LINC00973 Induces Proliferation Arrest of Drug-Treated Cancer Cells by Preventing p21 Degradation

 ,
,  , , , ,
, , , ,
Abstract
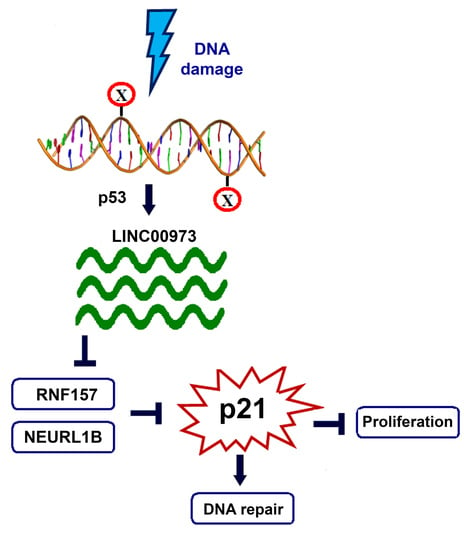
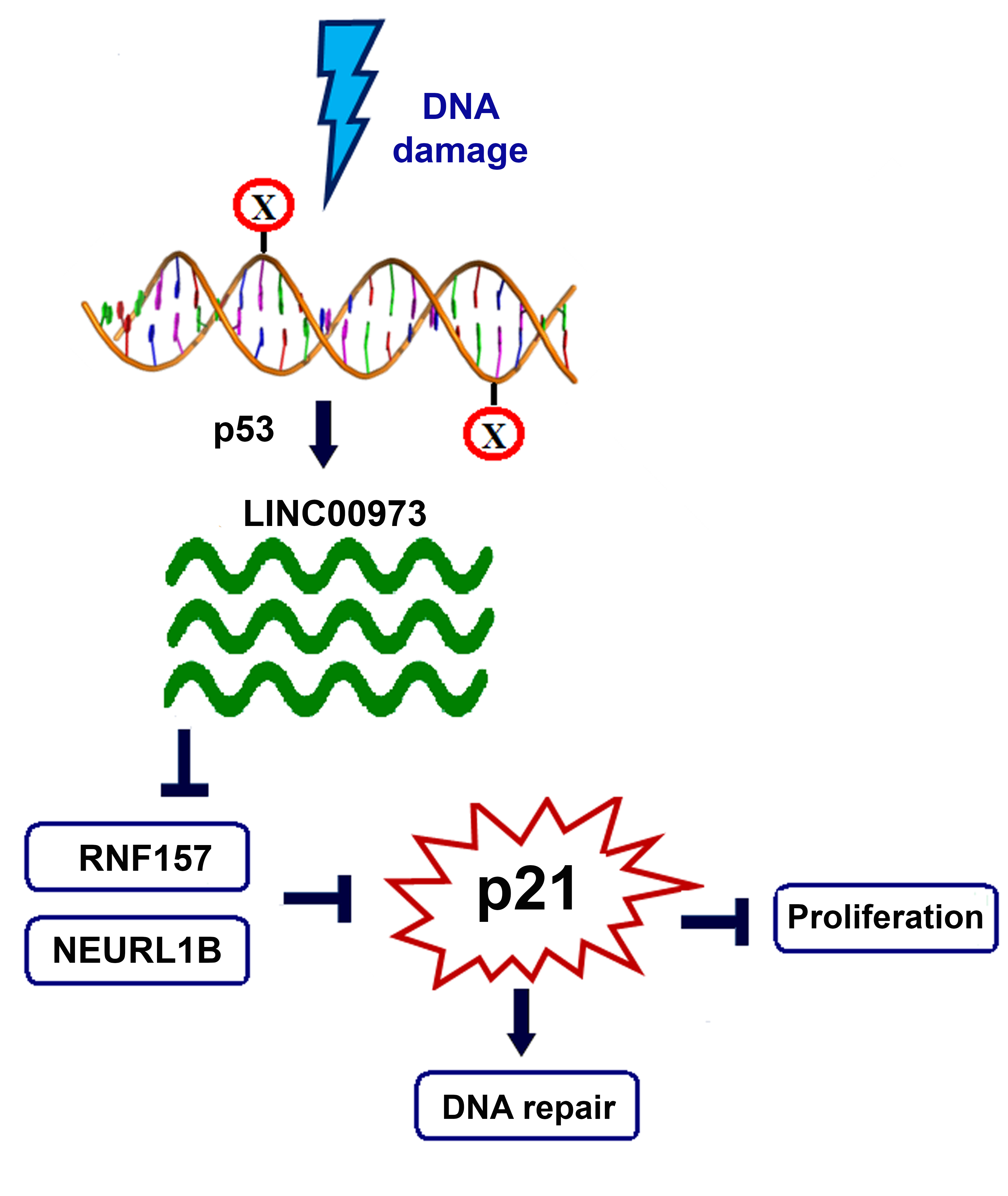
:
1. Introduction
2. Results
2.1. Bioinformatics Analysis of LINC00973 Expression in Cancer Cells Treated with Chemotherapeutics
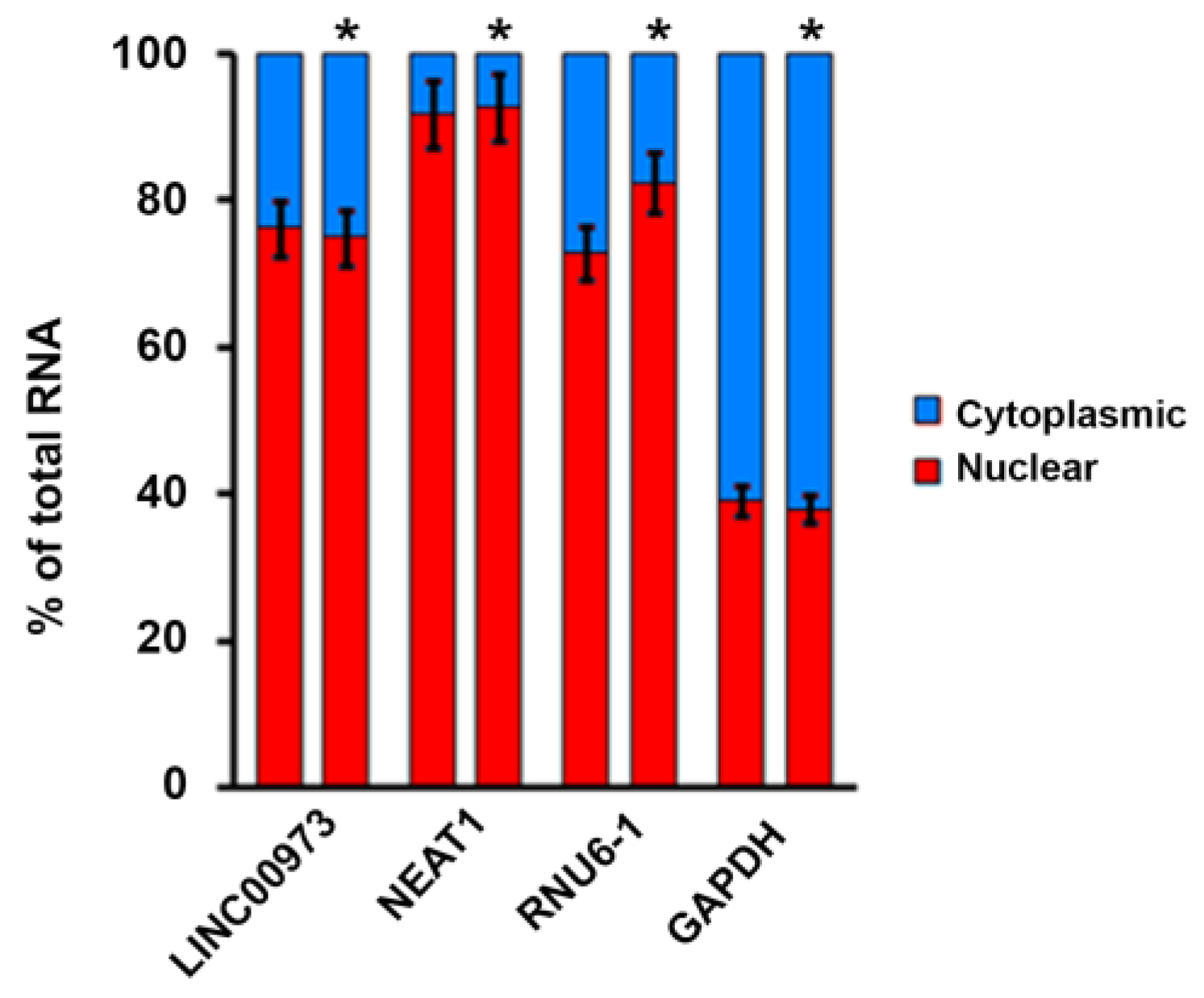
2.2. LINC00973 Subcellular Localization
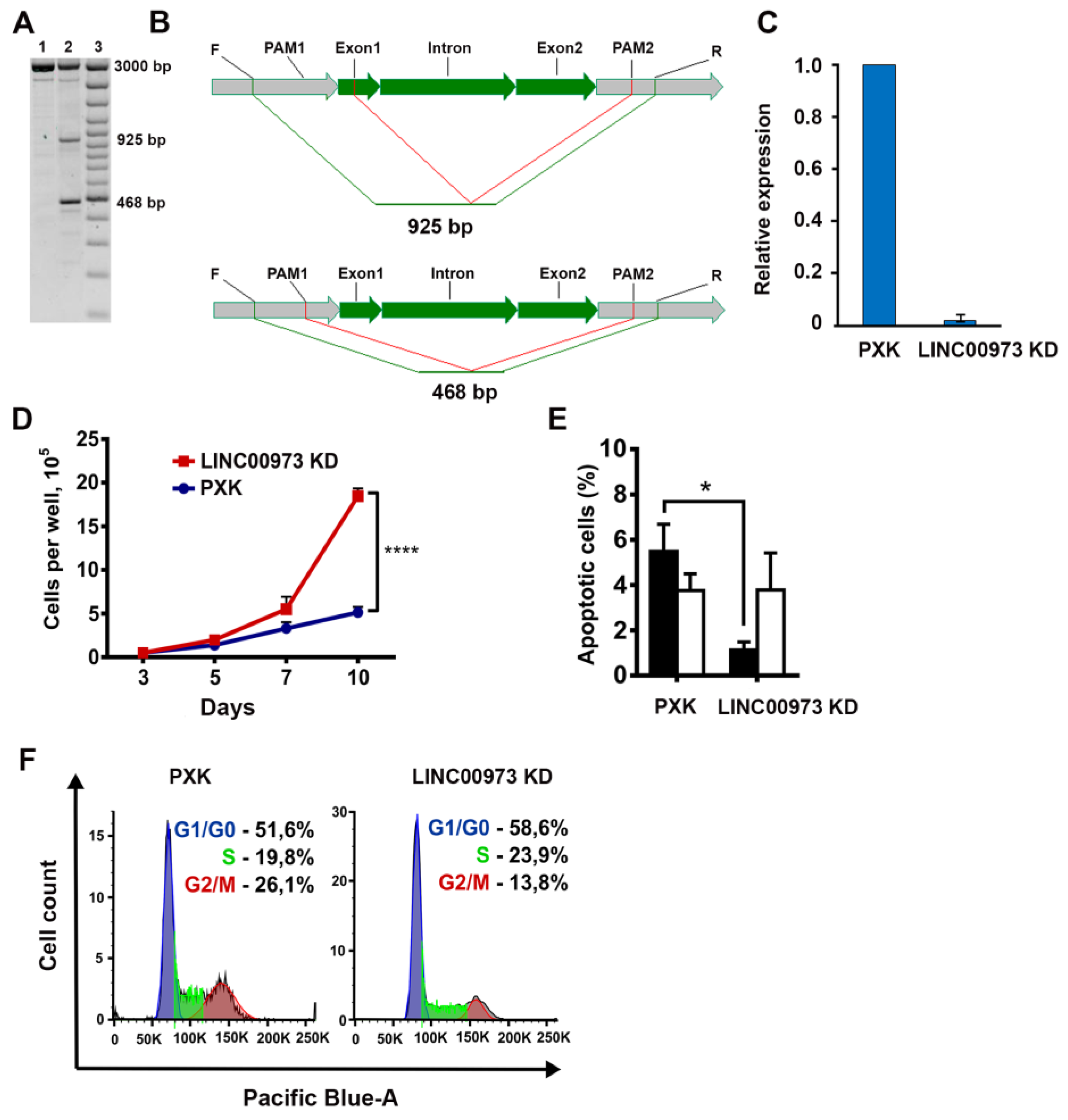
2.3. LINC00973 Knockdown Results in Dramatic Increase in HT-29 Cell Proliferation and 5-Fluorouracil Sensitivity, As Well As in Inhibition of Apoptosis
2.4. Analysis of Transcriptomic and Proteomic Changes in LINC00973 KD and LINC00973 Ectopically Over-Expressing Cells
3. Discussion
4. Materials and Methods
4.1. Analysis RNA-Seq Data and Data from the NCBI Sequence Read Archive
4.2. Cell Lines and Drug Treatment
4.3. RT-qPCR
4.4. LINC00973 Subcellular Localization
4.5. LINC00973 Knockdown
4.6. Viral Particle Production and Lentivirus Transduction
4.7. Analysis of Cellular Phenotypes
4.8. Determination of IC50 upon 5-FU Treatment
4.9. RNA Sequencing
4.10. Identification and Ranking of Differentially Expressed Transcripts
4.11. Western Blotting
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| LincRNA | Long intergenic non-coding RNA |
| DDR | DNA damage response |
| 5-FU | 5-Fluorouracil |
| OXP | Oxaliplatin |
| CIS | Cisplatin |
| CAR | Carboplatin |
| CSC | Cancer stem cell |
| PAC | Paclitaxel |
| ETO | Etoposide |
| DXR | Doxorubicin |
| RT-qPCR | Reverse transcription quantitative PCR |
| CPM | Counts per million |
| FC | Fold change |
| FDR | False discovery rate |
| CRC | Colorectal cancer |
| COAD | Colon adenocarcinoma |
| LUAD | Lung adenocarcinoma |
| NB | Neuroblastoma |
| OS | Osteosarcoma |
| ILF | Immortalized fetal lung fibroblasts |
| GEP-NET | Gastro-entero-pancreatic neuroendocrine tumor |
| LINC00973 KD | CRISPR-induced LINC00973 knockdown in PXK/HT-29 cells |
| PXK | HT-29 subclone derived from a single HT-29 cell, transfected with an empty plasmid PX459 |
References
- Hu, Y.; Zhu, Q.N.; Deng, J.L.; Li, Z.X.; Wang, G.; Zhu, Y.S. Emerging role of long non-coding RNAs in cisplatin resistance. Onco Targets Ther. 2018, 11, 3185–3194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, D.; Turchi, J.J. Chemotherapy induced DNA damage response: Convergence of drugs and pathways. Cancer Biol. Ther. 2013, 14, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Ryl, T.; Kuchen, E.E.; Bell, E.; Shao, C.; Florez, A.F.; Monke, G.; Gogolin, S.; Friedrich, M.; Lamprecht, F.; Westermann, F.; et al. Cell-Cycle Position of Single MYC-Driven Cancer Cells Dictates Their Susceptibility to a Chemotherapeutic Drug. Cell Syst. 2017, 5, 237–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Shan, B.; He, D.; Cheng, Y.; Li, B.; Zhang, C.; Duan, C. Recent Progress in Characterizing Long Noncoding RNAs in Cancer Drug Resistance. J. Cancer 2019, 10, 6693–6702. [Google Scholar] [CrossRef]
- Hsu, C.H.; Altschuler, S.J.; Wu, L.F. Patterns of Early p21 Dynamics Determine Proliferation-Senescence Cell Fate after Chemotherapy. Cell 2019, 178, 361–373.e12. [Google Scholar] [CrossRef]
- Bermudez, M.; Aguilar-Medina, M.; Lizarraga-Verdugo, E.; Avendano-Felix, M.; Silva-Benitez, E.; Lopez-Camarillo, C.; Ramos-Payan, R. LncRNAs as Regulators of Autophagy and Drug Resistance in Colorectal Cancer. Front. Oncol. 2019, 9, 1008. [Google Scholar] [CrossRef]
- Zinovieva, O.L.; Grineva, E.N.; Prokofjeva, M.M.; Karpov, D.S.; Zheltukhin, A.O.; Krasnov, G.S.; Snezhkina, A.V.; Kudryavtseva, A.V.; Chumakov, P.M.; Mashkova, T.D.; et al. Expression of long non-coding RNA LINC00973 is consistently increased upon treatment of colon cancer cells with different chemotherapeutic drugs. Biochimie 2018, 151, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Mitotic arrest and cell fate: Why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle 2007, 6, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Subramanian, M.; Jones, M.F.; Chaudhary, R.; Singh, D.K.; Zong, X.; Gryder, B.; Sindri, S.; Mo, M.; Schetter, A.; et al. Long Noncoding RNA PURPL Suppresses Basal p53 Levels and Promotes Tumorigenicity in Colorectal Cancer. Cell Rep. 2017, 20, 2408–2423. [Google Scholar] [CrossRef] [Green Version]
- Boeva, V.; Louis-Brennetot, C.; Peltier, A.; Durand, S.; Pierre-Eugene, C.; Raynal, V.; Etchevers, H.C.; Thomas, S.; Lermine, A.; Daudigeos-Dubus, E.; et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nat. Genet. 2017, 49, 1408–1413. [Google Scholar] [CrossRef]
- Zanotto-Filho, A.; Masamsetti, V.P.; Loranc, E.; Tonapi, S.S.; Gorthi, A.; Bernard, X.; Goncalves, R.M.; Moreira, J.C.; Chen, Y.; Bishop, A.J. Alkylating Agent-Induced NRF2 Blocks Endoplasmic Reticulum Stress-Mediated Apoptosis via Control of Glutathione Pools and Protein Thiol Homeostasis. Mol. Cancer Ther. 2016, 15, 3000–3014. [Google Scholar] [CrossRef] [Green Version]
- Guler, G.D.; Tindell, C.A.; Pitti, R.; Wilson, C.; Nichols, K.; KaiWai Cheung, T.; Kim, H.J.; Wongchenko, M.; Yan, Y.; Haley, B.; et al. Repression of Stress-Induced LINE-1 Expression Protects Cancer Cell Subpopulations from Lethal Drug Exposure. Cancer Cell 2017, 32, 221–237.e13. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Chen, D.; Zhang, W.; Zhao, J.; Zhi, L.; Huang, F.; Ji, H.; Zhang, J.; Liu, H.; Zou, L.; et al. Modulation of alternative splicing induced by paclitaxel in human lung cancer. Cell Death Dis. 2018, 9, 491. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef]
- Kim, S.K.; Kim, S.Y.; Kim, J.H.; Roh, S.A.; Cho, D.H.; Kim, Y.S.; Kim, J.C. A nineteen gene-based risk score classifier predicts prognosis of colorectal cancer patients. Mol. Oncol. 2014, 8, 1653–1666. [Google Scholar] [CrossRef]
- Jadaliha, M.; Gholamalamdari, O.; Tang, W.; Zhang, Y.; Petracovici, A.; Hao, Q.; Tariq, A.; Kim, T.G.; Holton, S.E.; Singh, D.K.; et al. A natural antisense lncRNA controls breast cancer progression by promoting tumor suppressor gene mRNA stability. PLoS Genet. 2018, 14, e1007802. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.J.; Subramaniam, P.S.; Tang, L.H.; Grunn, A.; Aburi, M.; Rieckhof, G.; Komissarova, E.V.; Hagan, E.A.; Bodei, L.; Clemons, P.A.; et al. A precision oncology approach to the pharmacological targeting of mechanistic dependencies in neuroendocrine tumors. Nat. Genet. 2018, 50, 979–989. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.Q.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Kopp, F.; Chang, T.C.; Sataluri, A.; Chen, B.; Sivakumar, S.; Yu, H.; Xie, Y.; Mendell, J.T. Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins. Cell 2016, 164, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Zinovieva, O.L.; Grineva, E.N.; Krasnov, G.S.; Karpov, D.S.; Zheltukhin, A.O.; Snezhkina, A.V.; Kudryavtseva, A.V.; Mashkova, T.D.; Lisitsyn, N.A. Treatment of cancer cells with chemotherapeutic drugs results in profound changes in expression of genes encoding aldehyde-metabolizing enzymes. J. Cancer 2019, 10, 4256–4263. [Google Scholar] [CrossRef] [PubMed]
- Contadini, C.; Monteonofrio, L.; Virdia, I.; Prodosmo, A.; Valente, D.; Chessa, L.; Musio, A.; Fava, L.L.; Rinaldo, C.; Di Rocco, G.; et al. p53 mitotic centrosome localization preserves centrosome integrity and works as sensor for the mitotic surveillance pathway. Cell Death Dis. 2019, 10, 850. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, X.; Zhang, C.; Zhang, H.; Huang, Y. LINC00973 is involved in cancer immune suppression through positive regulation of Siglec-15 in clear-cell renal cell carcinoma. Cancer Sci. 2020, 111, 3693. [Google Scholar] [CrossRef]
- Dogan, T.; Gnad, F.; Chan, J.; Phu, L.; Young, A.; Chen, M.J.; Doll, S.; Stokes, M.P.; Belvin, M.; Friedman, L.S.; et al. Role of the E3 ubiquitin ligase RNF157 as a novel downstream effector linking PI3K and MAPK signaling pathways to the cell cycle. J. Biol. Chem. 2017, 292, 14311–14324. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Liu, X.; Bai, J.; Pei, D.; Zheng, J. The emerging role of RUNX3 in cancer metastasis (Review). Oncol. Rep. 2016, 35, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Jing, C.; Ma, R.; Cao, H.; Wang, Z.; Liu, S.; Chen, D.; Wu, Y.; Zhang, J.; Wu, J. Long noncoding RNA and mRNA profiling in cetuximab-resistant colorectal cancer cells by RNA sequencing analysis. Cancer Med. 2019, 8, 1641–1651. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.G.; Nie, J.F.; Sicotte, H.; Li, Y.; Eckel-Passow, J.E.; Dasari, S.; Vedell, P.T.; Barman, P.; Wang, L.W.; Weinshiboum, R.; et al. Measure transcript integrity using RNA-seq data. BMC Bioinform. 2016, 17, 58. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Weber, K.; Bartsch, U.; Stocking, C.; Fehse, B. A multicolor panel of novel lentiviral “gene ontology” (LeGO) vectors for functional gene analysis. Mol. Ther. 2008, 16, 698–706. [Google Scholar] [CrossRef]
- Spirin, P.; Lebedev, T.; Orlova, N.; Morozov, A.; Poymenova, N.; Dmitriev, S.E.; Buzdin, A.; Stocking, C.; Kovalchuk, O.; Prassolov, V. Synergistic suppression of t(8;21)-positive leukemia cell growth by combining oridonin and MAPK1/ERK2 inhibitors. Oncotarget 2017, 8, 56991–57002. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Krasnov, G.S.; Dmitriev, A.A.; Kudryavtseva, A.V.; Shargunov, A.V.; Karpov, D.S.; Uroshlev, L.A.; Melnikova, N.V.; Blinov, V.M.; Poverennaya, E.V.; Archakov, A.I.; et al. PPLine: An Automated Pipeline for SNP, SAP, and Splice Variant Detection in the Context of Proteogenomics. J. Proteome Res. 2015, 14, 3729–3737. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
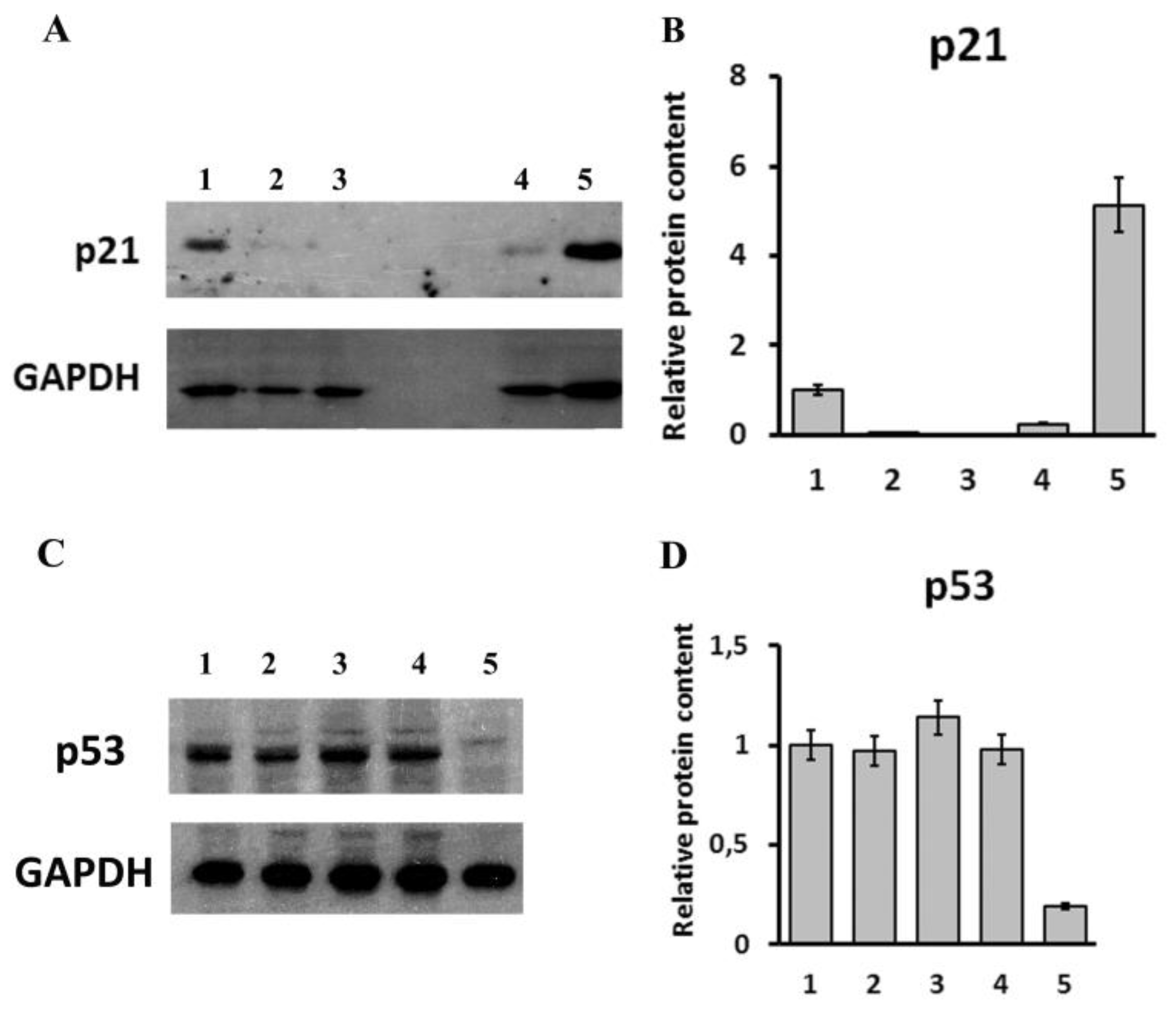{kind=link}
{kind=link}
| Cell Line | Cell Type 1 | Drug | Log2FC | P53 | Reference |
|---|---|---|---|---|---|
| Status | |||||
| HT-29 | COAD | 5-Fluorouracil | 7.2 | R273H | [7] |
| HT-29 | COAD | Oxaliplatin | 2.7 | R273H | [7] |
| HCT-116 | COAD | Oxaliplatin | 2.8 | wt | [7] |
| HCT-116 | COAD | Doxorubicin | 1.7 | wt | [9] |
| SK-N-SH | NB | Doxorubicin | 5.3 | wt | [10] |
| SK-N-SH | NB | Cisplatin | 5.2 | wt | [10] |
| IMR5/75 | NB | Doxorubicin | 4.4 | wt | [3] |
| U2OS | OS | Methyl methanesulfonate | −2.7 | wt | [11] |
| U2OS | OS | Etoposide | −1.5 | wt | [11] |
| pC9 | LUAD | Carboplatin | 2.5 | R248Q | [12] |
| A549 | LUAD | Paclitaxel | 1.1 | wt | [13] |
| IMR90 | ILF | Etoposide | 5.4 | wt | [14] |
| Cell Line | HCT116 | SW48 | ||
|---|---|---|---|---|
| p53 Status | WT | KO | WT | KO |
| LINC00973 | 1.73 | −0.17 | 0.68 | −0.1 |
| CDKN1A | 3.11 | 0.19 | 3.33 | −0.47 |
| GDF15 | 2.5 | −0.95 | 4.02 | −0.33 |
| BTG2 | 2.41 | 0.34 | 2.26 | −0.02 |
| BBC3 | 1.32 | −0.71 | 1.83 | −0.61 |
| PMAIP1 | 1.51 | 0.61 | 0.68 | −0.08 |
| GADD45 | 1.65 | −0.16 | 1.67 | −0.05 |
| Cell Line | HT-29 | SK-N-SH | IMR90 | pC9 | |||
|---|---|---|---|---|---|---|---|
| Cell Type | COAD | NB | ILF | LUAD | |||
| Drug | 5-FU | CIS | DXR | ETO | CAR | ||
| LINC00973 | LV2 | ||||||
| KD vs. PXK | vs. LV1 | ||||||
| LINC00973 | −5.64 | 4.51 | 7.15 | 5.24 | 5.3 | 5.39 | 2.49 |
| RNF157 | 2.39 | −2.11 | −2.86 | −2.86 | −2.38 | −1.19 | −0.43 |
| NEURL1B | 2.06 | 0.1 | −4.05 | −2.59 | −2.52 | −3.53 | 2.39 |
| CDKN2B | −1.51 | 0.23 | 2.19 | 3.26 | 3.43 | 2.13 | 2.5 |
| DCBLD2 | −2.01 | 0.1 | 1.9 | 2.29 | 2.74 | 0.18 | −0.49 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karpov, D.S.; Spirin, P.V.; Zheltukhin, A.O.; Tutyaeva, V.V.; Zinovieva, O.L.; Grineva, E.N.; Matrosova, V.A.; Krasnov, G.S.; Snezhkina, A.V.; Kudryavtseva, A.V.; et al. LINC00973 Induces Proliferation Arrest of Drug-Treated Cancer Cells by Preventing p21 Degradation. Int. J. Mol. Sci. 2020, 21, 8322. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218322
Karpov DS, Spirin PV, Zheltukhin AO, Tutyaeva VV, Zinovieva OL, Grineva EN, Matrosova VA, Krasnov GS, Snezhkina AV, Kudryavtseva AV, et al. LINC00973 Induces Proliferation Arrest of Drug-Treated Cancer Cells by Preventing p21 Degradation. International Journal of Molecular Sciences. 2020; 21(21):8322. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218322
Chicago/Turabian StyleKarpov, Dmitry S., Pavel V. Spirin, Andrey O. Zheltukhin, Vera V. Tutyaeva, Olga L. Zinovieva, Evgenia N. Grineva, Vera A. Matrosova, George S. Krasnov, Anastasiya V. Snezhkina, Anna V. Kudryavtseva, and et al. 2020. "LINC00973 Induces Proliferation Arrest of Drug-Treated Cancer Cells by Preventing p21 Degradation" International Journal of Molecular Sciences 21, no. 21: 8322. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21218322