HIV-1 Tat Dysregulates the Hypothalamic-Pituitary-Adrenal Stress Axis and Potentiates Oxycodone-Mediated Psychomotor and Anxiety-Like Behavior of Male Mice


Abstract
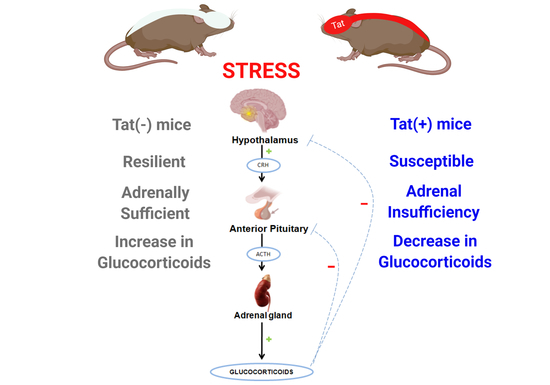
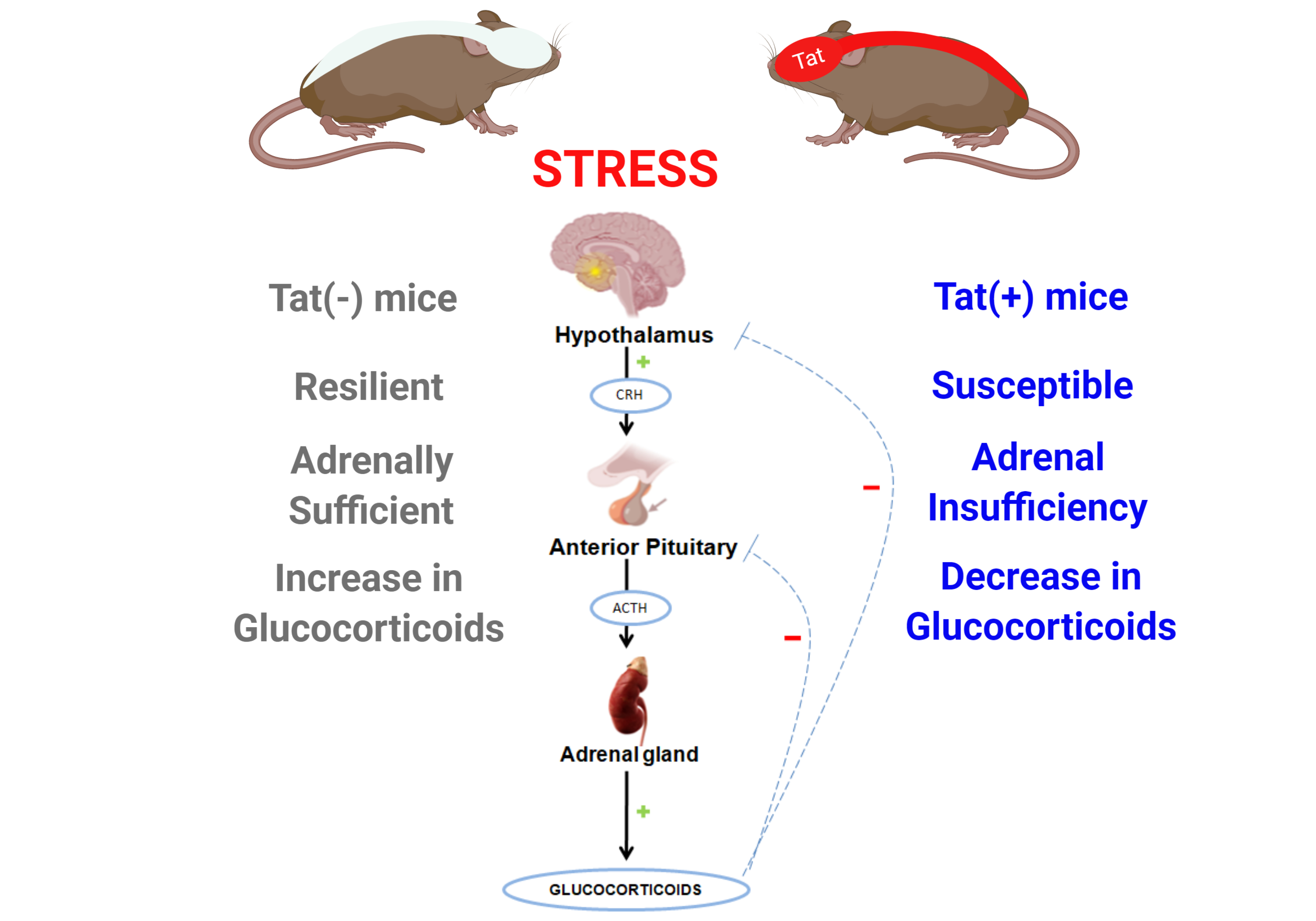
:
1. Introduction
2. Results
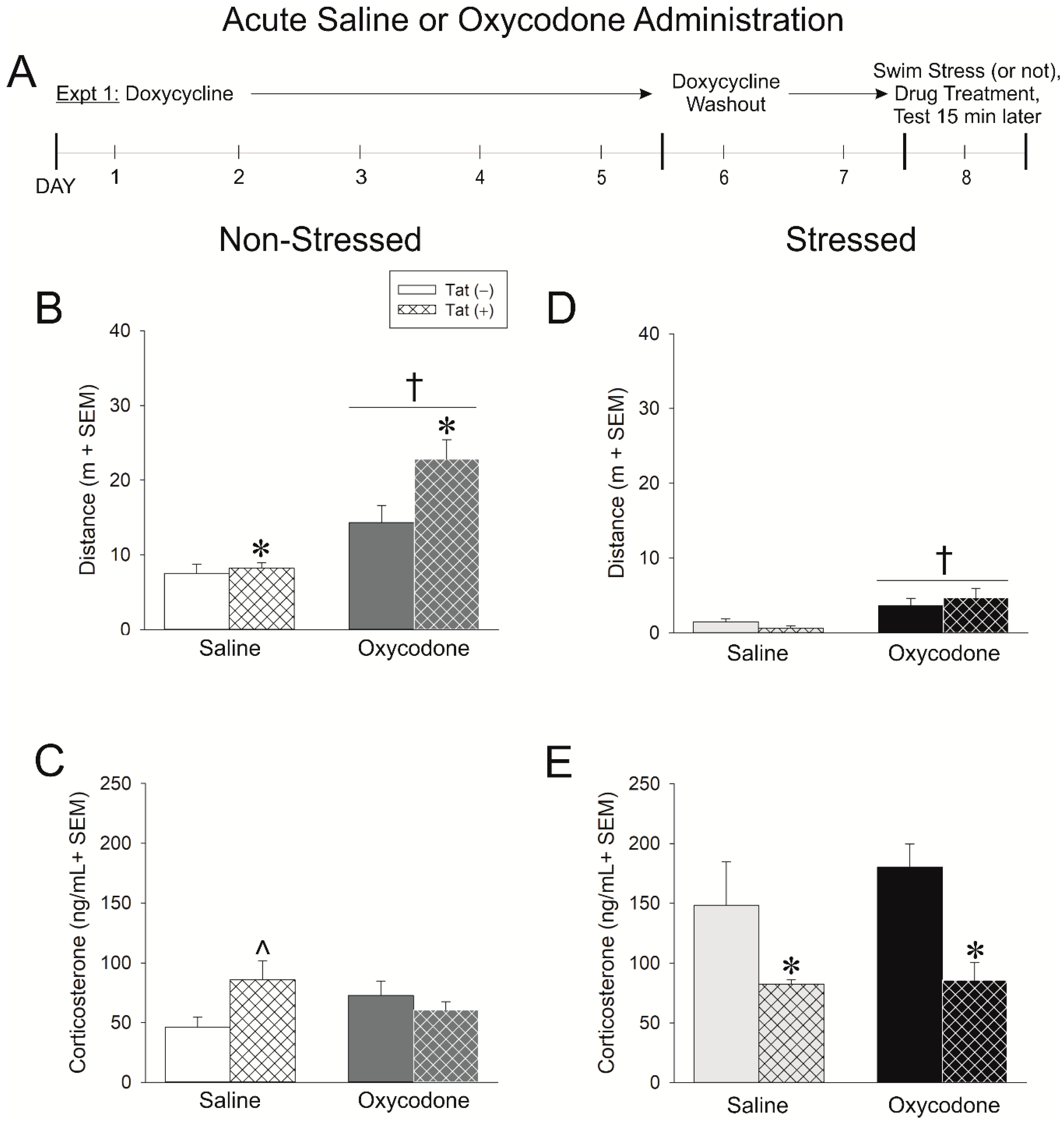
2.1. Experiment 1: HIV-1 Tat Expression Causes Adrenal Insufficiency and Potentiates Oxycodone’s Psychomotor Effects
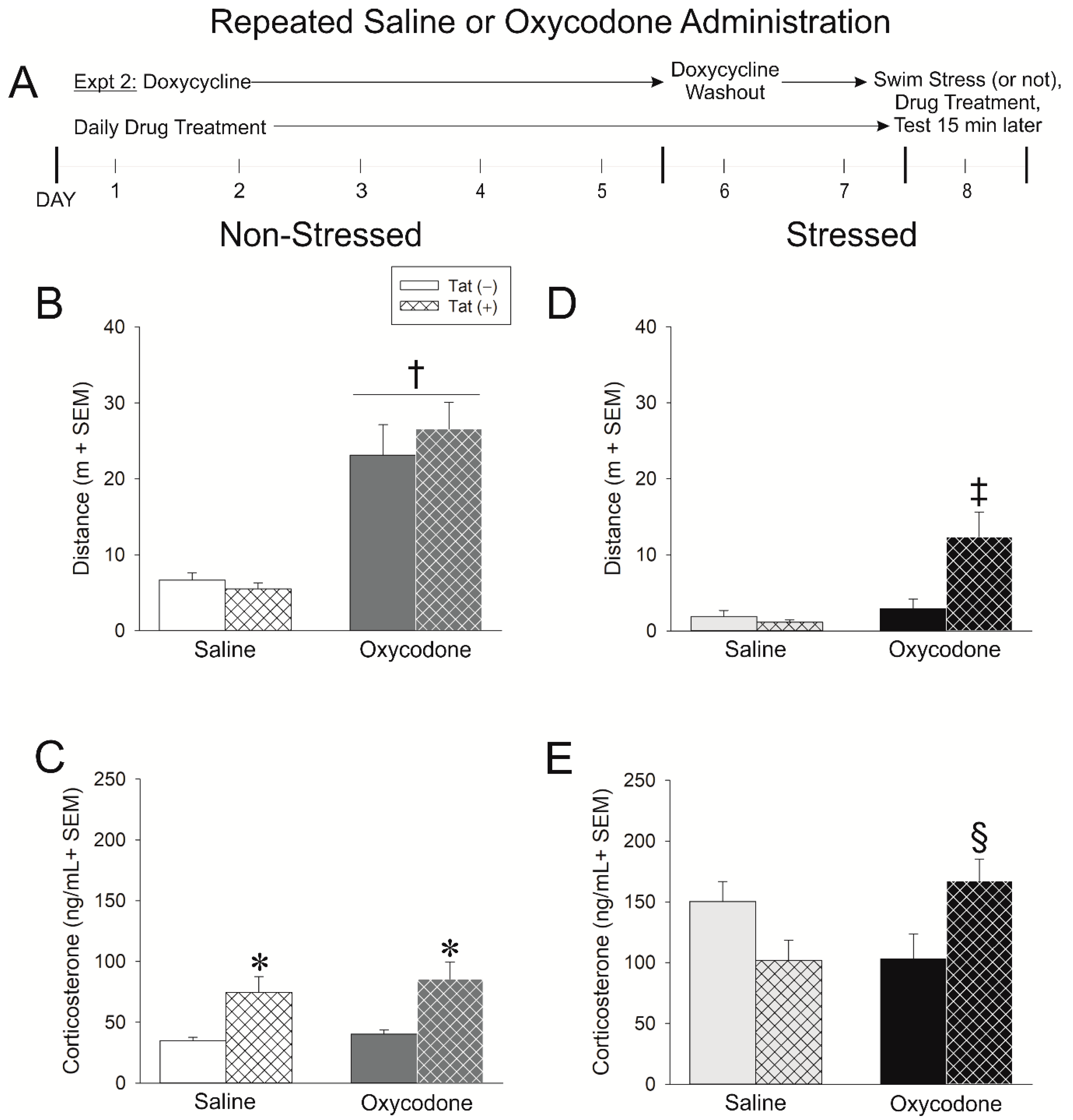
2.2. Experiment 2: Repeated Exposure to Oxycodone Increases the Tat-Mediated Psychomotor and Glucocorticoid Stress Response
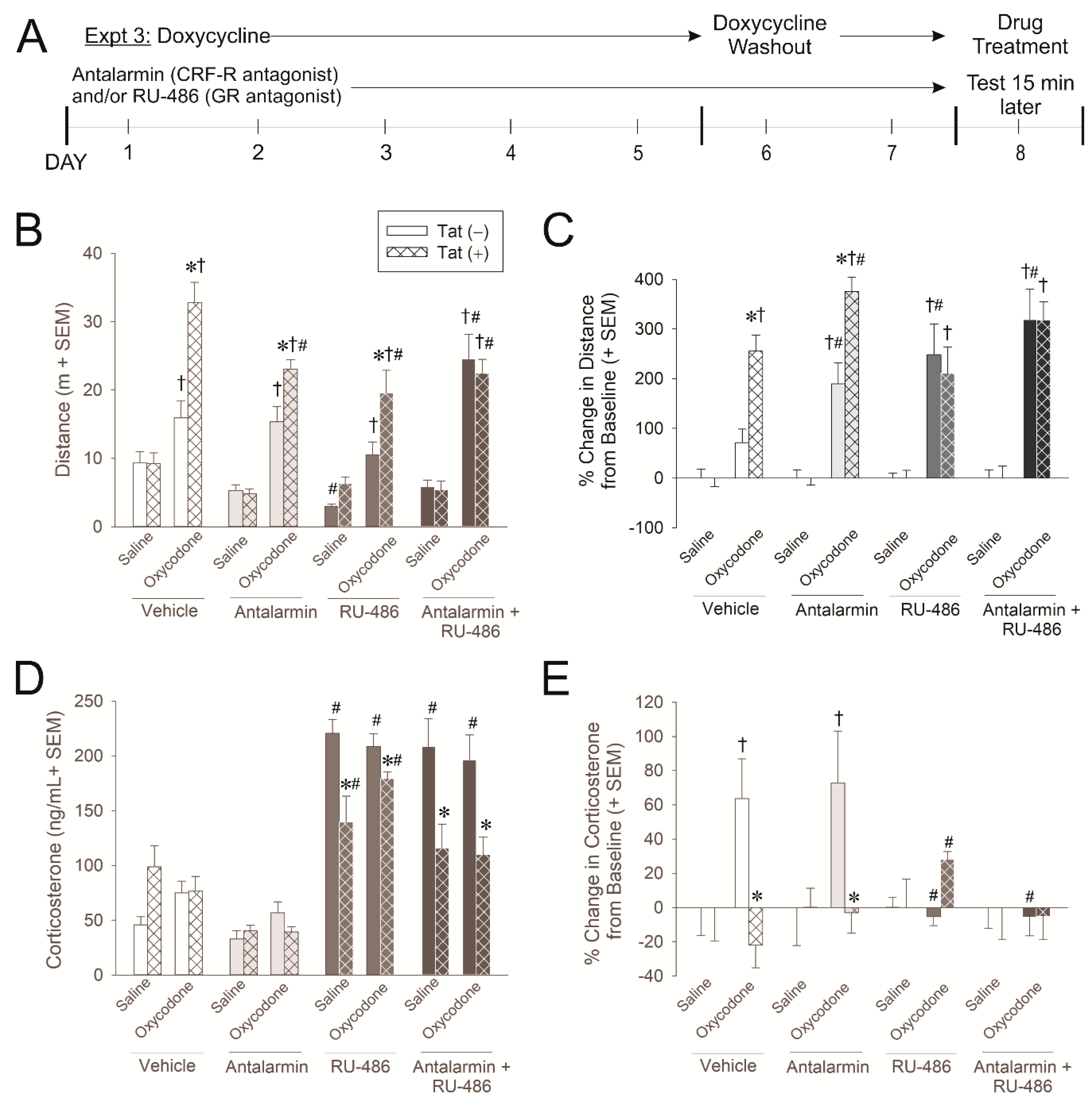
2.3. Experiment 3: Glucocorticoid and CRF Receptors Are Involved in the Psychomotor, Anxiety-Like, and HPA Axis Response to Oxycodone
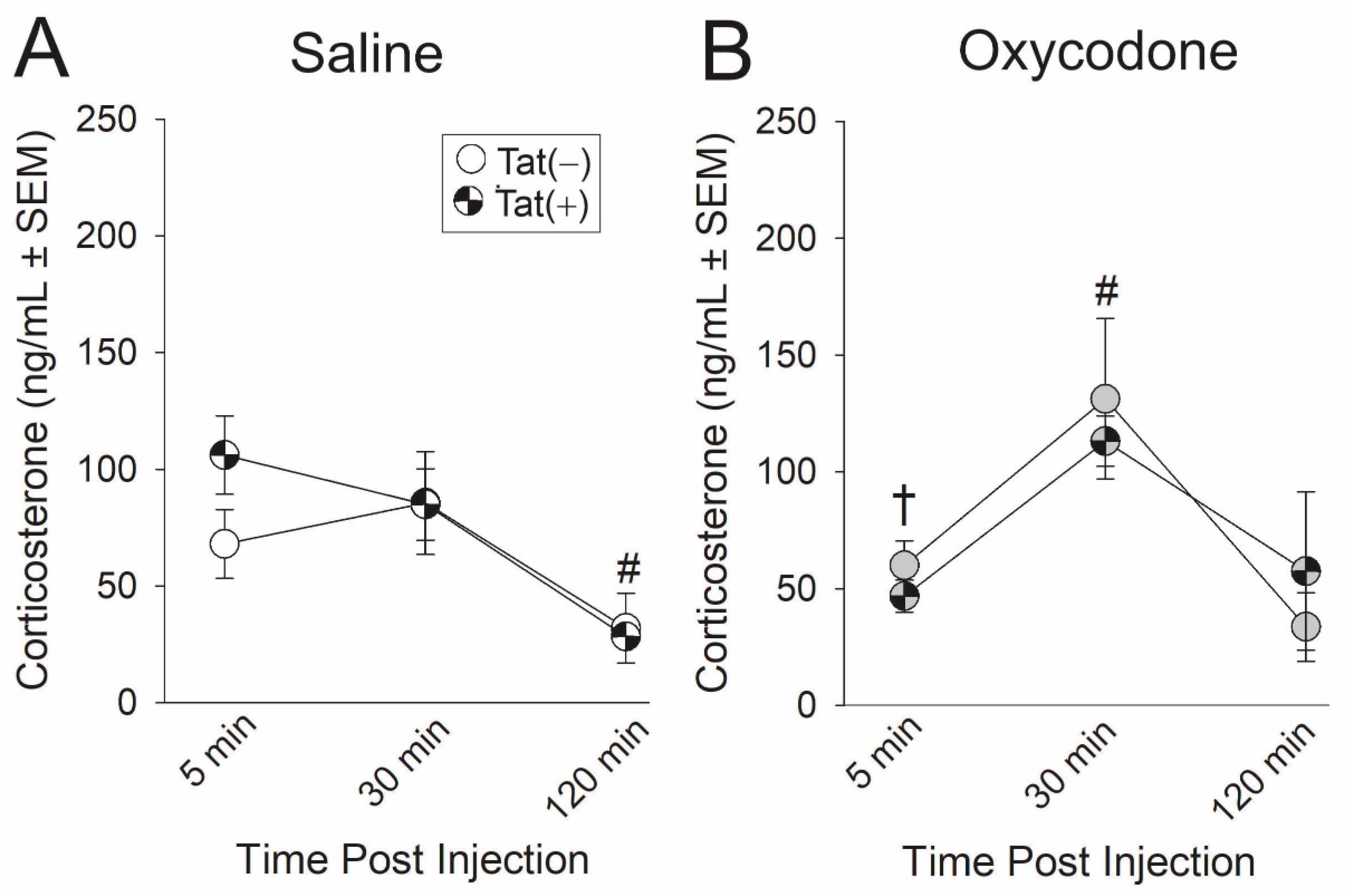
2.4. Experiment 4: The Time-Course of HPA Axis Activation Was Influenced by Oxycodone Exposure
3. Discussion
4. Materials and Methods
4.1. Subjects and Housing
4.2. Chemicals
4.3. Procedure
4.3.1. Experiment 1: Assessment of Acute Oxycodone Exposure in Non-Stressed and Stressed Mice
4.3.2. Experiment 2: Assessment of Repeated Oxycodone Exposure in Non-Stressed and Stressed Mice
4.3.3. Experiment 3: Assessment of Acute Oxycodone Exposure Following GR and/or CRF-R Blockade
4.3.4. Experiment 4: Determination of Peak HPA Activation Following Tat or Oxycodone Exposure
4.4. Behavioral Assessment
4.4.1. Forced Swim Stress Stimulus
4.4.2. Open Field
4.4.3. Light-Dark Transition Test
4.5. Corticosterone Assay
4.5.1. Tissue Collection
4.5.2. Steroid Extraction
4.5.3. Enzyme-Linked Immunosorbant Assay (ELISA)
4.6. Statistical Analyses
Author Contributions
Funding
Conflicts of Interest
References
- Centers for Disease Control and Prevention. HIV Surveillance Report (Updated); 2018; Volume 31. Available online: http://www.cdc.gov/hiv/library/reports/hiv-surveillance.html (accessed on 3 August 2020).
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder--pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.B.; Kaul, M. Neuronal Stress and Injury Caused by HIV-1, cART and Drug Abuse: Converging Contributions to HAND. Brain Sci. 2017, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Jeevanjee, S.; Penko, J.; Guzman, D.; Miaskowski, C.; Bangsberg, D.R.; Kushel, M.B. Opioid analgesic misuse is associated with incomplete antiretroviral adherence in a cohort of HIV-infected indigent adults in San Francisco. AIDS Behav. 2014, 18, 1352–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlin, J.S.; Tamhane, A.; Starrels, J.L.; Kertesz, S.; Saag, M.; Cropsey, K. Factors Associated with Prescription of Opioids and Co-prescription of Sedating Medications in Individuals with HIV. AIDS Behav. 2016, 20, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Silverberg, M.J.; Ray, G.T.; Saunders, K.; Rutter, C.M.; Campbell, C.I.; Merrill, J.O.; Sullivan, M.D.; Banta-Green, C.J.; Von Korff, M.; Weisner, C. Prescription long-term opioid use in HIV-infected patients. Clin. J. Pain 2012, 28, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrousos, G.P.; Zapanti, E.D. Hypothalamic-pituitary-adrenal axis in HIV infection and disease. Endocrinol. Metab. Clin. N. Am. 2014, 43, 791–806. [Google Scholar] [CrossRef] [PubMed]
- Marik, P.E.; Kiminyo, K.; Zaloga, G.P. Adrenal insufficiency in critically ill patients with human immunodeficiency virus. Crit. Care Med. 2002, 30, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Sharma, L.K.; Anand, A.; Gadpayle, A.K.; Gaurav, K.; Mukherjee, S.; Kulshreshtha, B.; Dutta, D. Presence, patterns & predictors of hypocortisolism in patients with HIV infection in India. Indian J. Med. Res. 2018, 147, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Frye, C.A.; Paris, J.J.; Osborne, D.M.; Campbell, J.C.; Kippin, T.E. Prenatal Stress Alters Progestogens to Mediate Susceptibility to Sex-Typical, Stress-Sensitive Disorders, such as Drug Abuse: A Review. Front. Psychiatry 2011, 2, 52. [Google Scholar] [CrossRef] [Green Version]
- Koob, G.F.; Volkow, N.D. Neurobiology of addiction: A neurocircuitry analysis. Lancet Psychiatry 2016, 3760–3773. [Google Scholar] [CrossRef]
- Kaul, M.; Zheng, J.; Okamoto, S.; Gendelman, H.E.; Lipton, S.A. HIV-1 infection and AIDS: Consequences for the central nervous system. Cell Death Differ. 2005, 1, 878–892. [Google Scholar] [CrossRef] [Green Version]
- Salahuddin, M.F.; Qrareya, A.N.; Mahdi, F.; Jackson, D.; Foster, M.; Vujanovic, T.; Box, J.G.; Paris, J.J. Combined HIV-1 Tat and oxycodone activate the hypothalamic-pituitary-adrenal and -gonadal axes and promote psychomotor, affective, and cognitive dysfunction in female mice. Horm. Behav. 2020, 119, 104649. [Google Scholar] [CrossRef]
- Bokhari, S.M.; Yao, H.; Bethel-Brown, C.; Fuwang, P.; Williams, R.; Dhillon, N.K.; Hegde, R.; Kumar, A.; Buch, S.J. Morphine enhances Tat-induced activation in murine microglia. J. Neurovirol. 2009, 15, 219–228. [Google Scholar] [CrossRef]
- Gonek, M.; McLane, V.D.; Stevens, D.L.; Lippold, K.; Akbarali, H.I.; Knapp, P.E.; Dewey, W.L.; Hauser, K.F.; Paris, J.J. CCR5 mediates HIV-1 Tat-induced neuroinflammation and influences morphine tolerance, dependence, and reward. Brain Behav. Immun. 2018, 69, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Fitting, S.; Zou, S.; Chen, W.; Vo, P.; Hauser, K.F.; Knapp, P.E. Regional heterogeneity and diversity in cytokine and chemokine production by astroglia: Differential responses to HIV-1 Tat, gp120, and morphine revealed by multiplex analysis. J. Proteome Res. 2010, 9, 1795–1804. [Google Scholar] [CrossRef] [Green Version]
- Pace, T.W.; Miller, A.H. Cytokines and glucocorticoid receptor signaling. Relevance to major depression. Ann. N. Y. Acad. Sci. 2009, 1179, 86–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goleva, E.; Kisich, K.O.; Leung, D.Y. A role for STAT5 in the pathogenesis of IL-2-induced glucocorticoid resistance. J. Immunol. 2002, 169, 5934–5940. [Google Scholar] [CrossRef] [Green Version]
- Pariante, C.M.; Pearce, B.D.; Pisell, T.L.; Sanchez, C.I.; Po, C.; Su, C.; Miller, A.H. The proinflammatory cytokine, interleukin-1alpha, reduces glucocorticoid receptor translocation and function. Endocrinology 1999, 140, 4359–4366. [Google Scholar] [CrossRef] [PubMed]
- Raddatz, D.; Toth, S.; Schwörer, H.; Ramadori, G. Glucocorticoid receptor signaling in the intestinal epithelial cell lines IEC-6 and Caco-2: Evidence of inhibition by interleukin-1beta. Int. J. Colorectal Dis. 2001, 16, 377–383. [Google Scholar] [CrossRef]
- Bellavance, M.A.; Rivest, S. The HPA—Immune Axis and the Immunomodulatory Actions of Glucocorticoids in the Brain. Front. Immunol. 2014, 5, 136. [Google Scholar] [CrossRef] [Green Version]
- Franchimont, D. Overview of the actions of glucocorticoids on the immune response: A good model to characterize new pathways of immunosuppression for new treatment strategies. Ann. N. Y. Acad. Sci. 2004, 1024, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y.; Martin, R.J.; Szefler, S.J.; Sher, E.R.; Ying, S.; Kay, A.B.; Hamid, Q. Dysregulation of interleukin 4, interleukin 5, and interferon gamma gene expression in steroid-resistant asthma. J. Exp. Med. 1995, 181, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Baschant, U.; Tuckermann, J. The role of the glucocorticoid receptor in inflammation and immunity. J. Steroid Biochem. Mol. Biol. 2010, 120, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Zapanti, E.; Terzidis, K.; Chrousos, G. Dysfunction of the hypothalamic-pituitary-adrenal axis in HIV infection and disease. Hormones 2008, 7, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Mukerji, S.S.; Misra, V.; Lorenz, D.R.; Chettimada, S.; Keller, K.; Letendre, S.; Ellis, R.J.; Morgello, S.; Parker, R.A.; Gabuzda, D. Low Neuroactive Steroids Identifies a Biological Subtype of Depression in Adults with Human Immunodeficiency Virus on Suppressive Antiretroviral Therapy. J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [Green Version]
- Qiao, S.; Li, X.; Zilioli, S.; Chen, Z.; Deng, H.; Pan, J.; Guo, W. Hair Measurements of Cortisol, DHEA, and DHEA to Cortisol Ratio as Biomarkers of Chronic Stress among People Living with HIV in China: Known-Group Validation. PLoS ONE 2017, 12, e0169827. [Google Scholar] [CrossRef]
- Christeff, N.; Gherbi, N.; Mammes, O.; Dalle, M.T.; Gharakhanian, S.; Lortholary, O.; Melchior, J.C.; Nunez, E.A. Serum cortisol and DHEA concentrations during HIV infection. Psychoneuroendocrinology 1997, 22, S11–S18. [Google Scholar] [CrossRef]
- Schürmeyer, T.H.; Müller, V.; von zurMühlen, A.; Schmidt, R.E. Thyroid and adrenal function in HIV-infected outpatients. Eur. J. Med. Res. 1997, 2, 220–226. [Google Scholar]
- Norbiato, G.; Bevilacqua, M.; Vago, T.; Baldi, G.; Chebat, E.; Bertora, P.; Moroni, M.; Galli, M.; Oldenburg, N. Cortisol resistance in acquired immunodeficiency syndrome. J. Clin. Endocrinol. Metab. 1992, 74, 608–613. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.P.; Adams, D.; Prewitt, C. Regulatory changes in neuroendocrine stress-integrative circuitry produced by a variable stress paradigm. Neuroendocrinology 1995, 61, 180–190. [Google Scholar] [CrossRef]
- Kitraki, E.; Karandrea, D.; Kittas, C. Long-lasting effects of stress on glucocorticoid receptor gene expression in the rat brain. Neuroendocrinology 1999, 69, 331–338. [Google Scholar] [CrossRef]
- Irusen, E.; Matthews, J.G.; Takahashi, A.; Barnes, P.J.; Chung, K.F.; Adcock, I.M. p38 Mitogen-activated protein kinase-induced glucocorticoid receptor phosphorylation reduces its activity: Role in steroid-insensitive asthma. J. Allergy Clin. Immunol. 2002, 109, 649–657. [Google Scholar] [CrossRef]
- Paris, J.J.; Liere, P.; Kim, S.; Mahdi, F.; Buchanan, M.E.; Nass, S.R.; Qrareya, A.N.; Salahuddin, M.F.; Pianos, A.; Fernandez, N.; et al. Pregnane steroidogenesis is altered by HIV-1 Tat and morphine: Physiological allopregnanolone is protective against neurotoxic and psychomotor effects. Neurobiol. Stress 2020, 12, 100211. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, N.C.; Galata, Z.; Kino, T.; Chrousos, G.P.; Charmandari, E. The human glucocorticoid receptor: Molecular basis of biologic function. Steroids 2010, 75, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, Y.; Iwasaki, Y.; Tsugita, M.; Nishiyama, M.; Taguchi, T.; Okazaki, M.; Nakayama, S.; Kambayashi, M.; Hashimoto, K.; Terada, Y. Glucocorticoid receptor-beta and receptor-gamma exert dominant negative effect on gene repression but not on gene induction. Endocrinology 2010, 151, 3204–3213. [Google Scholar] [CrossRef] [Green Version]
- Webster, J.C.; Oakley, R.H.; Jewell, C.M.; Cidlowski, J.A. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: A mechanism for the generation of glucocorticoid resistance. Proc. Natl. Acad. Sci. USA 2001, 98, 6865–6870. [Google Scholar] [CrossRef] [Green Version]
- Bekhbat, M.; Mehta, C.C.; Kelly, S.D.; Vester, A.; Ofotokun, I.; Felger, J.; Wingood, G.; Anastos, K.; Gustafson, D.R.; Kassaye, S.; et al. HIV and symptoms of depression are independently associated with impaired glucocorticoid signaling. Psychoneuroendocrinology 2018, 96, 118–125. [Google Scholar] [CrossRef]
- Bandaru, V.V.; Mielke, M.M.; Sacktor, N.; McArthur, J.C.; Grant, I.; Letendre, S.; Chang, L.; Wojna, V.; Pardo, C.; Calabresi, P.; et al. A lipid storage-like disorder contributes to cognitive decline in HIV-infected subjects. Neurology 2013, 81, 1492–1499. [Google Scholar] [CrossRef] [Green Version]
- Cotto, B.; Natarajaseenivasan, K.; Ferrero, K.; Wesley, L.; Sayre, M.; Langford, D. Cocaine and HIV-1 Tat disrupt cholesterol homeostasis in astrocytes: Implications for HIV-associated neurocognitive disorders in cocaine user patients. Glia 2018, 66, 889–902. [Google Scholar] [CrossRef]
- MohseniAhooyi, T.; Shekarabi, M.; Torkzaban, B.; Langford, T.D.; Burdo, T.H.; Gordon, J.; Datta, P.K.; Amini, S.; Khalili, K. Dysregulation of Neuronal Cholesterol Homeostasis upon Exposure to HIV-1 Tat and Cocaine Revealed by RNA-Sequencing. Sci. Rep. 2018, 8, 16300. [Google Scholar] [CrossRef]
- Haughey, N.J.; Cutler, R.G.; Tamara, A.; McArthur, J.C.; Vargas, D.L.; Pardo, C.A.; Turchan, J.; Nath, A.; Mattson, M.P. Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann. Neurol. 2004, 55, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.A.; Ellis, R.J. HIV in the cART era and the mitochondrial: Immune interface in the CNS. Int. Rev. Neurobiol. 2019, 145, 29–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesby, J.P.; Najera, J.A.; Romoli, B.; Fang, Y.; Basova, L.; Birmingham, A.; Marcondes, M.C.G.; Dulcis, D.; Semenova, S. HIV-1 TAT protein enhances sensitization to methamphetamine by affecting dopaminergic function. Brain Behav. Immun. 2017, 65, 210–221. [Google Scholar] [CrossRef] [Green Version]
- De Guglielmo, G.; Fu, Y.; Chen, J.; Larrosa, E.; Hoang, I.; Kawamura, T.; Lorrai, I.; Zorman, B.; Bryant, J.; George, O.; et al. Increases in compulsivity, inflammation, and neural injury in HIV transgenic rats with escalated methamphetamine self-administration under extended-access conditions. Brain Res. 2020, 1726, 146502. [Google Scholar] [CrossRef]
- McIntosh, S.; Sexton, T.; Pattison, L.P.; Childers, S.R.; Hemby, S.E. Increased Sensitivity to Cocaine Self-Administration in HIV-1 Transgenic Rats is Associated with Changes in Striatal Dopamine Transporter Binding. J. Neuroimmune Pharmacol. 2015, 10, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Huynh, Y.W.; Thompson, B.M.; Larsen, C.E.; Buch, S.; Guo, M.L.; Bevins, R.A.; Murray, J.E. Male HIV-1 transgenic rats show reduced cocaine-maintained lever-pressing compared to F344 wildtype rats despite similar baseline locomotion. J. Exp. Anal. Behav. 2020, 113, 468–484. [Google Scholar] [CrossRef]
- Kesby, J.P.; Chang, A.; Najera, J.A.; Marcondes, M.C.G.; Semenova, S. Brain Reward Function after Chronic and Binge Methamphetamine Regimens in Mice Expressing the HIV-1 TAT Protein. Curr. HIV Res. 2019, 17, 126–133. [Google Scholar] [CrossRef]
- Wayman, W.N.; Chen, L.; Hu, X.T.; Napier, T.C. HIV-1 Transgenic Rat Prefrontal Cortex Hyper-Excitability is Enhanced by Cocaine Self-Administration. Neuropsychopharmacology 2016, 41, 1965–1973. [Google Scholar] [CrossRef] [Green Version]
- Bali, A.; Randhawa, P.K.; Jaggi, A.S. Stress and opioids: Role of opioids in modulating stress-related behavior and effect of stress on morphine conditioned place preference. Neurosci. Biobehav. Rev. 2015, 51, 138–150. [Google Scholar] [CrossRef]
- Gaskill, P.J.; Miller, D.R.; Gamble-George, J.; Yano, H.; Khoshbouei, H. HIV, Tat and dopamine transmission. Neurobiol. Dis. 2017, 105, 51–73. [Google Scholar] [CrossRef] [PubMed]
- Raber, J.; Toggas, S.M.; Lee, S.; Bloom, F.E.; Epstein, C.J.; Mucke, L. Central nervous system expression of HIV-1 Gp120 activates the hypothalamic-pituitary-adrenal axis: Evidence for involvement of NMDA receptors and nitric oxide synthase. Virology 1996, 226, 362–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.; Nappi, R.E.; Polatti, F.; Poma, A.; Grossman, A.B.; Nappi, G. Stimulating effect of HIV-1 coat protein gp120 on corticotropin-releasing hormone and arginine vasopressin in the rat hypothalamus: Involvement of nitric oxide. Exp. Neurol. 2000, 166, 376–384. [Google Scholar] [CrossRef]
- Kino, T.; Gragerov, A.; Kopp, J.B.; Stauber, R.H.; Pavlakis, G.N.; Chrousos, G.P. The HIV-1 virion-associated protein vpr is a coactivator of the human glucocorticoid receptor. J. Exp. Med. 1999, 189, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Sawaya, B.E.; Khalili, K.; Gordon, J.; Taube, R.; Amini, S. Cooperative interaction between HIV-1 regulatory proteins Tat and Vpr modulates transcription of the viral genome. J. Biol. Chem. 2000, 275, 35209–35214. [Google Scholar] [CrossRef] [Green Version]
- Kino, T.; Slobodskaya, O.; Pavlakis, G.N.; Chrousos, G.P. Nuclear receptor coactivator p160 proteins enhance the HIV-1 long terminal repeat promoter by bridging promoter-bound factors and the Tat-P-TEFb complex. J. Biol. Chem. 2002, 277, 2396–2405. [Google Scholar] [CrossRef] [Green Version]
- Bruce-Keller, A.J.; Turchan-Cholewo, J.; Smart, E.J.; Geurin, T.; Chauhan, A.; Reid, R.; Xu, R.; Nath, A.; Knapp, P.E.; Hauser, K.F. Morphine causes rapid increases in glial activation and neuronal injury in the striatum of inducible HIV-1 Tat transgenic mice. Glia 2008, 56, 1414–1427. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Wang, S.; Zeng, Z.; Li, F.; Montalvo-Ortiz, J.; Tucker, C.; Akhtar, S.; Shi, J.; Meltzer, H.Y.; Rice, K.C.; et al. Effects of corticotrophin-releasing factor receptor 1 antagonists on amyloid-β and behavior in Tg2576 mice. Psychopharmacology 2014, 231, 4711–4722. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wang, S.; Rice, K.C.; Munro, C.A.; Wand, G.S. Restraint stress and ethanol consumption in two mouse strains. Alcohol. Clin. Exp. Res. 2008, 32, 840–852. [Google Scholar] [CrossRef] [Green Version]
- Hofford, R.S.; Prendergast, M.A.; Bardo, M.T. Pharmacological manipulation of glucocorticoid receptors differentially affects cocaine self-administration in environmentally enriched and isolated rats. Behav. Brain Res. 2015, 283, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefevre, E.M.; Medley, G.A.; Reeks, T.; Alexander, S.; Burne, T.H.J.; Eyles, D.W. Effect of the glucocorticoid receptor antagonist RU486 on MK-801 induced behavioural sensitisation. PLoS ONE 2017, 12, e0176156. [Google Scholar] [CrossRef]
- Leite, L.M.; Carvalho, A.G.; Ferreira, P.L.; Pessoa, I.X.; Gonçalves, D.O.; Lopes Ade, A.; Góes, J.G.; Alves, V.C.; Leal, L.K.; Brito, G.A.; et al. Anti-inflammatory properties of doxycycline and minocycline in experimental models: An in vivo and in vitro comparative study. Inflammopharmacology 2011, 19, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Paris, J.J.; Singh, H.D.; Ganno, M.L.; Jackson, P.; McLaughlin, J.P. Anxiety-like behavior of mice produced by conditional central expression of the HIV-1 regulatory protein, Tat. Psychopharmacology 2014, 231, 2349–2360. [Google Scholar] [CrossRef] [Green Version]
- Fitting, S.; Scoggins, K.L.; Xu, R.; Dever, S.M.; Knapp, P.E.; Dewey, W.L.; Hauser, K.F. Morphine efficacy is altered in conditional HIV-1 Tat transgenic mice. Eur. J. Pharmacol. 2012, 689, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Porsolt, R.D.; Bertin, A.; Jalfre, M. Behavioral despair in mice: A primary screening test for antidepressants. Arch. Int. Pharmacodyn. Ther. 1977, 229, 327–336. [Google Scholar]
- Bourin, M.; Hascoët, M. The mouse light/dark box test. Eur. J. Pharmacol. 2003, 463, 55–65. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Behavioral Measure | Non-Stressed | Stressed | ||||||
|---|---|---|---|---|---|---|---|---|
| Saline (0.9% w/v) | Oxycodone (3 mg/kg) | Saline (0.9% w/v) | Oxycodone (3 mg/kg) | |||||
| Tat(−) (n = 8–9) | Tat(+) (n = 7–8) | Tat(−) (n = 12) | Tat(+) (n = 8–9) | Tat(−) (n = 8) | Tat(+) (n = 9) | Tat(−) (n = 8) | Tat(+) (n = 9) | |
| Mean Velocity (m/s) | 0.025 ± 0.004 | 0.027 ± 0.002 * | 0.048 ± 0.008 † | 0.076 ± 0.009 †* | 0.005 ± 0.001 | 0.002 ± 0.001 | 0.012 ± 0.003 † | 0.015 ± 0.004 † |
| Rearing number | 39.4 ± 6.2 | 23.8 ± 3.2 | 18.1 ± 11.4 † | 6.8 ± 2.2 † | 4.5 ± 2.4 | 0.9 ± 0.7 | 0.8 ± 0.3 | 0.7 ± 0.2 |
| Rearing Time (s) | 31.13 ± 4.74 | 20.31 ± 2.58 * | 3.66 ± 1.56 † | 2.14 ± 0.92 †* | 2.61 ± 1.53 | 0.63 ± 0.60 | 0.29 ± 0.15 | 0.19 ± 0.09 |
| Latency to first enter dark (s) | 61 ± 32 | 28 ± 19 | 14 ± 4 | 22 ± 9 | 77 ± 45 | 89 ± 36 | 63 ± 38 | 37 ± 33 |
| Light zone time (s) | 106 ± 32 | 17 ± 5 * | 75 ± 17 | 43 ± 9 * | 102 ± 40 | 121 ± 27 | 112 ± 32 | 30 ± 5 |
| Number of transitions | 9 ± 3 | 5 ± 1 * | 13 ± 2 | 8 ± 1 * | 9 ± 3 | 7 ± 1 | 13 ± 3 | 11 ± 3 |
| Behavioral Measure | Non-Stressed | Stressed | ||||||
|---|---|---|---|---|---|---|---|---|
| Saline (0.9% w/v) | Oxycodone (3 mg/kg) | Saline (0.9% w/v) | Oxycodone (3 mg/kg) | |||||
| Tat(−) (n = 8) | Tat(+) (n = 10) | Tat(−) (n = 8) | Tat(+) (n = 10) | Tat(−) (n = 8) | Tat(+) (n = 8) | Tat(−) (n = 8) | Tat(+) (n = 9) | |
| Mean Velocity (m/s) | 0.022 ± 0.003 | 0.018 ± 0.003 | 0.077 ± 0.013 † | 0.089 ± 0.012 † | 0.007 ± 0.003 | 0.004 ± 0.001 | 0.01 ± 0.004 | 0.041 ± 0.011 § |
| Rearing number | 34.8 ± 6.6 | 25.3 ± 6.2 | 13.8 ± 4.0 † | 19.7 ± 4.1 † | 1.1 ± 0.6 | 1.6 ± 0.5 | 1.4 ± 0.9 | 2.0 ± 1.2 |
| Rearing Time (s) | 23.3 ± 5.6 | 19.5 ± 5.5 | 5.2 ± 1.8 † | 9.2 ± 2.3 † | 0.4 ± 0.3 | 0.6 ± 0.2 | 0.6 ± 0.5 | 1.0 ± 0.6 |
| Latency to first enter dark (s) | 30 ± 12 | 81 ± 32 | 38 ± 17 | 8 ± 2 ‡ | 44 ± 37 | 38 ± 19 | 17 ± 9 | 14 ± 5 |
| Light zone time (s) | 116 ± 32 | 91 ± 25 * | 119 ± 25 | 46 ± 12 * | 102± 38 | 68 ± 19 | 53 ± 12 † | 33± 7 † |
| Number of transitions | 10 ± 2 | 9 ± 2 | 17 ± 3 † | 11 ± 1 † | 10 ± 2 | 11± 2 | 15 ± 3 | 11 ± 2 |
| Behavioral Measure | Vehicle | Antalarmin | ||||||
| Saline (0.9% w/v) | Oxycodone (3 mg/kg) | Saline (0.9% w/v) | Oxycodone (3 mg/kg) | |||||
| Tat(−) (n = 8) | Tat(+) (n = 7–8) | Tat(−) (n = 8–9) | Tat(+) (n = 9) | Tat(−) (n = 8) | Tat(+) (n = 8–9) | Tat(−) (n = 8–9) | Tat(+) (n = 10) | |
| Mean Velocity (m/s) | 0.031 ± 0.006 | 0.031 ± 0.005 | 0.053 ± 0.008 † | 0.11 ± 0.01 †* | 0.018 ± 0.003 | 0.016 ± 0.002 | 0.051 ± 0.007 † | 0.077 ± 0.005 †*# |
| Rearing number | 34 ± 5 | 32 ± 8 | 8 ± 4 | 38 ± 12 | 16 ± 3 | 21 ± 6 | 10 ± 4 | 12 ± 5 |
| Rearing Time (s) | 27.3 ± 4.5 | 27.7 ± 7.5 | 1.9 ± 0.7 † | 8.6 ± 2.0 † | 11 ± 2.4 # | 13.6 ± 4.5 # | 6.6 ± 3.3 † | 2.7 ± 0.9 † |
| Latency to first entry to dark zone (s) | 40.7 ± 14.2 | 5.3 ± 1.8 | 10.7 ± 4.9 | 7.3 ± 1.7 | 86.9 ± 43.3 | 125.5 ± 36.4 | 7.0 ± 1.3 † | 6.8 ± 1.9 † |
| Light zone time (s) | 177 ± 31 | 38 ± 11 * | 32. ± 7 † | 32 ± 5 | 104 ± 40 | 148 ± 34 # | 98 ± 31 | 74 ± 26 † |
| Number of transitions | 14.9 ± 3.6 | 11.4 ± 2.9 | 8.4 ± 1.2 | 9.1 ± 1.3 | 6.0 ± 2.5 | 6.1 ± 1.3 | 14.2 ± 3.5 | 11.8 ± 2.3 |
| RU-486 | Antalarmin + RU-486 | |||||||
| Saline (0.9% w/v) | Oxycodone (3 mg/kg) | Saline (0.9% w/v) | Oxycodone (3 mg/kg) | |||||
| Tat(−) (n = 8–9) | Tat(+) (n = 7–8) | Tat(−) (n = 8–9) | Tat(+) (n = 7–8) | Tat(−) (n = 9) | Tat(+) (n = 8) | Tat(−) (n = 9–10) | Tat(+) (n = 9–10) | |
| Mean Velocity (m/s) | 0.01 ± 0.001 # | 0.021 ± 0.003 | 0.035 ± 0.006 † | 0.065 ± 0.011 †*# | 0.020 ± 0.003 | 0.018 ± 0.004 | 0.082 ± 0.012 †# | 0.075 ± 0.007 †# |
| Rearing number | 9 ± 2 | 17 ± 4 | 25 ± 20 | 15 ± 7 | 12 ± 3 | 16 ± 5 | 57 ± 29 | 11 ± 4 |
| Rearing Time (s) | 7.8 ± 2.1 # | 13.8 ± 3.5 # | 2.9 ± 1.9 † | 4.0 ± 1.2 † | 8.6 ± 1.9 # | 11.0 ± 4.0 # | 9.1 ± 2.9 † | 1.8 ± 0.4 † |
| Latency to first entry to dark zone (s) | 78.0 ± 35.5 | 97.3 ± 52.4 | 16.1 ± 4.5 | 3.2 ± 0.7 | 110.0 ± 47.9 | 23.9 ± 6.4 | 14.8 ± 4.0 † | 3.8 ± 0.9 † |
| Light zone time (s) | 120 ± 43 | 107 ± 43 | 116 ± 378 # | 8 ± 1 †* | 169 ± 40 | 34 ± 5 * | 200 ± 17 # | 61 ± 13 * |
| Number of transitions | 3.4 ± 0.7 # | 3.1 ± 0.9 # | 9.0 ± 2.1 | 5.0 ± 0.9 | 7.4 ± 2.4 | 4.0 ± 0.5 | 27.4 ± 6.0 †# | 33.2 ± 15.5 †# |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salahuddin, M.F.; Mahdi, F.; Paris, J.J. HIV-1 Tat Dysregulates the Hypothalamic-Pituitary-Adrenal Stress Axis and Potentiates Oxycodone-Mediated Psychomotor and Anxiety-Like Behavior of Male Mice. Int. J. Mol. Sci. 2020, 21, 8212. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218212
Salahuddin MF, Mahdi F, Paris JJ. HIV-1 Tat Dysregulates the Hypothalamic-Pituitary-Adrenal Stress Axis and Potentiates Oxycodone-Mediated Psychomotor and Anxiety-Like Behavior of Male Mice. International Journal of Molecular Sciences. 2020; 21(21):8212. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218212
Chicago/Turabian StyleSalahuddin, Mohammed F., Fakhri Mahdi, and Jason J. Paris. 2020. "HIV-1 Tat Dysregulates the Hypothalamic-Pituitary-Adrenal Stress Axis and Potentiates Oxycodone-Mediated Psychomotor and Anxiety-Like Behavior of Male Mice" International Journal of Molecular Sciences 21, no. 21: 8212. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218212