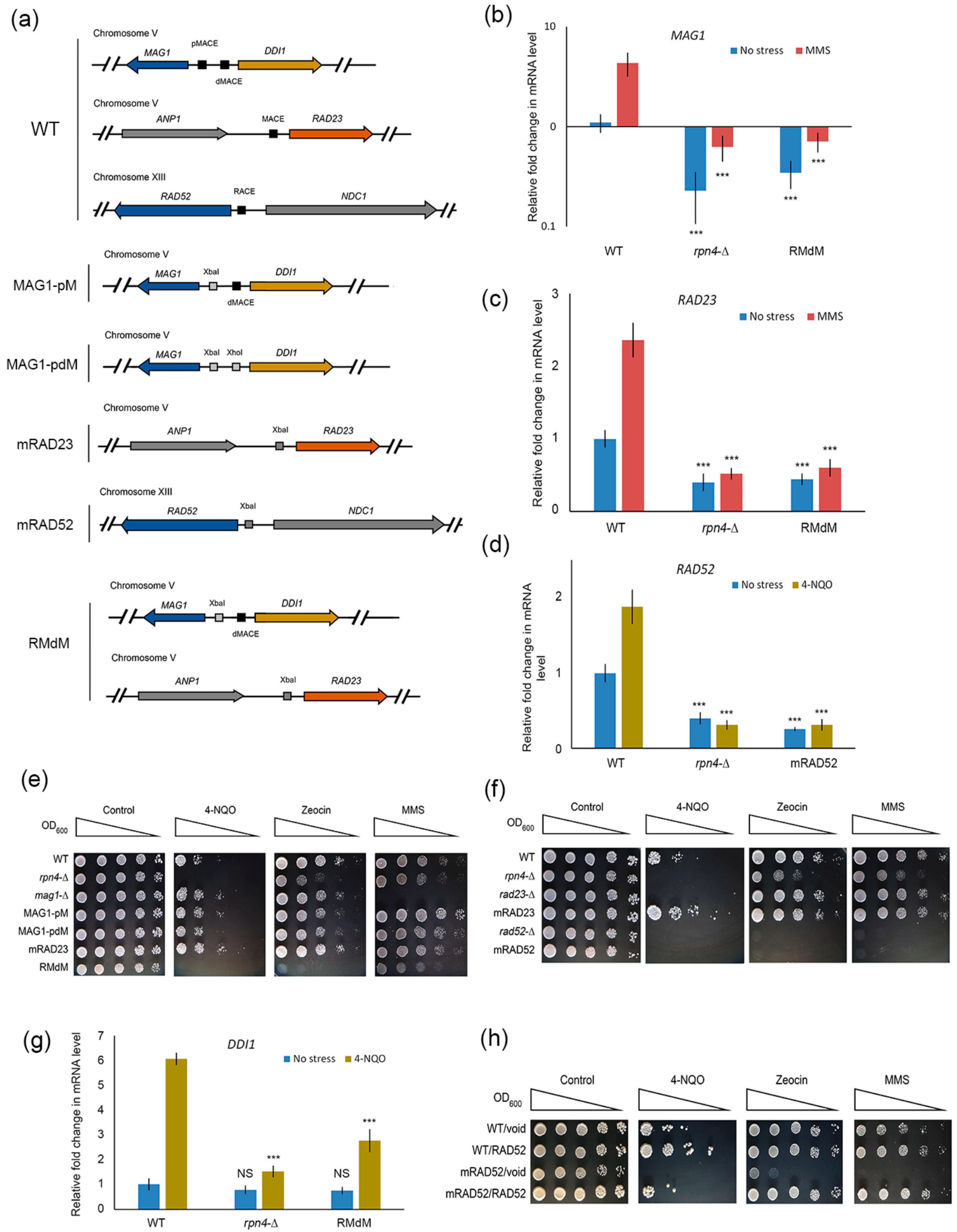{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Deregulation of Essential Proteasomal Genes Induces Hyper-Resistance to DNA Damage
2.2. Identification of Rpn4-Dependent Genes Involved in the Cellular Response to DNA Damage
2.3. Deregulation of Rpn4-Dependent DNA Repair Genes Sensitizes Mutant Yeast to DNA Damage
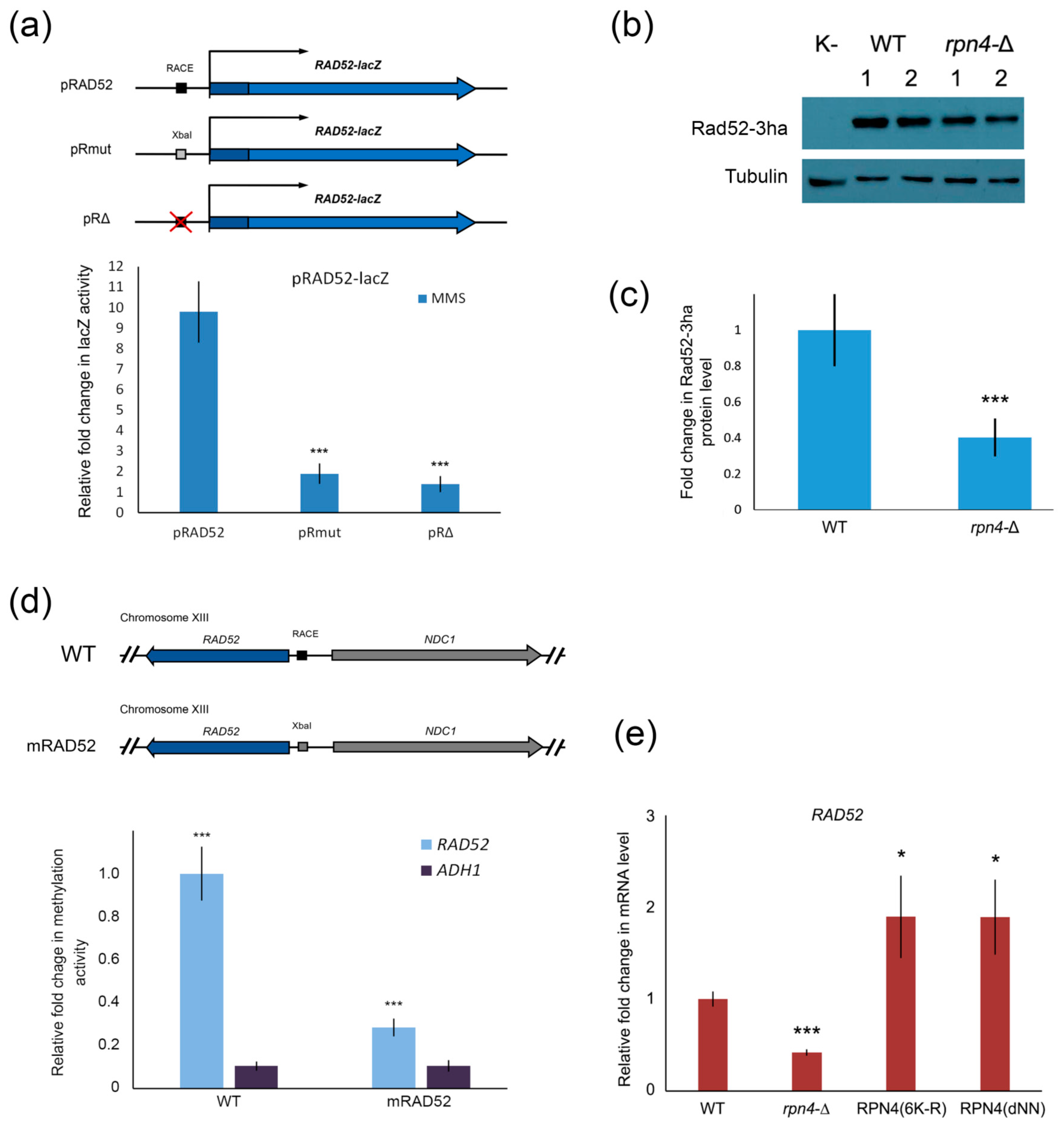
2.4. Rpn4 Directly Regulates RAD52 via a PACE Variant
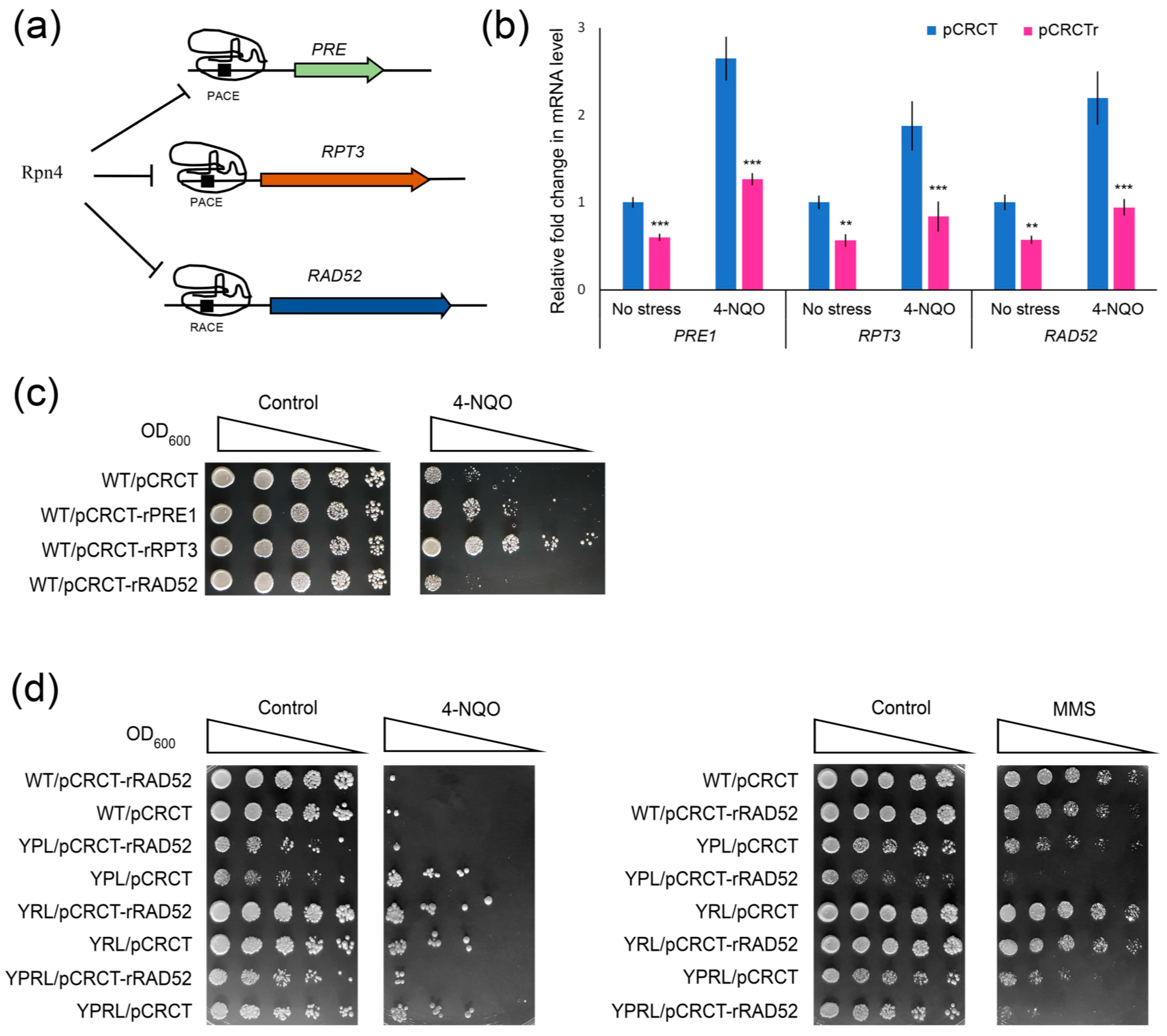
2.5. Impaired Rpn4-Dependent Regulation of DNA Repair Genes Restores the Sensitivity of Proteasome Mutants to DNA Damage
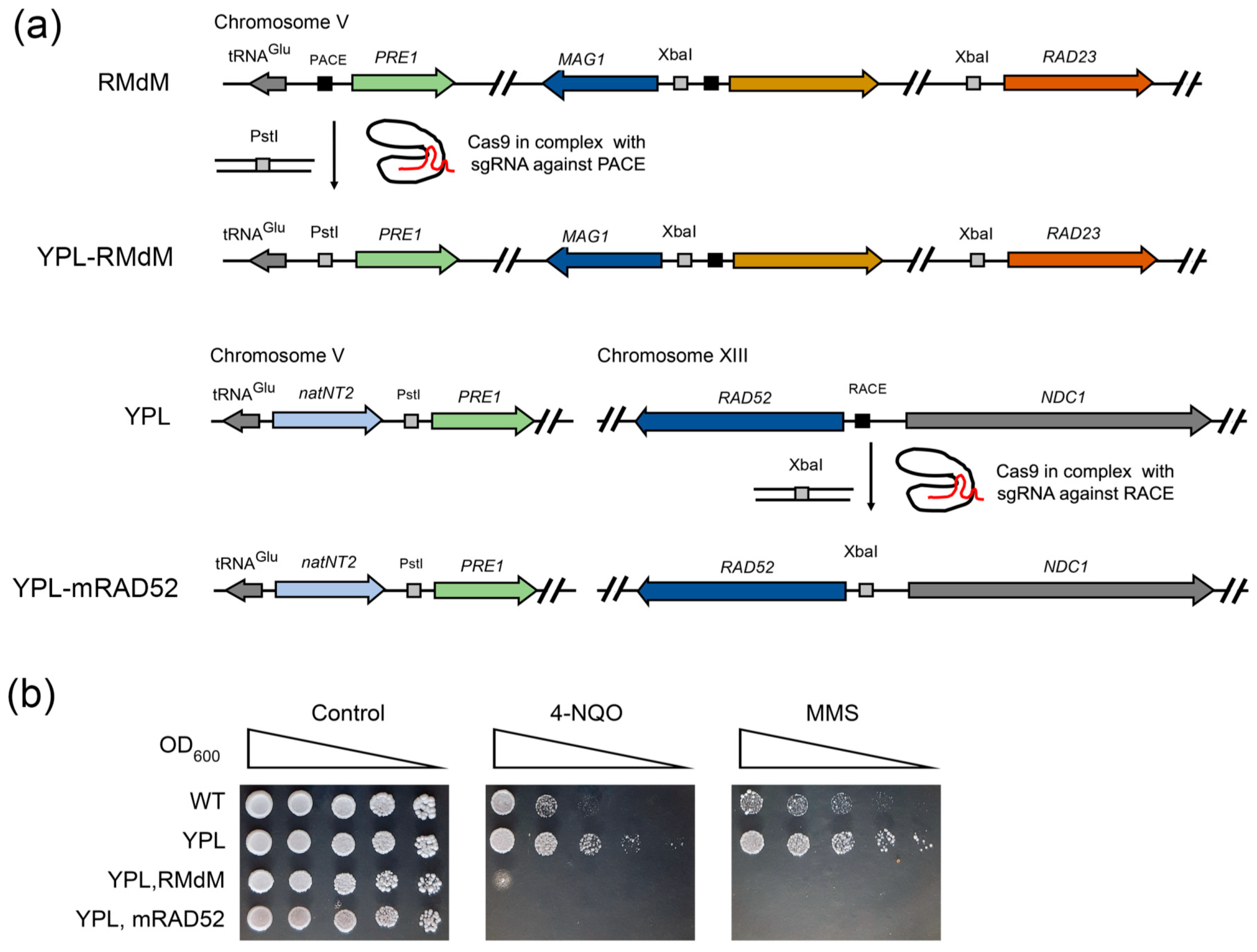
2.6. CRISPR-Mediated RAD52 Repression Decreases the Resistance of Proteasome Mutants to DNA Damage
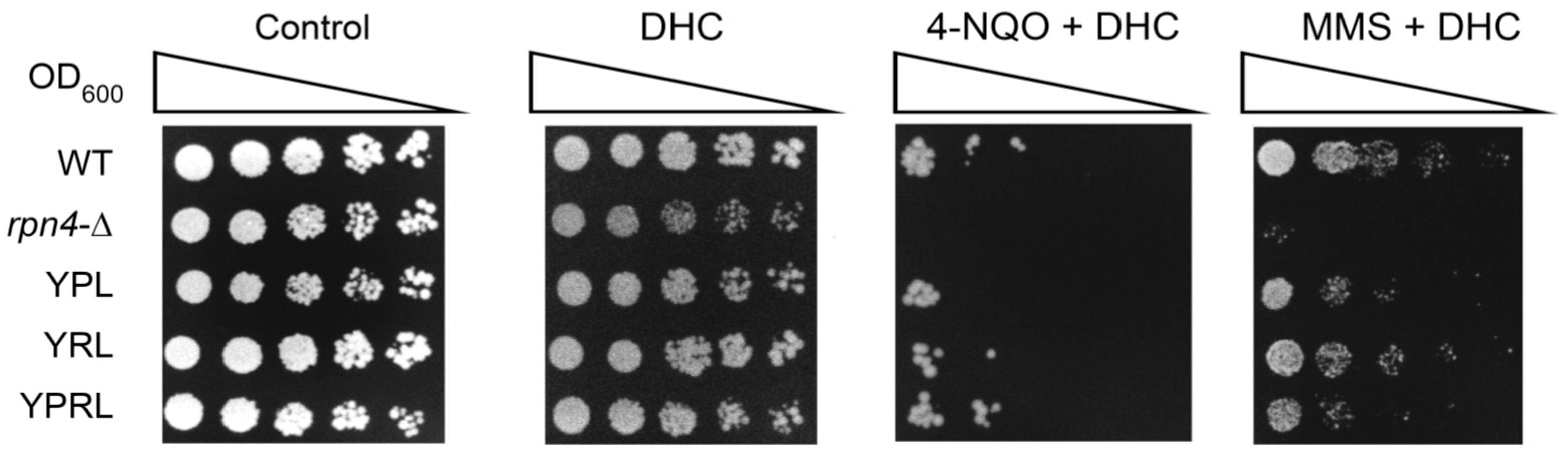
2.7. Dihydrocoumarin (DHC) Reverses the DNA Damage Hyper-Resistance Phenotype of Proteasome Mutants
3. Discussion
4. Materials and Methods
4.1. Yeast Strains
4.2. RT-PCR
4.3. β-. Galactosidase Assay
4.4. DamID Assay
4.5. Stress Resistance Test
4.6. Western Blot Analysis
4.7. Proteasome Activity Measurement
4.8. Mutation of Rpn4 Binding Sites Using the CRISPR/Cas9 System
4.9. Gene Repression with the CRISPR/Cas9 System
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-NQO | 4-Nitroquinoline-1-oxide |
| AZE | Azetidine-2-carboxylic acid |
| MMS | Methyl methanesulfonate |
| PACE | Proteasome-associated control element |
| MACE | MAG1-associated control element |
| RACE | RPN8-associated control element |
| SSBs | DNA single-strand breaks |
| DSBs | DNA double-strand breaks |
| DDR | DNA damage response |
| BER | Base excision repair |
| NER | Nucleotide excision repair |
| MMR | Mismatch repair |
| HDR | Homology-directed repair |
| NHEJ | Non-homologous end joining |
| MMEJ | Microhomology-mediated end joining |
| SUMO | Small ubiquitin-like modifier |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| PAM | Protospacer-adjacent motif |
| Cas9 | CRISPR-associated protein 9 |
| sgRNA | Single guide RNA |
| RT-PCR | Real-time polymerase chain reaction |
| UV | Ultraviolet |
| DHC | Dihydrocoumarin |
| TopIcc | Topoisomerase I-DNA covalent complex |
| MM | Multiple myeloma |
| VMP | Bortezomib, melphalan, and prednisone regimen |
| HDAC | Histone deacetylase |
| FA | Fanconi anaemia |
References
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Cannan, W.J.; Pederson, D.S. Mechanisms and consequences of double-strand DNA break formation in chromatin. J. Cell. Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Giglia-Mari, G.; Zotter, A.; Vermeulen, W. DNA damage response. Cold Spring Harb. Perspect. Biol. 2011, 3, a000745. [Google Scholar] [CrossRef]
- Frosina, G. Overexpression of enzymes that repair endogenous damage to DNA. Eur. J. Biochem. 2000, 267, 2135–2149. [Google Scholar] [CrossRef]
- Ortolan, T.G.; Chen, L.; Tongaonkar, P.; Madura, K. Rad23 stabilizes Rad4 from degradation by the Ub/proteasome pathway. Nucleic Acids Res. 2004, 32, 6490–6500. [Google Scholar] [CrossRef] [Green Version]
- Skoneczna, A.; McIntyre, J.; Skoneczny, M.; Policinska, Z.; Sledziewska-Gojska, E. Polymerase eta is a short-lived, proteasomally degraded protein that is temporarily stabilized following UV irradiation in Saccharomyces cerevisiae. J. Mol. Biol. 2007, 366, 1074–1086. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ruggiero, C.; Li, S. Yeast Rpb9 plays an important role in ubiquitylation and degradation of Rpb1 in response to UV-induced DNA damage. Mol. Cell. Biol. 2007, 27, 4617–4625. [Google Scholar] [CrossRef] [Green Version]
- Ribar, B.; Prakash, L.; Prakash, S. Requirement of ELC1 for RNA polymerase II polyubiquitylation and degradation in response to DNA damage in Saccharomyces cerevisiae. Mol. Cell. Biol. 2006, 26, 3999–4005. [Google Scholar] [CrossRef] [Green Version]
- Hauer, M.H.; Seeber, A.; Singh, V.; Thierry, R.; Sack, R.; Amitai, A.; Kryzhanovska, M.; Eglinger, J.; Holcman, D.; Owen-Hughes, T.; et al. Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat. Struct. Mol. Biol. 2017, 24, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Rodriguez, N.; Wong, R.P.; Ulrich, H.D. Functions of ubiquitin and SUMO in DNA replication and replication stress. Front. Genet. 2016, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Varshavsky, A. RPN4 is a ligand, substrate, and transcriptional regulator of the 26S proteasome: A negative feedback circuit. Proc. Natl. Acad. Sci. USA 2001, 98, 3056–3061. [Google Scholar] [CrossRef] [Green Version]
- Mannhaupt, G.; Schnall, R.; Karpov, V.; Vetter, I.; Feldmann, H. Rpn4p acts as a transcription factor by binding to PACE, a nonamer box found upstream of 26S proteasomal and other genes in yeast. FEBS Lett. 1999, 450, 27–34. [Google Scholar] [CrossRef] [Green Version]
- London, M.K.; Keck, B.I.; Ramos, P.C.; Dohmen, R.J. Regulatory mechanisms controlling biogenesis of ubiquitin and the proteasome. FEBS Lett. 2004, 567, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Hahn, J.S.; Neef, D.W.; Thiele, D.J. A stress regulatory network for co-ordinated activation of proteasome expression mediated by yeast heat shock transcription factor. Mol. Microbiol. 2006, 60, 240–251. [Google Scholar] [CrossRef]
- Owsianik, G.; Balzi, L.; Ghislain, M. Control of 26S proteasome expression by transcription factors regulating multidrug resistance in Saccharomyces cerevisiae. Mol. Microbiol. 2002, 43, 1295–1308. [Google Scholar] [CrossRef]
- Karpov, D.S.; Osipov, S.A.; Preobrazhenskaya, O.V.; Karpov, V.L. Rpn4p is a positive and negative transcriptional regulator of the ubiquitin-proteasome system. Mol. Biol. 2008, 42, 456–462. [Google Scholar] [CrossRef]
- Karpov, D.S.; Spasskaya, D.S.; Tutyaeva, V.V.; Mironov, A.S.; Karpov, V.L. Proteasome inhibition enhances resistance to DNA damage via upregulation of Rpn4-dependent DNA repair genes. FEBS Lett. 2013, 587, 3108–3114. [Google Scholar] [CrossRef] [Green Version]
- Jelinsky, S.A.; Estep, P.; Church, G.M.; Samson, L.D. Regulatory networks revealed by transcriptional profiling of damaged Saccharomyces cerevisiae cells: Rpn4 links base excision repair with proteasomes. Mol. Cell. Biol. 2000, 20, 8157–8167. [Google Scholar] [CrossRef]
- Le Tallec, B.; Barrault, M.B.; Courbeyrette, R.; Guerois, R.; Marsolier-Kergoat, M.C.; Peyroche, A. 20S proteasome assembly is orchestrated by two distinct pairs of chaperones in yeast and in mammals. Mol. Cell 2007, 27, 660–674. [Google Scholar] [CrossRef]
- Le Tallec, B.; Barrault, M.B.; Guerois, R.; Carre, T.; Peyroche, A. Hsm3/S5b participates in the assembly pathway of the 19S regulatory particle of the proteasome. Mol. Cell 2009, 33, 389–399. [Google Scholar] [CrossRef]
- Tallec, B.L.; Peyroche, A. Using DNA damage sensitivity phenotypes to characterize mutations affecting proteasome function. Methods Mol. Biol. 2012, 832, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Karpov, D.S.; Spasskaya, D.S.; Nadolinskaia, N.I.; Tutyaeva, V.V.; Lysov, Y.P.; Karpov, V.L. Deregulation of the 19S proteasome complex increases yeast resistance to 4-NQO and oxidative stress via upregulation of Rpn4- and proteasome-dependent stress responsive genes. FEMS Yeast Res. 2019, 19, foz002. [Google Scholar] [CrossRef]
- Karpov, D.S.; Tutyaeva, V.V.; Karpov, V.L. Mapping of yeast Rpn4p transactivation domains. FEBS Lett. 2008, 582, 3459–3464. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xu, H.; Ha, S.W.; Ju, D.; Xie, Y. Proteasomal degradation of Rpn4 in Saccharomyces cerevisiae is critical for cell viability under stressed conditions. Genetics 2010, 184, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Gasch, A.P.; Moses, A.M.; Chiang, D.Y.; Fraser, H.B.; Berardini, M.; Eisen, M.B. Conservation and evolution of cis-regulatory systems in ascomycete fungi. PLoS Biol. 2004, 2, e398. [Google Scholar] [CrossRef]
- Shirozu, R.; Yashiroda, H.; Murata, S. Identification of minimum Rpn4-responsive elements in genes related to proteasome functions. FEBS Lett. 2015, 589, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Gasch, A.P.; Huang, M.; Metzner, S.; Botstein, D.; Elledge, S.J.; Brown, P.O. Genomic expression responses to DNA-damaging agents and the regulatory role of the yeast ATR homolog Mec1p. Mol. Biol. Cell 2001, 12, 2987–3003. [Google Scholar] [CrossRef] [Green Version]
- Fry, R.C.; Sambandan, T.G.; Rha, C. DNA damage and stress transcripts in Saccharomyces cerevisiae mutant sgs1. Mech. Ageing Dev. 2003, 124, 839–846. [Google Scholar] [CrossRef]
- Benton, M.G.; Somasundaram, S.; Glasner, J.D.; Palecek, S.P. Analyzing the dose-dependence of the Saccharomyces cerevisiae global transcriptional response to methyl methanesulfonate and ionizing radiation. BMC Genom. 2006, 7, 305. [Google Scholar] [CrossRef] [Green Version]
- Jaehnig, E.J.; Kuo, D.; Hombauer, H.; Ideker, T.G.; Kolodner, R.D. Checkpoint kinases regulate a global network of transcription factors in response to DNA damage. Cell Rep. 2013, 4, 174–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapranov, A.B.; Kuryatova, M.V.; Preobrazhenskaya, O.V.; Tutyaeva, V.V.; Stucka, R.; Feldmann, H.; Karpov, V.L. Isolation and identification of PACE-binding protein Rpn4, a new transcriptional activator regulating 26S-proteasomal and other genes. Mol. Biol. 2001, 35, 356–364. [Google Scholar] [CrossRef]
- Spasskaya, D.S.; Karpov, D.S.; Karpov, V.L. Escherichia coli Dam-methylase as a molecular tool for mapping binding sites of the yeast transcription factor Rpn4. Mol. Biol. 2011, 45, 591–599. [Google Scholar] [CrossRef]
- Ju, D.; Wang, X.; Ha, S.W.; Fu, J.; Xie, Y. Inhibition of proteasomal degradation of rpn4 impairs nonhomologous end-joining repair of DNA double-strand breaks. PLoS ONE 2010, 5, e9877. [Google Scholar] [CrossRef] [Green Version]
- Jansen, L.E.; Verhage, R.A.; Brouwer, J. Preferential binding of yeast Rad4.Rad23 complex to damaged DNA. J. Biol. Chem. 1998, 273, 33111–33114. [Google Scholar] [CrossRef] [Green Version]
- Guzder, S.N.; Habraken, Y.; Sung, P.; Prakash, L.; Prakash, S. Reconstitution of yeast nucleotide excision repair with purified Rad proteins, replication protein A, and transcription factor TFIIH. J. Biol. Chem. 1995, 270, 12973–12976. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Xiao, W. Pdr3 is required for DNA damage induction of MAG1 and DDI1 via a bi-directional promoter element. Nucleic Acids Res. 2004, 32, 5066–5075. [Google Scholar] [CrossRef]
- Spasskaya, D.S.; Karpov, D.S.; Mironov, A.S.; Karpov, V.L. Transcription factor Rpn4 promotes a complex antistress response in Saccharomyces cerevisiae cells exposed to methyl methanesulfonate. Mol. Biol. 2014, 48, 141–149. [Google Scholar] [CrossRef]
- Krogh, B.O.; Symington, L.S. Recombination proteins in yeast. Annu. Rev. Genet. 2004, 38, 233–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannhaupt, G.; Feldmann, H. Genomic evolution of the proteasome system among hemiascomycetous yeasts. J. Mol. Evol. 2007, 65, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Karpov, D.S.; Lysov, Y.P.; Karpov, V.L. Evolution of the system of coordinate regulation of proteasomal gene expression in the yeast class Saccharomycetes. Mol. Biol. 2019, 53, 1029–1037. [Google Scholar] [CrossRef]
- Krol, K.; Brozda, I.; Skoneczny, M.; Bretner, M.; Skoneczna, A. A genomic screen revealing the importance of vesicular trafficking pathways in genome maintenance and protection against genotoxic stress in diploid Saccharomyces cerevisiae cells. PLoS ONE 2015, 10, e0120702. [Google Scholar] [CrossRef] [Green Version]
- Flores-Rozas, H.; Jaafar, L.; Xia, L. The role of DNA mismatch repair and recombination in the processing of DNA alkylating damage in living yeast cells. Adv. Biosci. Biotechnol. 2015, 6, 408–418. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, M.; Umezu, K. Homologous recombination is required for recovery from oxidative DNA damage. Genes Genet. Syst. 2017, 92, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arima, Y.; Nishigori, C.; Takeuchi, T.; Oka, S.; Morimoto, K.; Utani, A.; Miyachi, Y. 4-Nitroquinoline 1-oxide forms 8-hydroxydeoxyguanosine in human fibroblasts through reactive oxygen species. Toxicol. Sci. 2006, 91, 382–392. [Google Scholar] [CrossRef]
- Yip, M.C.J.; Bodnar, N.O.; Rapoport, T.A. Ddi1 is a ubiquitin-dependent protease. Proc. Natl. Acad. Sci. USA 2020, 117, 7776–7781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serbyn, N.; Noireterre, A.; Bagdiul, I.; Plank, M.; Michel, A.H.; Loewith, R.; Kornmann, B.; Stutz, F. The aspartic protease Ddi1 contributes to DNA-protein crosslink repair in yeast. Mol. Cell 2020, 77, 1066–1079. [Google Scholar] [CrossRef]
- Svoboda, M.; Konvalinka, J.; Trempe, J.F.; Grantz Saskova, K. The yeast proteases Ddi1 and Wss1 are both involved in the DNA replication stress response. DNA Repair (Amst) 2019, 80, 45–51. [Google Scholar] [CrossRef]
- Bubis, J.A.; Spasskaya, D.S.; Gorshkov, V.A.; Kjeldsen, F.; Kofanova, A.M.; Lekanov, D.S.; Gorshkov, M.V.; Karpov, V.L.; Tarasova, I.A.; Karpov, D.S. Rpn4 and proteasome-mediated yeast resistance to ethanol includes regulation of autophagy. Appl. Microbiol. Biotechnol. 2020, 104, 4027–4041. [Google Scholar] [CrossRef]
- Russell, S.J.; Reed, S.H.; Huang, W.; Friedberg, E.C.; Johnston, S.A. The 19S regulatory complex of the proteasome functions independently of proteolysis in nucleotide excision repair. Mol. Cell 1999, 3, 687–695. [Google Scholar] [CrossRef]
- Gillette, T.G.; Huang, W.; Russell, S.J.; Reed, S.H.; Johnston, S.A.; Friedberg, E.C. The 19S complex of the proteasome regulates nucleotide excision repair in yeast. Genes Dev. 2001, 15, 1528–1539. [Google Scholar] [CrossRef] [Green Version]
- Olaharski, A.J.; Rine, J.; Marshall, B.L.; Babiarz, J.; Zhang, L.; Verdin, E.; Smith, M.T. The flavoring agent dihydrocoumarin reverses epigenetic silencing and inhibits sirtuin deacetylases. PLoS Genet. 2005, 1, e77. [Google Scholar] [CrossRef]
- Chen, C.C.; Huang, J.S.; Wang, T.H.; Kuo, C.H.; Wang, C.J.; Wang, S.H.; Leu, Y.L. Dihydrocoumarin, an HDAC inhibitor, increases DNA damage sensitivity by inhibiting Rad52. Int. J. Mol. Sci. 2017, 18, 2655. [Google Scholar] [CrossRef] [Green Version]
- Maynard, S.; Fang, E.F.; Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. DNA damage, DNA repair, aging, and neurodegeneration. Cold Spring Harb. Perspect. Biol. 2015, 5, a025130. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.D.; Richardson, C.D.; Corn, J.E. Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol. 2019, 21, 1468–1478. [Google Scholar] [CrossRef]
- Dantuma, N.P.; Heinen, C.; Hoogstraten, D. The ubiquitin receptor Rad23: At the crossroads of nucleotide excision repair and proteasomal degradation. DNA Repair (Amst) 2009, 8, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Kaplun, L.; Tzirkin, R.; Bakhrat, A.; Shabek, N.; Ivantsiv, Y.; Raveh, D. The DNA damage-inducible UbL-UbA protein Ddi1 participates in Mec1-mediated degradation of Ho endonuclease. Mol. Cell. Biol. 2005, 25, 5355–5362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolaet, B.L.; Clarke, D.J.; Wolff, M.; Watson, M.H.; Henze, M.; Divita, G.; Reed, S.I. UBA domains mediate protein-protein interactions between two DNA damage-inducible proteins. J. Mol. Biol. 2001, 313, 955–963. [Google Scholar] [CrossRef]
- de Graaf, B.; Clore, A.; McCullough, A.K. Cellular pathways for DNA repair and damage tolerance of formaldehyde-induced DNA-protein crosslinks. DNA Repair (Amst) 2009, 8, 1207–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, T.; Katafuchi, A.; Matsubara, M.; Terato, H.; Tsuboi, T.; Masuda, T.; Tatsumoto, T.; Pack, S.P.; Makino, K.; Croteau, D.L.; et al. Homologous recombination but not nucleotide excision repair plays a pivotal role in tolerance of DNA-protein cross-links in mammalian cells. J. Biol. Chem. 2009, 284, 27065–27076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohsawa, K.-I.; Furihata, C.; Mori, M.; Ikui, E. Ability of N-methyl-N′-nitro-N-nitrosoguanidine, 4-nitroquinoline 1-oxide, dimethylnitrosome, and NaCl to induce inscheduled DNA synthesis, stimulate replicative DNA synthesis, and produce DNA single-strand breaks in pyloric mucosa of rat stomach. Mutat. Res. 1993, 287, 307–319. [Google Scholar] [CrossRef]
- Miao, Z.H.; Rao, V.A.; Agama, K.; Antony, S.; Kohn, K.W.; Pommier, Y. 4-nitroquinoline-1-oxide induces the formation of cellular topoisomerase I-DNA cleavage complexes. Cancer Res. 2006, 66, 6540–6545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; Redon, C.; Rao, V.A.; Seiler, J.A.; Sordet, O.; Takemura, H.; Antony, S.; Meng, L.; Liao, Z.; Kohlhagen, G.; et al. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat. Res. 2003, 532, 173–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Sacher, M.; Pfander, B.; Hoege, C.; Jentsch, S. Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat. Cell Biol. 2006, 8, 1284–1290. [Google Scholar] [CrossRef]
- Cole, G.M.; Schild, D.; Mortimer, R.K. Two DNA repair and recombination genes in Saccharomyces cerevisiae, RAD52 and RAD54, are induced during meiosis. Mol. Cell. Biol. 1989, 9, 3101–3104. [Google Scholar] [CrossRef] [Green Version]
- Cole, G.M.; Schild, D.; Lovett, S.T.; Mortimer, R.K. Regulation of RAD54- and RAD52-lacZ gene fusions in Saccharomyces cerevisiae in response to DNA damage. Mol. Cell. Biol. 1987, 7, 1078–1084. [Google Scholar] [CrossRef] [Green Version]
- Asleson, E.N.; Livingston, D.M. Investigation of the stability of yeast rad52 mutant proteins uncovers post-translational and transcriptional regulation of Rad52p. Genetics 2003, 163, 91–101. [Google Scholar]
- Dornfeld, K.J.; Livingston, D.M. Effects of controlled RAD52 expression on repair and recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 1991, 11, 2013–2017. [Google Scholar] [CrossRef] [Green Version]
- Matuo, R.; Sousa, F.G.; Soares, D.G.; Bonatto, D.; Saffi, J.; Escargueil, A.E.; Larsen, A.K.; Henriques, J.A. Saccharomyces cerevisiae as a model system to study the response to anticancer agents. Cancer Chemother. Pharmacol. 2012, 70, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Lin, P.Y.; Chiu, Y.C.; Huang, J.S.; Kuo, Y.T.; Wu, J.C.; Chen, C.C. Curcumin-mediated HDAC inhibition suppresses the DNA damage response and contributes to increased DNA damage sensitivity. PLoS ONE 2015, 10, e0134110. [Google Scholar] [CrossRef] [PubMed]
- San Miguel, J.F.; Schlag, R.; Khuageva, N.K.; Dimopoulos, M.A.; Shpilberg, O.; Kropff, M.; Spicka, I.; Petrucci, M.T.; Palumbo, A.; Samoilova, O.S.; et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N. Engl. J. Med. 2008, 359, 906–917. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Van der Sluis, P.C.; Boulware, D.; Hazlehurst, L.A.; Dalton, W.S. The FA/BRCA pathway is involved in melphalan-induced DNA interstrand cross-link repair and accounts for melphalan resistance in multiple myeloma cells. Blood 2005, 106, 698–705. [Google Scholar] [CrossRef]
- Bharti, A.C.; Donato, N.; Singh, S.; Aggarwal, B.B. Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor-kappa B and IkappaBalpha kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood 2003, 101, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Xiao, Q.; Zhang, K.; Zuo, X.; Shrestha, U.K. Reversal of multidrug resistance by curcumin through FA/BRCA pathway in multiple myeloma cell line MOLP-2/R. Ann. Hematol. 2010, 89, 399–404. [Google Scholar] [CrossRef]
- Sung, B.; Kunnumakkara, A.B.; Sethi, G.; Anand, P.; Guha, S.; Aggarwal, B.B. Curcumin circumvents chemoresistance in vitro and potentiates the effect of thalidomide and bortezomib against human multiple myeloma in nude mice model. Mol. Cancer Ther. 2009, 8, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Lyu, J.; Yang, E.J.; Liu, Y.; Wu, C.; Pardeshi, L.; Tan, K.; Chen, Q.; Xu, X.; Deng, C.X.; et al. Class I histone deacetylase inhibition is synthetic lethal with BRCA1 deficiency in breast cancer cells. Acta Pharm. Sin. B 2020, 10, 615–627. [Google Scholar] [CrossRef]
- van Leeuwen, J.; Andrews, B.; Boone, C.; Tan, G. Rapid and efficient plasmid construction by homologous recombination in yeast. Cold Spring Harb. Protoc. 2015, 2015, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Bao, Z.; Xiao, H.; Liang, J.; Zhang, L.; Xiong, X.; Sun, N.; Si, T.; Zhao, H. Homology-integrated CRISPR-Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae. ACS Synth. Biol. 2015, 4, 585–594. [Google Scholar] [CrossRef]
- Dahlman, J.E.; Abudayyeh, O.O.; Joung, J.; Gootenberg, J.S.; Zhang, F.; Konermann, S. Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat. Biotechnol. 2015, 33, 1159–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spasskaya, D.S.; Nadolinskaia, N.I.; Tutyaeva, V.V.; Lysov, Y.P.; Karpov, V.L.; Karpov, D.S. Yeast Rpn4 Links the Proteasome and DNA Repair via RAD52 Regulation. Int. J. Mol. Sci. 2020, 21, 8097. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218097
Spasskaya DS, Nadolinskaia NI, Tutyaeva VV, Lysov YP, Karpov VL, Karpov DS. Yeast Rpn4 Links the Proteasome and DNA Repair via RAD52 Regulation. International Journal of Molecular Sciences. 2020; 21(21):8097. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218097
Chicago/Turabian StyleSpasskaya, Daria S., Nonna I. Nadolinskaia, Vera V. Tutyaeva, Yuriy P. Lysov, Vadim L. Karpov, and Dmitry S. Karpov. 2020. "Yeast Rpn4 Links the Proteasome and DNA Repair via RAD52 Regulation" International Journal of Molecular Sciences 21, no. 21: 8097. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218097