Stearoyl-CoA Desaturase-2 in Murine Development, Metabolism, and Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
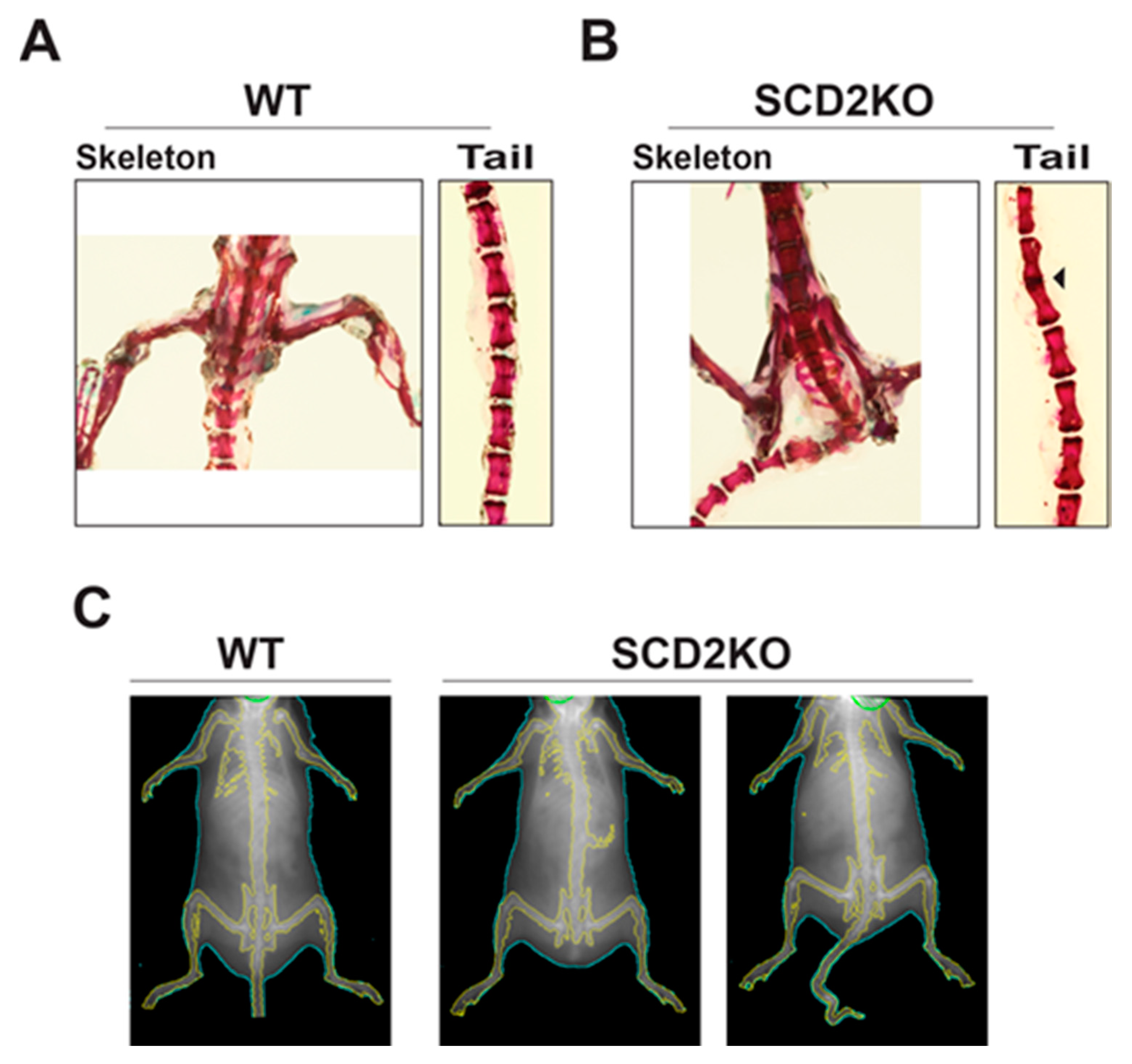
2.1. The Role of SCD2 in Developing Mice
2.2. SCD2 and Bone Density
2.3. The Relationship between SCD2, Adiposity, and Energy Expenditure
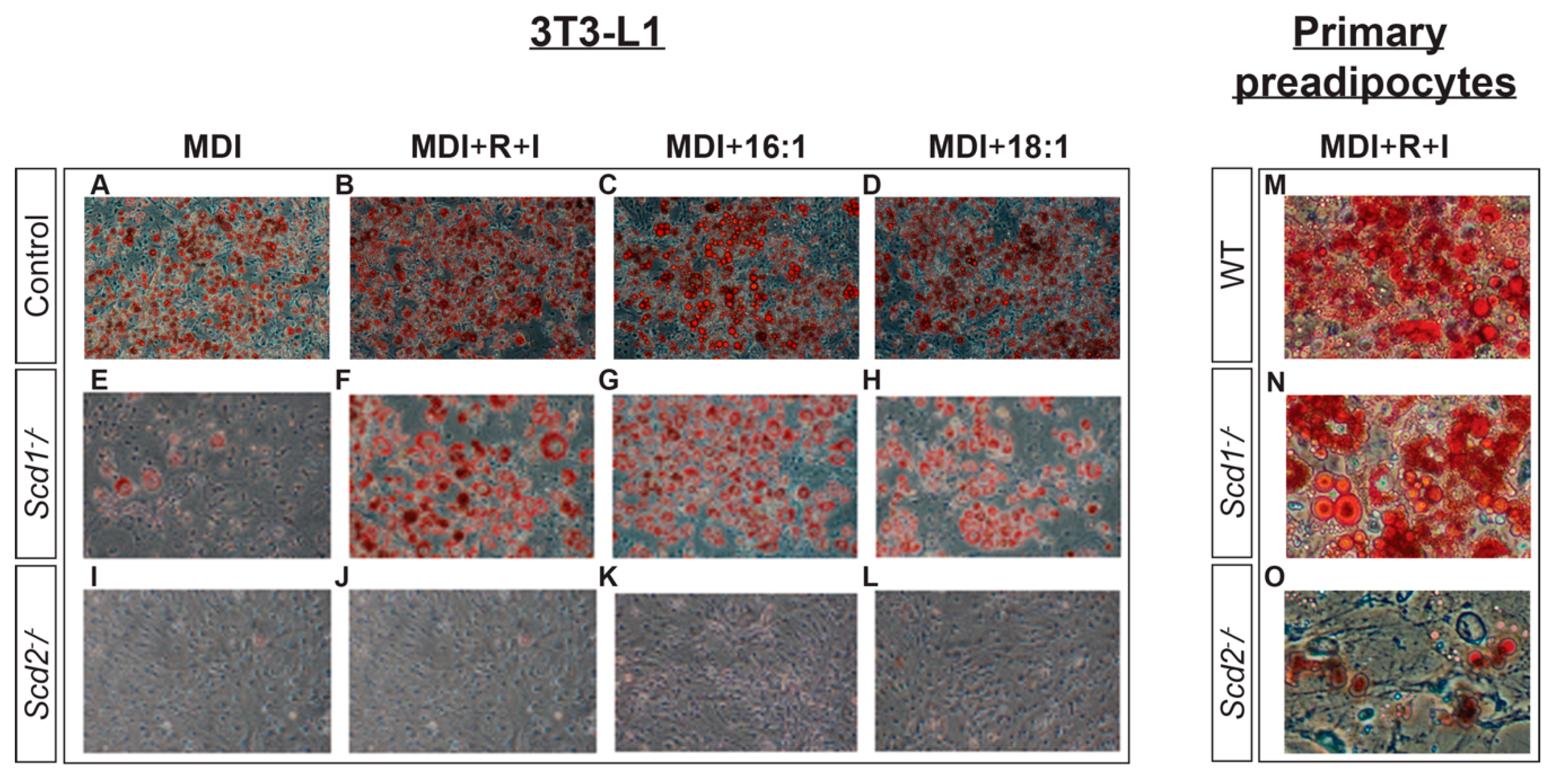
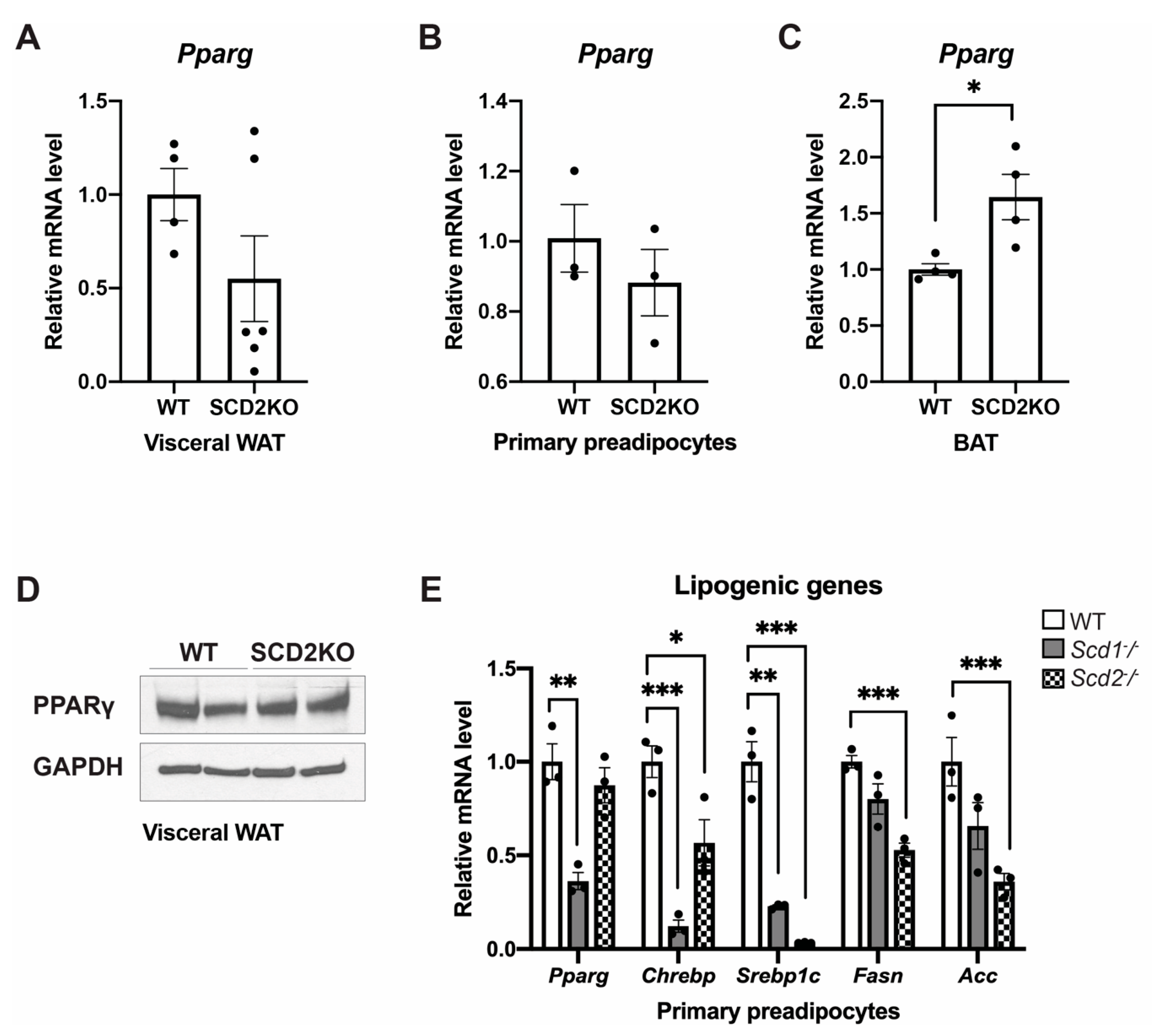
2.4. The Role of SCD2 in Preadipocyte Differentiation In Vitro and In Vivo
2.5. SCD2 and Vascular Calcification
2.6. SCD2 and Central Nervous System Diseases
2.7. Human Relevance
3. Materials and Methods
3.1. Animals and Diets
3.2. Skeletal Staining
3.3. Dual-Energy X-ray Absorptiometry (DEXA)
3.4. Generation of SCD1 and SCD2 Knockout 3T3-L1 Cells
3.5. Primary Preadipocyte Isolation
3.6. Cell Culture and Treatments
3.7. Oil Red O Staining
3.8. Quantitative Real-Time PCR
3.9. Western Blot Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | Acylceramide |
| ACC | Acetyl-CoA carboxylase |
| AD | Alzheimer′s disease |
| ATF4 | Activating transcription factor 4 |
| BAT | Brown adipose tissue |
| CE | Cholesterol ester |
| CEBP | CCAAT/Enhancer binding protein |
| ChREBP | Carbohydrate response element-binding protein |
| CKD | Chronic kidney disease |
| CNS | Central nervous system |
| DGAT2 | Acyl-CoA:diacylglycerol acyltransferase-2 |
| ER | Endoplasmic reticulum |
| FA | Fatty acid |
| FAS | Fatty acid synthase |
| FFA | Free fatty acid |
| GBA | β-cerebrosidase |
| hSCD1 | Human stearoyl-CoA desaturase-1 |
| hSCD5 | Human stearoyl-CoA desatuarse-5 |
| MS | Multiple sclerosis |
| mTOR | Mammalian target of rapamycin |
| MUFA | Monounsaturated fatty acid |
| PA | Phosphatidic acids |
| PD | Parkinson′s Disease |
| PGC1α | Peroxisome proliferator-activated receptor gamma, coactivator 1 alpha |
| PL | Phospholipid |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| SCD | Stearoyl-CoA desaturase |
| SCD1 | Stearoyl-CoA desaturase-1 |
| SCD2 | Stearoyl-CoA Desaturase-2 |
| SCD3 | Stearoyl-CoA desaturase-3 |
| SCD4 | Stearoyl-CoA desaturase-4 |
| SFA | Saturated fatty acid |
| SREBP1c | Sterol regulatory element-binding protein-1c |
| TG | Triglyceride |
| UCP1 | Uncoupling protein-1 |
| VC | Vascular calcification |
| WAT | White adipose tissue |
References
- Paton, C.M.; Ntambi, J.M. Biochemical and physiological function of stearoyl-CoA desaturase. Am. J. Physiol. Metab. 2009, 297, E28–E37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ntambi, J.M.; Miyazaki, M.; Dobrzyń, A. Regulation of stearoyl-CoA desaturase expression. Lipids 2004, 39, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Bruggink, S.M.; Ntambi, J.M. Identification of mouse palmitoyl-coenzyme A Δ9-desaturase. J. Lipid Res. 2006, 47, 700–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, W.C.; Miyazaki, M.; Chu, K.; Ntambi, J.M. Membrane Topology of Mouse Stearoyl-CoA Desaturase 1. J. Biol. Chem. 2005, 281, 1251–1260. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; McCoy, J.G.; Levin, E.J.; Zhou, M. X-Ray Structure of a Mammalian Stearoyl-Coa Desaturase-1. Biophys. J. 2015, 108, 534a. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Klein, M.G.; Zou, H.; Lane, W.; Snell, G.; Levin, I.; Li, K.; Sang, B.-C. Crystal structure of human stearoyl–coenzyme A desaturase in complex with substrate. Nat. Struct. Mol. Biol. 2015, 22, 581–585. [Google Scholar] [CrossRef]
- Miyazaki, M.; Enrique, G.F.; Ntambi, J.M. Lack of stearoyl-CoA desaturase-1 function induces a palmitoyl-CoA Δ6 desaturase and represses the stearoyl-CoA desaturase-3 gene in the preputial glands of the mouse. J. Lipid Res. 2002, 43, 2146–2154. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Gerhold, K.; Mayers, J.R.; Wiest, M.M.; Watkins, S.M.; Hotamisligil, G.S. Identification of a Lipokine, a Lipid Hormone Linking Adipose Tissue to Systemic Metabolism. Cell 2008, 134, 933–944. [Google Scholar] [CrossRef] [Green Version]
- Lounis, M.A.; Bergeron, K.-F.; Burhans, M.S.; Ntambi, J.M.; Mounier, C. Oleate activates SREBP-1 signaling activity in SCD1-deficient hepatocytes. Am. J. Physiol. Metab. 2017, 313, E710–E720. [Google Scholar] [CrossRef]
- Aljohani, A.; Khan, M.I.; Syed, D.N.; Abram, B.; Lewis, S.; Neill, L.O.; Mukhtar, H.; Ntambi, J.M. Hepatic Stearoyl-CoA desaturase-1 deficiency-mediated activation of mTORC1- PGC-1α axis regulates ER stress during high-carbohydrate feeding. Sci. Rep. 2019, 9, 15761–15769. [Google Scholar] [CrossRef]
- Obici, S.; Feng, Z.; Morgan, K.; Stein, D.; Karkanias, G.; Rossetti, L. Central Administration of Oleic Acid Inhibits Glucose Production and Food Intake. Diabetes 2001, 51, 271–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaestner, K.H.; Ntambi, J.M.; Kelly, T.J.; Lane, M.D. Differentiation-induced gene expression in 3t3-l1 preadipocytes. A second differentially expressed gene encoding stearoyl-coa desaturase. J. Biol. Chem. 1989, 264, 14755–14761. [Google Scholar] [PubMed]
- Christianson, J.L.; Nicoloro, S.; Straubhaar, J.; Czech, M.P. Stearoyl-coa desaturase 2 is required for peroxisome proliferator-activated receptor gamma expression and adipogenesis in cultured 3t3-l1 cells. J. Biol. Chem. 2008, 283, 2906–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, M.; Dobrzyn, A.; Elias, P.M.; Ntambi, J.M. Stearoyl-CoA Desaturase-2 gene expression is required for lipid synthesis during early skin and liver development. Proc. Natl. Acad. Sci. USA 2005, 102, 12501–12506. [Google Scholar] [CrossRef] [Green Version]
- Cantley, J.L.; O′Neill, L.M.; Ntambi, J.M.; Czech, M.P. The cellular function of Stearoyl-CoA Desaturase-2 in development and differentiation. In Stearoyl-coa Desaturase Genes in Lipid Metabolism; Ntambi, P.D.J.M., Ed.; Springer: New York, NY, USA, 2013; pp. 119–130. [Google Scholar]
- Moreau, C.; Froment, P.; Tosca, L.; Moreau, V.; Dupont, J. Expression and Regulation of the SCD2 Desaturase in the Rat Ovary. Biol. Reprod. 2006, 74, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Tebbey, P.W.; Buttke, T.M. Stearoyl-CoA desaturase gene expression in lymphocytes. Biochem. Biophys. Res. Commun. 1992, 186, 531–536. [Google Scholar] [CrossRef]
- Saether, T.; Tran, T.N.; Rootwelt, H.; Christophersen, B.O.; Haugen, T.B. Expression and regulation of delta5-desaturase, delta6-desaturase, stearoyl-coenzyme a (coa) desaturase 1, and stearoyl-coa desaturase 2 in rat testis. Biol. Reprod. 2003, 69, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Madison, K.C. Barrier Function of the Skin: “La Raison d’Être” of the Epidermis. J. Investig. Dermatol. 2003, 121, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Hansen, H.S.; Jensen, B. Essential function of linoleic acid esterified in acylglucosylceramide and acylceramide in maintaining the epidermal water permeability barrier. Evidence from feeding studies with oleate, linoleate, arachidonate, columbinate and α-linolenate. Biochim. Biophys. Acta 1985, 834, 357–363. [Google Scholar] [CrossRef]
- Wertz, P.W.; Downing, D.T. Metabolism of linoleic acid in porcine epidermis. J. Lipid Res. 1990, 31, 1839–1844. [Google Scholar]
- Stone, S.J.; Myers, H.M.; Watkins, S.M.; Brown, B.E.; Feingold, K.R.; Elias, P.M.; Farese, R.V., Jr. Lipopenia and Skin Barrier Abnormalities in DGAT2-deficient Mice. J. Biol. Chem. 2003, 279, 11767–11776. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, G.; Jevon, G.; Colobong, K.E.; Randall, D.R.; Choy, F.Y.; Clarke, L.A. Generation of a conditional knockout of murine glucocerebrosidase: Utility for the study of Gaucher disease. Mol. Genet. Metab. 2007, 90, 148–156. [Google Scholar] [CrossRef]
- O’Neill, L.M.; Phang, Y.X.; Matango, M.; Shamsuzzaman, S.; Guo, C.-A.; Nelson, D.W.; Yen, C.-L.E.; Ntambi, J.M. Global deficiency of Stearoyl-CoA Desaturase-2 protects against diet-induced adiposity. Biochem. Biophys. Res. Commun. 2020, 527, 589–595. [Google Scholar] [CrossRef]
- Sawin, E.A.; Stroup, B.M.; Murali, S.G.; O’Neill, L.M.; Ntambi, J.M.; Ney, D.M. Differential Effects of Dietary Fat Content and Protein Source on Bone Phenotype and Fatty Acid Oxidation in Female C57Bl/6 Mice. PLoS ONE 2016, 11, e0163234. [Google Scholar] [CrossRef] [Green Version]
- Reid, I.R. Fat and bone. Arch. Biochem. Biophys. 2010, 503, 20–27. [Google Scholar] [CrossRef]
- De Moura, R.F.; Nascimento, L.F.; Ignacio-Souza, L.M.; Morari, J.; Razolli, D.S.; Solon, C.; De Souza, G.F.P.; Festuccia, W.T.; A Velloso, L. Hypothalamic Stearoyl-CoA Desaturase-2 (SCD2) controls whole-body energy expenditure. Int. J. Obes. 2015, 40, 471–478. [Google Scholar] [CrossRef]
- Miyazaki, M.; Sampath, H.; Liu, X.; Flowers, M.T.; Chu, K.; Dobrzyn, A.; Ntambi, J.M. Stearoyl-coa desaturase-1 deficiency attenuates obesity and insulin resistance in leptin-resistant obese mice. Biochem. Biophys. Res. Commun. 2009, 380, 818–822. [Google Scholar] [CrossRef] [Green Version]
- Ntambi, J.M.; Miyazaki, M.; Stoehr, J.P.; Lan, H.; Kendziorski, C.M.; Yandell, B.S.; Song, Y.; Cohen, P.; Friedman, J.M.; Attie, A.D. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl. Acad. Sci. USA 2002, 99, 11482–11486. [Google Scholar] [CrossRef] [Green Version]
- Sampath, H.; Flowers, M.T.; Liu, X.; Paton, C.M.; Sullivan, R.; Chu, K.; Zhao, M.; Ntambi, J.M. Skin-specific deletion of stearoyl-coa desaturase-1 alters skin lipid composition and protects mice from high fat diet-induced obesity. J. Biol. Chem. 2009, 284, 19961–19973. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-H.; Dobrzyn, P.; Rahman, S.M.; Miyazaki, M.; Ntambi, J.M. Lack of stearoyl-CoA desaturase 1 upregulates basal thermogenesis but causes hypothermia in a cold environment. J. Lipid Res. 2004, 45, 1674–1682. [Google Scholar] [CrossRef] [Green Version]
- Ntambi, J.M.; Buhrow, S.A.; Kaestner, K.H.; Christy, R.J.; Sibley, E.; Kelly, T.J.; Lane, M.D. Differentiation-induced gene expression in 3t3-l1 preadipocytes. Characterization of a differentially expressed gene encoding stearoyl-coa desaturase. J. Biol. Chem. 1988, 263, 17291–17300. [Google Scholar] [PubMed]
- Man, W.C.; Miyazaki, M.; Chu, K.; Ntambi, J. Colocalization of SCD1 and DGAT2: Implying preference for endogenous monounsaturated fatty acids in triglyceride synthesis. J. Lipid Res. 2006, 47, 1928–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tontonoz, P.; Spiegelman, B.M. Fat and Beyond: The Diverse Biology of PPARgamma. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, C.J.; Liu, X.S.; et al. Ppargamma and c/ebp factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Wang, D.; Zhao, W.; Xu, L. Deciphering the Roles of PPARγ in Adipocytes via Dynamic Change of Transcription Complex. Front. Endocrinol. 2018, 9, 473. [Google Scholar] [CrossRef] [Green Version]
- Gregoire, F.M.; Smas, C.M.; Sul, H.S. Understanding Adipocyte Differentiation. Physiol. Rev. 1998, 78, 783–809. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Gomez, F.E.; Fox, B.G.; Ntambi, J.M. Differential regulation of the stearoyl-coa desaturase genes by thiazolidinediones in 3t3-l1 adipocytes. J. Lipid Res. 2000, 41, 1310–1316. [Google Scholar]
- Mizobuchi, M.; Towler, D.; Slatopolsky, E. Vascular Calcification: The Killer of Patients with Chronic Kidney Disease. J. Am. Soc. Nephrol. 2009, 20, 1453–1464. [Google Scholar] [CrossRef] [Green Version]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial Calcification in Chronic Kidney Disease: Key Roles for Calcium and Phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Demer, L.L.; Tintut, Y. Inflammatory, Metabolic, and Genetic Mechanisms of Vascular Calcification. Arter. Thromb. Vasc. Biol. 2014, 34, 715–723. [Google Scholar] [CrossRef] [Green Version]
- Towler, D.A. Molecular and cellular aspects of calcific aortic valve disease. Circ. Res. 2013, 113, 198–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, M.; Miyazaki-Anzai, S.; Keenan, A.L.; Okamura, K.; Kendrick, J.; Chonchol, M.; Offermanns, S.; Ntambi, J.M.; Kuro-O, M.; Miyazaki, M. Saturated phosphatidic acids mediate saturated fatty acid-induced vascular calcification and lipotoxicity. J. Clin. Investig. 2015, 125, 4544–4558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, M.; Ting, T.C.; Levi, M.; Saunders, S.J.; Miyazaki-Anzai, S.; Miyazaki, M. Activating transcription factor 4 regulates stearate-induced vascular calcification. J. Lipid Res. 2012, 53, 1543–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uto, Y. Recent progress in the discovery and development of stearoyl CoA desaturase inhibitors. Chem. Phys. Lipids 2016, 197, 3–12. [Google Scholar] [CrossRef]
- Hayes, C.E.; Ntambi, J.M. Multiple Sclerosis: Lipids, Lymphocytes, and Vitamin D. Immunometabolism 2020, 2, 2. [Google Scholar] [CrossRef]
- Vincent, B.M.; Tardiff, D.F.; Piotrowski, J.S.; Aron, R.; Lucas, M.C.; Chung, C.Y.; Bacherman, H.; Chen, Y.; Pires, M.; Subramaniam, R.; et al. Inhibiting stearoyl-coa desaturase ameliorates α-synuclein cytotoxicity. Cell Rep. 2018, 25, 2742–2754. [Google Scholar] [CrossRef] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer′s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Astarita, G.; Jung, K.M.; Vasilevko, V.; Dipatrizio, N.V.; Martin, S.K.; Cribbs, D.H.; Head, E.; Cotman, C.W.; Piomelli, D. Elevated stearoyl-coa desaturase in brains of patients with alzheimer′s disease. PLoS ONE 2011, 6, e24777. [Google Scholar] [CrossRef] [Green Version]
- Bartel, P.L.; Roch, J.-M. Sugiyama Therapeutic methods, compounds and compositions. U.S. Patent Application No 11/523,767, 19 April 2007. [Google Scholar]
- Ferreira, H.B.; Neves, B.; Guerra, I.M.; Moreira, A.; Melo, T.; Paiva, A.; Domingues, M.R.M. An overview of lipidomic analysis in different human matrices of multiple sclerosis. Mult. Scler. Relat. Disord. 2020, 44, 102189. [Google Scholar] [CrossRef]
- Rahmanzadeh, R.; Bruck, W.; Minagar, A.; Sahraian, M.A. Multiple sclerosis pathogenesis: Missing pieces of an old puzzle. Rev. Neurosci. 2018, 30, 67–83. [Google Scholar] [CrossRef]
- Love, S. Demyelinating diseases. J. Clin. Pathol. 2006, 59, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A Transcriptome Database for Astrocytes, Neurons, and Oligodendrocytes: A New Resource for Understanding Brain Development and Function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cermenati, G.; Mitro, N.; Audano, M.; Melcangi, R.C.; Crestani, M.; De Fabiani, E.; Caruso, D. Lipids in the nervous system: From biochemistry and molecular biology to patho-physiology. Biochim. Biophys. Acta 2015, 1851, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.; Kristiansen, K.; Boggs, J.M.; Husted, C.; Zasadzinski, J.A.; Israelachvili, J.N. Interaction forces and adhesion of supported myelin lipid bilayers modulated by myelin basic protein. Proc. Natl. Acad. Sci. USA 2009, 106, 3154–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewkowicz, N.; Piatek, P.; Namiecinska, M.; Domowicz, M.; Bonikowski, R.; Szemraj, J.; Przygodzka, P.; Stasiołek, M.; Lewkowicz, P. Naturally Occurring Nervonic Acid Ester Improves Myelin Synthesis by Human Oligodendrocytes. Cells 2019, 8, 786. [Google Scholar] [CrossRef] [Green Version]
- Sargent, J.; Coupland, K.; Wilson, R. Nervonic acid and demyelinating disease. Med. Hypotheses 1994, 42, 237–242. [Google Scholar] [CrossRef]
- Bogie, J.F.J.; Grajchen, E.; Wouters, E.; Corrales, A.G.; Dierckx, T.; Vanherle, S.; Mailleux, J.; Gervois, P.; Wolfs, E.; Dehairs, J.; et al. Stearoyl-coa desaturase-1 impairs the reparative properties of macrophages and microglia in the brain. J. Exp. Med. 2020, 217, e20191660. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-H.; Kim, D.-H.; Kwon, D.-Y.; Choi, M.; Kim, S.; Jung, J.-H.; Han, K.; Park, Y.-G. Trends in the incidence and prevalence of Parkinson’s disease in Korea: A nationwide, population-based study. BMC Geriatr. 2019, 19, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Rubin, J.E.; McIntyre, C.C.; Turner, R.S.; Wichmann, T. Basal ganglia activity patterns in parkinsonism and computational modeling of their downstream effects. Eur. J. Neurosci. 2012, 36, 2213–2228. [Google Scholar] [CrossRef]
- Hidalgo-Agudo, R.D.; Lucena-Antón, D.; Luque-Moreno, C.; Heredia-Rizo, A.M.; Moral-Munoz, J.A. Additional Physical Interventions to Conventional Physical Therapy in Parkinson’s Disease: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. J. Clin. Med. 2020, 9, 1038. [Google Scholar] [CrossRef] [Green Version]
- Outeiro, T.F. Yeast Cells Provide Insight into Alpha-Synuclein Biology and Pathobiology. Science 2003, 302, 1772–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanning, S.; Haque, A.; Imberdis, T.; Baru, V.; Barrasa, M.I.; Nuber, S.; Termine, D.; Ramalingam, N.; Ho, G.P.; Noble, T.; et al. Lipidomic Analysis of α-Synuclein Neurotoxicity Identifies Stearoyl CoA Desaturase as a Target for Parkinson Treatment. Mol. Cell 2019, 73, 1001–1014.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuber, S.; Nam, A.Y.; Rajsombath, M.M.; Cirka, H.; Hronowski, X.; Wang, J.; Hodgetts, K.; Kalinichenko, L.S.; Müller, C.P.; Lambrecht, V.; et al. A stearoyl-coenzyme a desaturase inhibitor prevents multiple parkinson disease phenotypes in α-synuclein mice. Ann. Neurol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yu, L.; Schmidt, R.E.; Su, C.; Huang, X.; Gould, K.; Cao, G. Characterization of hscd5, a novel human stearoyl-coa desaturase unique to primates. Biochem. Biophys. Res. Commun. 2005, 332, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Beiraghi, S.; Zhou, M.; Talmadge, C.B.; Went-Sumegi, N.; Davis, J.R.; Huang, D.; Saal, H.; Seemayer, T.A.; Sumegi, J. Identification and characterization of a novel gene disrupted by a pericentric inversion inv(4)(p13.1q21.1) in a family with cleft lip. Gene 2003, 309, 11–21. [Google Scholar] [CrossRef]
- Ovchinnikov, D. Alcian Blue/Alizarin Red Staining of Cartilage and Bone in Mouse. Cold Spring Harb. Protoc. 2009, 2009, 5170. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Ding, F.; Yuan, X.; Li, Q.; Sun, W.; Gan, C.; He, H.; Song, C.; Wang, J. Cloning, characterization and expression of Peking duck fatty acid synthase during adipocyte differentiation. Electron. J. Biotechnol. 2014, 17, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Aljohani, A.; Khan, M.I.; Bonneville, A.; Guo, C.; Jeffery, J.; O’Neill, L.; Syed, D.N.; Lewis, S.A.; Burhans, M.; Mukhtar, H.; et al. Hepatic stearoyl CoA desaturase 1 deficiency increases glucose uptake in adipose tissue partially through the PGC-1α-FGF21 axis in mice. J. Biol. Chem. 2019, 294, 19475–19485. [Google Scholar] [CrossRef]
- Zhang, Z.; Dales, N.A.; Winther, M.D. Opportunities and Challenges in Developing Stearoyl-Coenzyme A Desaturase-1 Inhibitors as Novel Therapeutics for Human Disease. J. Med. Chem. 2014, 57, 5039–5056. [Google Scholar] [CrossRef]
- Theodoropoulos, P.C.; Gonzales, S.S.; Winterton, S.E.; Rodriguez-Navas, C.; McKnight, J.S.; Morlock, L.K.; Hanson, J.M.; Cross, B.; Owen, A.E.; Duan, Y.; et al. Discovery of tumor-specific irreversible inhibitors of stearoyl CoA desaturase. Nat. Chem. Biol. 2016, 12, 218–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Neill, L.M.; Guo, C.-A.; Ding, F.; Phang, Y.X.; Liu, Z.; Shamsuzzaman, S.; Ntambi, J.M. Stearoyl-CoA Desaturase-2 in Murine Development, Metabolism, and Disease. Int. J. Mol. Sci. 2020, 21, 8619. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228619
O’Neill LM, Guo C-A, Ding F, Phang YX, Liu Z, Shamsuzzaman S, Ntambi JM. Stearoyl-CoA Desaturase-2 in Murine Development, Metabolism, and Disease. International Journal of Molecular Sciences. 2020; 21(22):8619. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228619
Chicago/Turabian StyleO’Neill, Lucas M., Chang-An Guo, Fang Ding, Yar Xin Phang, Zhaojin Liu, Sohel Shamsuzzaman, and James M. Ntambi. 2020. "Stearoyl-CoA Desaturase-2 in Murine Development, Metabolism, and Disease" International Journal of Molecular Sciences 21, no. 22: 8619. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228619