Unraveling the Role of Epicardial Adipose Tissue in Coronary Artery Disease: Partners in Crime?

 , and
, and
Abstract
:1. Introduction
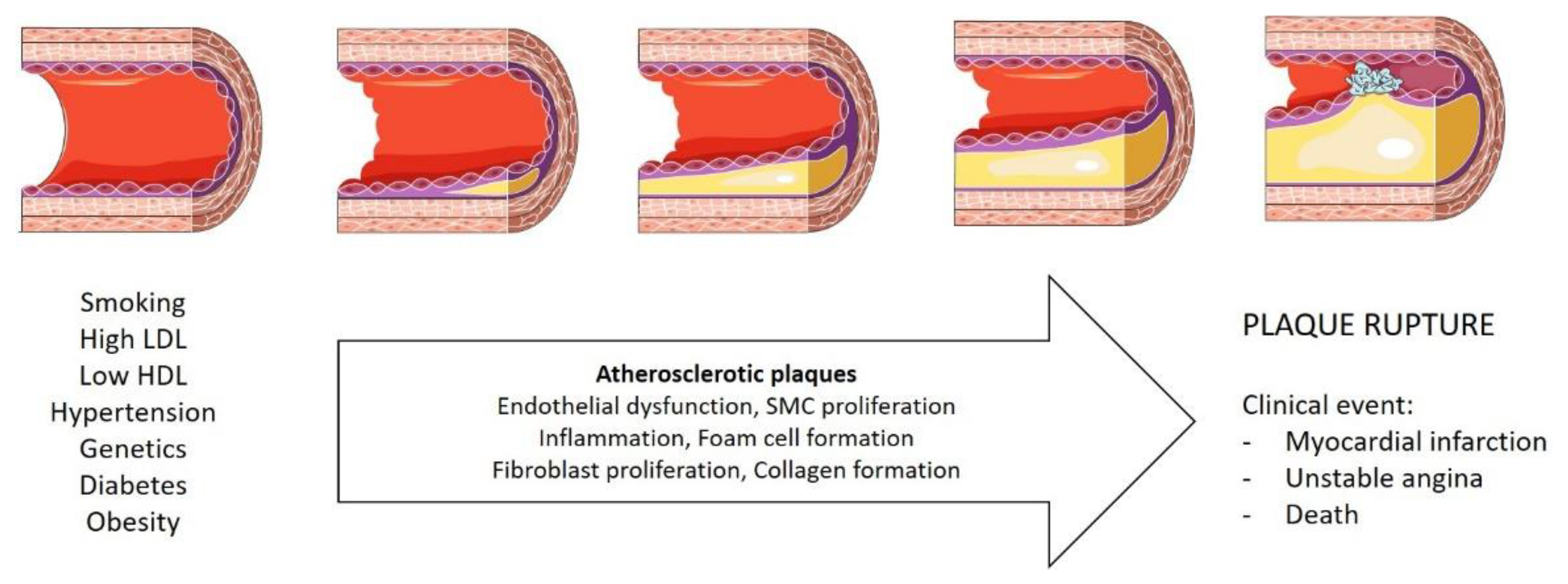
1.1. Coronary Artery Disease
1.2. Obesity as a Risk Factor for Coronary Artery Disease
1.3. Epicardial Adipose Tissue
1.4. Epicardial Adipose Tissue and Coronary Artery Disease (CAD)
2. Methods
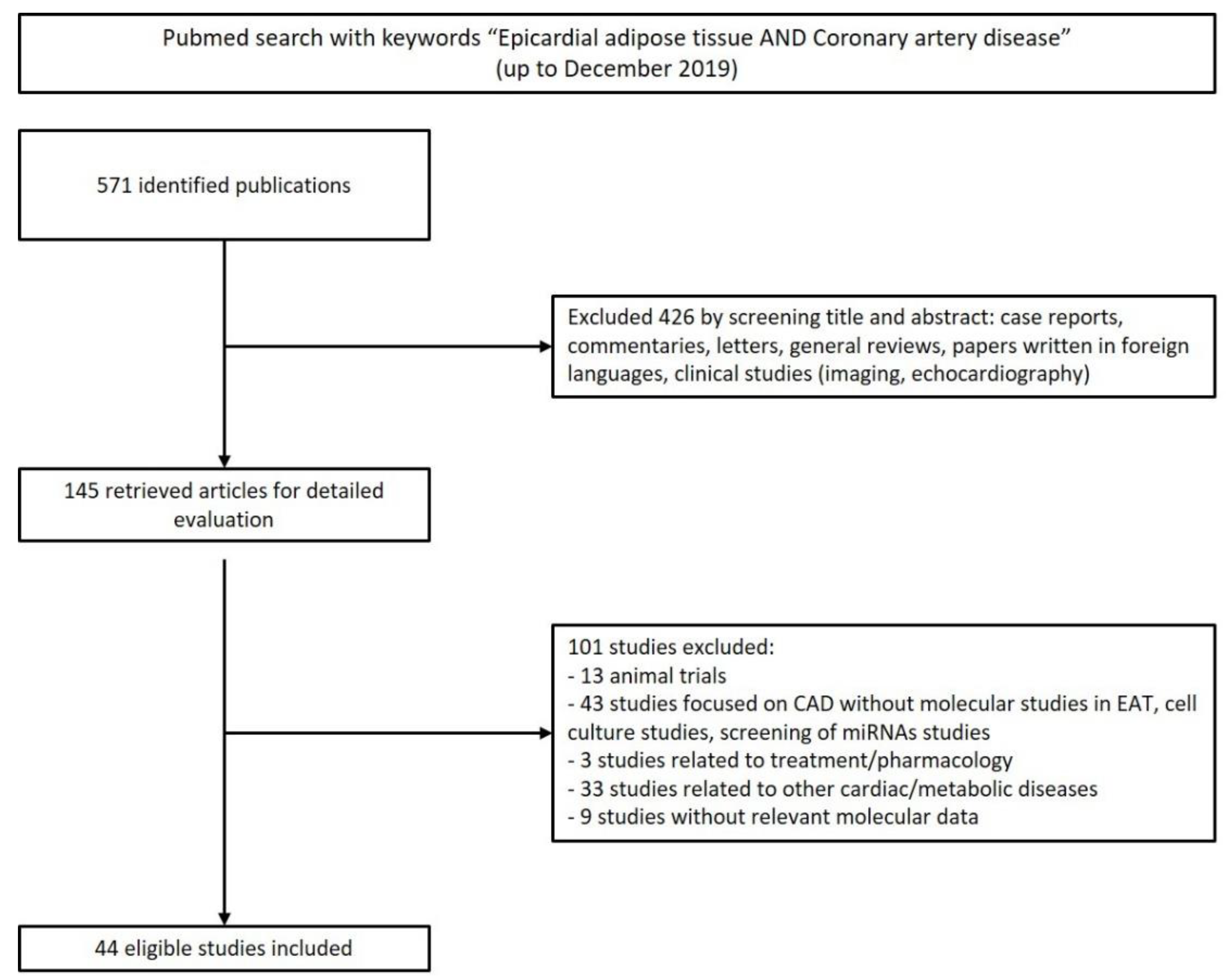
2.1. Search Strategy
2.2. Inclusion and Exclusion Criteria of Study Selection
2.3. Data Extraction
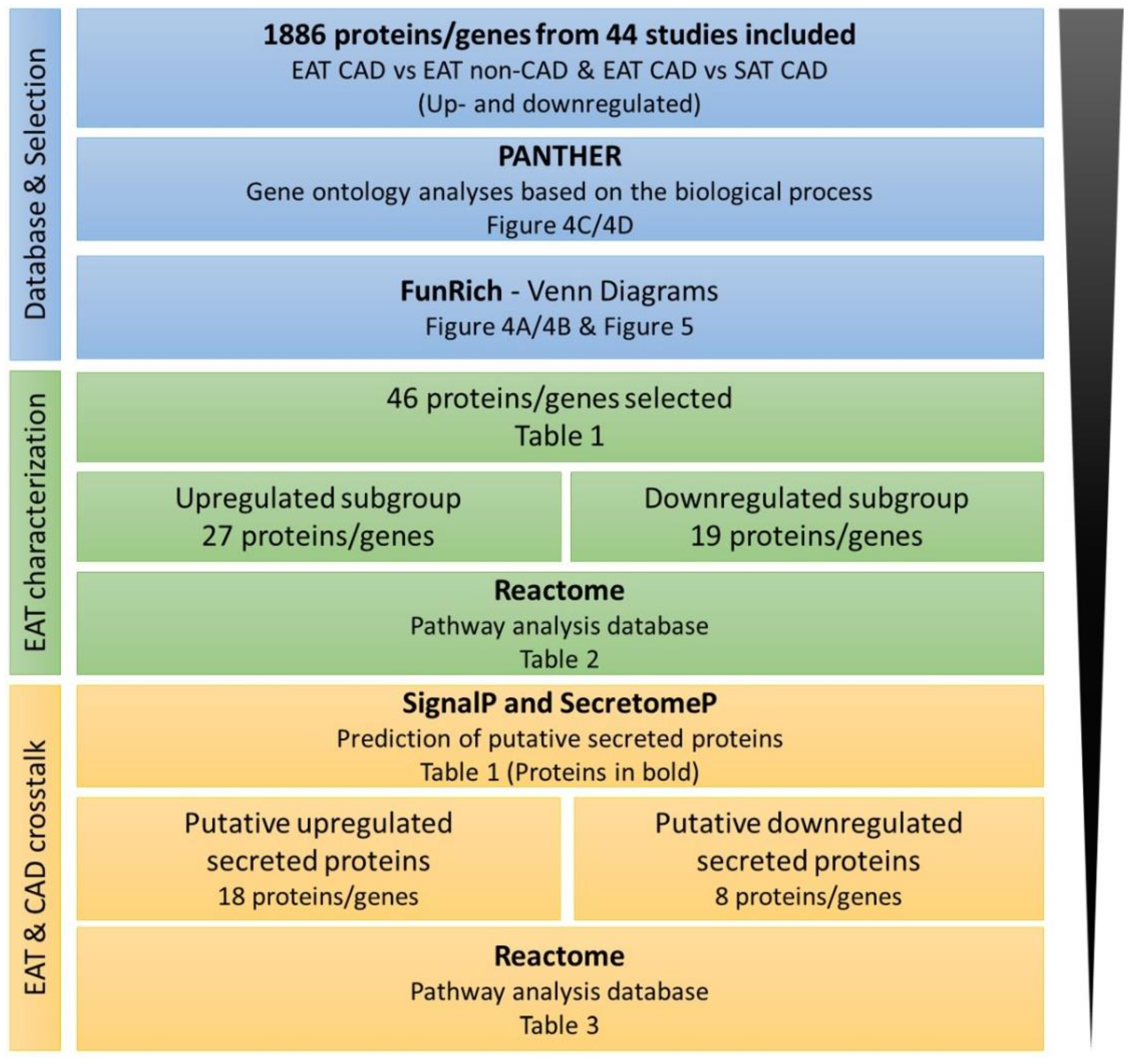
2.4. Bioinformatic Analysis
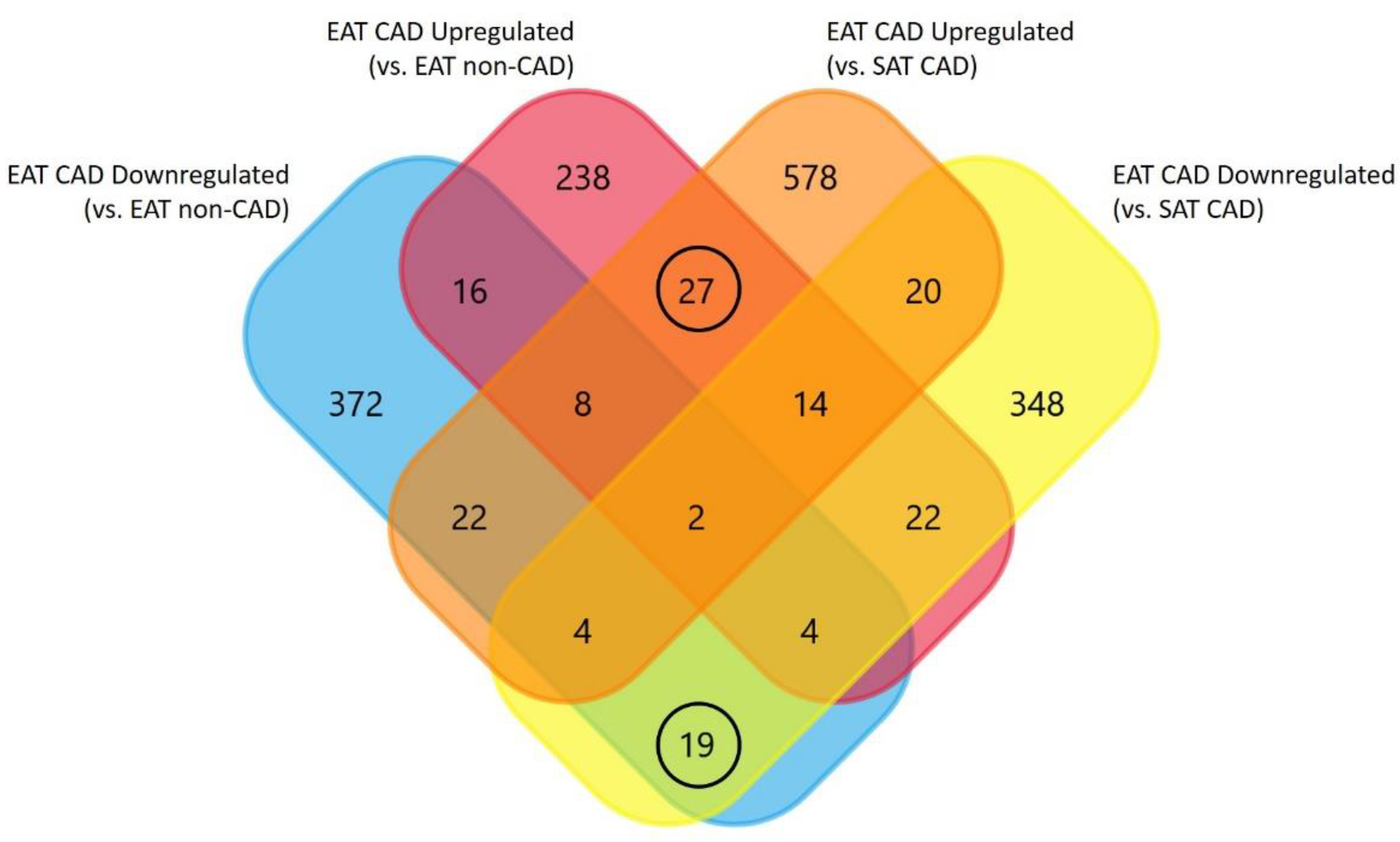
3. Results
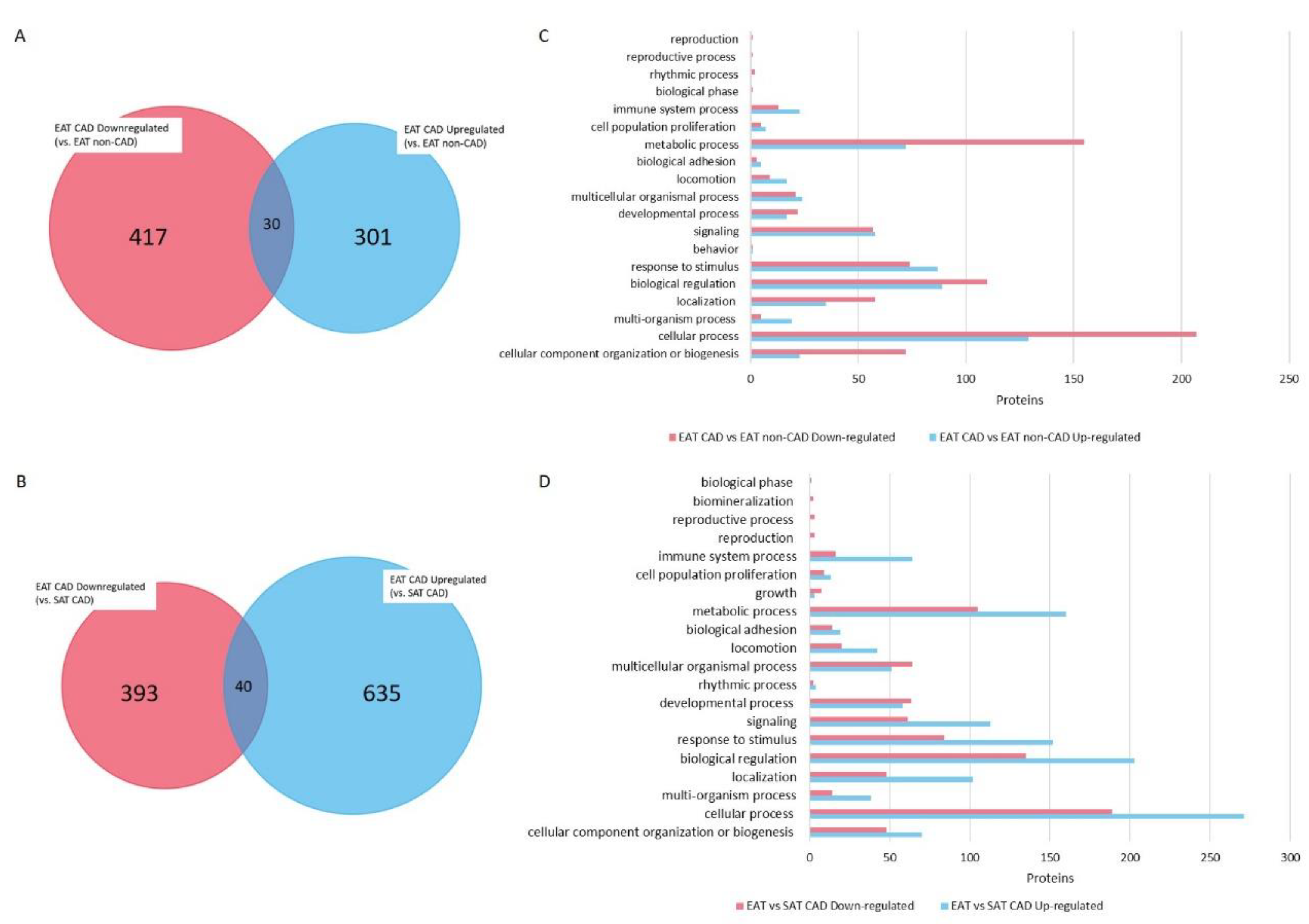
4. Bioinformatics Analysis Provides a Protein Network Overview from CAD Epicardial Adipose Tissue
4.1. Functional Protein Categorization and Integrative Analysis with REACTOME
4.2. Predictions of Putative Secreted Proteins
5. Discussion
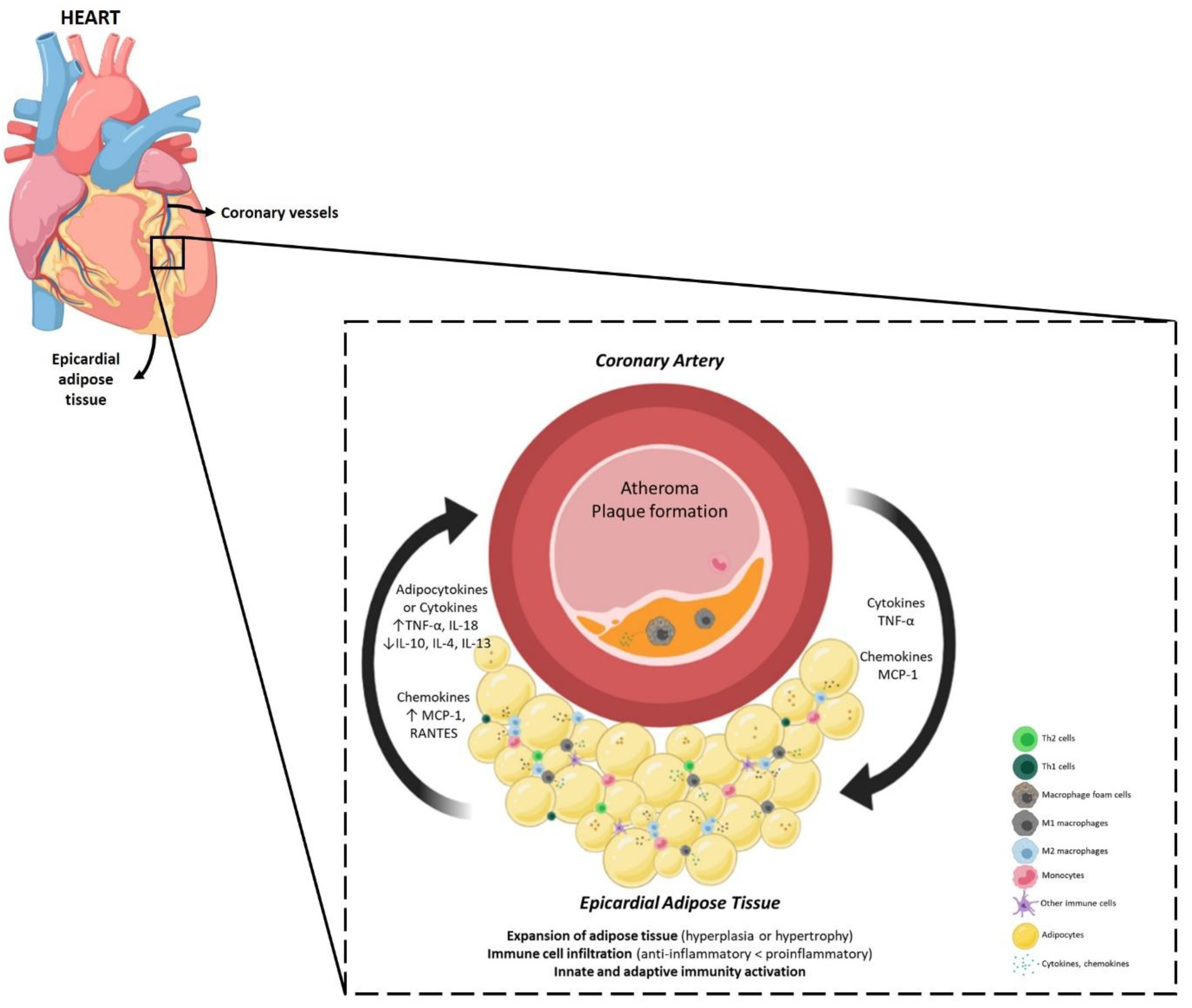
5.1. The Contribution of Epicardial Adipose Tissue to the Proinflammatory Profile of CAD
5.2. The Contribution of Epicardial Adipose Tissue to the Atherosclerotic Plaque Formation in CAD
5.3. The Epicardial Adipose Tissue and Atheroma Communication
5.4. Influence of Epicardial Adipose Tissue Proteome on Cardiac Function and Structure
5.5. Therapeutic Strategies against Inflammation-Related Coronary Artery Disease
6. Conclusions
Supplementary Materials
Funding
Conflicts of Interest
References
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef] [Green Version]
- Manno, C.; Campobasso, N.; Nardecchia, A.; Triggiani, V.; Zupo, R.; Gesualdo, L.; Silvestris, F.; De Pergola, G. Relationship of para- and perirenal fat and epicardial fat with metabolic parameters in overweight and obese subjects. Eat. Weight Disord. 2019, 24, 24–67. [Google Scholar] [CrossRef] [PubMed]
- Iacobellis, G. Local and systemic effects of the multifaceted epicardial adipose tissue depot. Nat. Rev. Endocrinol. 2015, 11, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, R.; Takasu, J.; Itani, Y.; Yamamoto, R.; Yokoyama, K.; Watanabe, S.; Masuda, Y. Pericardial fat accumulation in men as a risk factor for coronary artery disease. Atherosclerosis 2001, 157, 203–209. [Google Scholar] [CrossRef]
- Bettencourt, N.; Toschke, A.M.; Leite, D.; Rocha, J.; Carvalho, M.; Sampaio, F.; Xará, S.; Leite-Moreira, A.; Nagel, E.; Gama, V. Epicardial adipose tissue is an independent predictor of coronary atherosclerotic burden. Int. J. Cardiol. 2012, 158, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Cassar, A.; Holmes, D.R., Jr.; Rihal, C.S.; Gersh, B.J. Chronic coronary artery disease: Diagnosis and management. Mayo Clin. Proc. 2009, 84, 1130–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Ades, P.A.; Savage, P.D. Obesity in coronary heart disease: An unaddressed behavioral risk factor. Prev. Med. 2017, 104, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Piche, M.E.; Poirier, P.; Lemieux, I.; Despres, J.P. Overview of Epidemiology and Contribution of Obesity and Body Fat Distribution to Cardiovascular Disease: An Update. Prog. Cardiovasc. Dis. 2018, 61, 103–113. [Google Scholar] [CrossRef]
- Libby, P.; Theroux, P. Pathophysiology of coronary artery disease. Circulation 2005, 111, 3481–3488. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Leducq Transatlantic Network on A. Inflammation in atherosclerosis: From pathophysiology to practice. J. Am. Coll. Cardiol. 2009, 54, 2129–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prospective Studies, C.; Whitlock, G.; Lewington, S.; Sherliker, P.; Clarke, R.; Emberson, J.; Halsey, J.; Qizilbash, N.; Collins, R.; Peto, R. Body-mass index and cause-specific mortality in 900 000 adults: Collaborative analyses of 57 prospective studies. Lancet 2009, 373, 1083–1096. [Google Scholar] [CrossRef] [Green Version]
- Fuster, J.J.; Ouchi, N.; Gokce, N.; Walsh, K. Obesity-Induced Changes in Adipose Tissue Microenvironment and Their Impact on Cardiovascular Disease. Circ. Res. 2016, 118, 1786–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, K.; Shibata, R.; Ouchi, N.; Tokuda, Y.; Funakubo, H.; Suzuki, M.; Kataoka, T.; Nagao, T.; Okumura, S.; Shinoda, N.; et al. Increased expression of the adipocytokine omentin in the epicardial adipose tissue of coronary artery disease patients. Atherosclerosis 2016, 251, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Sicari, R.; Sironi, A.M.; Petz, R.; Frassi, F.; Chubuchny, V.; De Marchi, D.; Positano, V.; Lombardi, M.; Picano, E.; Gastaldelli, A.; et al. Pericardial rather than epicardial fat is a cardiometabolic risk marker: An MRI vs echo study. J. Am. Soc. Echocardiogr. 2011, 24, 1156–1162. [Google Scholar] [CrossRef] [Green Version]
- Iacobellis, G.; Lonn, E.; Lamy, A.; Singh, N.; Sharma, A.M. Epicardial fat thickness and coronary artery disease correlate independently of obesity. Int. J. Cardiol. 2011, 146, 452–454. [Google Scholar] [CrossRef]
- Villasante Fricke, A.C.; Iacobellis, G. Epicardial Adipose Tissue: Clinical Biomarker of Cardio-Metabolic Risk. Int. J. Mol. Sci. 2019, 20, 5989. [Google Scholar] [CrossRef] [Green Version]
- Bachar, G.N.; Dicker, D.; Kornowski, R.; Atar, E. Epicardial adipose tissue as a predictor of coronary artery disease in asymptomatic subjects. Am. J. Cardiol. 2012, 110, 534–538. [Google Scholar] [CrossRef]
- Clauser, M.; Altenberger, J. Obesity and cardiac cachexia in chronic heart failure. Herz 2013, 38, 610–617. [Google Scholar] [CrossRef]
- Mancio, J.; Barros, A.S.; Conceicao, G.; Pessoa-Amorim, G.; Santa, C.; Bartosch, C.; Ferreira, W.; Carvalho, M.; Ferreira, N.; Vouga, L.; et al. Epicardial adipose tissue volume and annexin A2/fetuin-A signalling are linked to coronary calcification in advanced coronary artery disease: Computed tomography and proteomic biomarkers from the EPICHEART study. Atherosclerosis 2020, 292, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.X.; Zhu, H.J.; Pan, H.; Liu, X.M.; Wang, L.J.; Yang, H.B.; Li, N.; Gong, F.; Sun, W.; Zeng, Y. Comparative Proteome Analysis of Epicardial and Subcutaneous Adipose Tissues from Patients with or without Coronary Artery Disease. Int. J. Endocrinol. 2019, 2019, 6976712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conceicao, G.; Matos, J.; Miranda-Silva, D.; Goncalves, N.; Sousa-Mendes, C.; Goncalves, A.; Ferreira, R.; Leite-Moreira, A.F.; Vitorino, R.; Falcao-Pires, I. Fat Quality Matters: Distinct Proteomic Signatures Between Lean and Obese Cardiac Visceral Adipose Tissue Underlie its Differential Myocardial Impact. Cell Physiol. Biochem. 2020, 54, 384–400. [Google Scholar] [PubMed] [Green Version]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.W.; Dixit, V.D. Adipose tissue as an immunological organ. Obesity (Silver Spring) 2015, 23, 512–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mraz, M.; Haluzik, M. The role of adipose tissue immune cells in obesity and low-grade inflammation. J. Endocrinol. 2014, 222, R113–R127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacy, P. Editorial: Secretion of cytokines and chemokines by innate immune cells. Front. Immunol. 2015, 6, 190. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Mazurek, T.; Zhang, L.; Zalewski, A.; Mannion, J.D.; Diehl, J.T.; Arafat, H.; Sarov-Blat, L.; O’Brien, S.; Keiper, E.A.; Johnson, A.G.; et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation 2003, 108, 2460–2466. [Google Scholar] [CrossRef] [Green Version]
- Hirata, Y.; Tabata, M.; Kurobe, H.; Motoki, T.; Akaike, M.; Nishio, C.; Higashida, M.; Mikasa, H.; Nakaya, Y.; Takanashi, S.; et al. Coronary atherosclerosis is associated with macrophage polarization in epicardial adipose tissue. J. Am. Coll. Cardiol. 2011, 58, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Vishvanath, L.; Gupta, R.K. Contribution of adipogenesis to healthy adipose tissue expansion in obesity. J. Clin. Investig. 2019, 129, 4022–4031. [Google Scholar] [CrossRef]
- Sadler, D.; Mattacks, C.A.; Pond, C.M. Changes in adipocytes and dendritic cells in lymph node containing adipose depots during and after many weeks of mild inflammation. J. Anat. 2005, 207, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Wernstedt Asterholm, I.; Tao, C.; Morley, T.S.; Wang, Q.A.; Delgado-Lopez, F.; Wang, Z.V.; Scherer, P.E. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014, 20, 103–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClain, J.; Hsu, F.; Brown, E.; Burke, G.; Carr, J.; Harris, T.; Kritchevsky, S.; Szklo, M.; Tracy, R.; Ding, J. Pericardial adipose tissue and coronary artery calcification in the Multi-ethnic Study of Atherosclerosis (MESA). Obesity (Silver Spring) 2013, 21, 1056–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raggi, P.; Genest, J.; Giles, J.T.; Rayner, K.J.; Dwivedi, G.; Beanlands, R.S.; Gupta, M. Role of inflammation in the pathogenesis of atherosclerosis and therapeutic interventions. Atherosclerosis 2018, 276, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Antoniades, C.; Kotanidis, C.P.; Berman, D.S. State-of-the-art review article. Atherosclerosis affecting fat: What can we learn by imaging perivascular adipose tissue? J. Cardiovasc. Comput. Tomogr. 2019, 13, 288–296. [Google Scholar] [CrossRef] [Green Version]
- Barrett, T.J. Macrophages in Atherosclerosis Regression. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 20–33. [Google Scholar] [CrossRef]
- Nishihira, K.; Imamura, T.; Yamashita, A.; Hatakeyama, K.; Shibata, Y.; Nagatomo, Y.; Date, H.; Kita, T.; Eto, T.; Asada, Y. Increased expression of interleukin-10 in unstable plaque obtained by directional coronary atherectomy. Eur. Heart J. 2006, 27, 1685–1689. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Qin, X.; Wang, S.; Sun, X.; Dong, B. Decreased cathepsin K levels in human atherosclerotic plaques are associated with plaque instability. Exp. Ther. Med. 2017, 14, 3471–3476. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Liu, Z.; Wang, R.; Zheng, Y.; Li, H.; Yang, L. Galectin-3 Is a Potential Mediator for Atherosclerosis. J. Immunol. Res. 2020, 2020, 5284728. [Google Scholar] [CrossRef] [Green Version]
- MacKinnon, A.C.; Liu, X.; Hadoke, P.W.; Miller, M.R.; Newby, D.E.; Sethi, T. Inhibition of galectin-3 reduces atherosclerosis in apolipoprotein E-deficient mice. Glycobiology 2013, 23, 654–663. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern Med. 2015, 278, 483–493. [Google Scholar] [CrossRef] [PubMed]
- de Winther, M.P.; van Dijk, K.W.; Havekes, L.M.; Hofker, M.H. Macrophage scavenger receptor class A: A multifunctional receptor in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Zani, I.A.; Stephen, S.L.; Mughal, N.A.; Russell, D.; Homer-Vanniasinkam, S.; Wheatcroft, S.B.; Ponnambalam, S. Scavenger receptor structure and function in health and disease. Cells 2015, 4, 178–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexopoulos, A.; Peroukides, S.; Bravou, V.; Varakis, J.; Pyrgakis, V.; Papadaki, H. Implication of bone regulatory factors in human coronary artery calcification. Artery Res. 2011, 5, 101–108. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Li, X.; Qian, S.W.; Guo, L.; Huang, H.Y.; He, Q.; Liu, Y.; Ma, C.-G.; Tang, Q.-Q. Down-regulation of type I Runx2 mediated by dexamethasone is required for 3T3-L1 adipogenesis. Mol. Endocrinol. 2012, 26, 798–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonopoulos, A.S.; Margaritis, M.; Coutinho, P.; Shirodaria, C.; Psarros, C.; Herdman, L.; Sanna, F.; De Silva, R.; Petrou, M.; Sayeed, R.; et al. Adiponectin as a link between type 2 diabetes and vascular NADPH oxidase activity in the human arterial wall: The regulatory role of perivascular adipose tissue. Diabetes 2015, 64, 2207–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaoka, M.; Suzuki, H.; Shioda, S.; Sekikawa, K.; Saito, Y.; Nagai, R.; Sata, M. Endovascular injury induces rapid phenotypic changes in perivascular adipose tissue. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1576–1582. [Google Scholar] [CrossRef]
- Ohyama, K.; Matsumoto, Y.; Amamizu, H.; Uzuka, H.; Nishimiya, K.; Morosawa, S.; Hirano, M.; Watabe, H.; Funaki, Y.; Miyata, S.; et al. Association of Coronary Perivascular Adipose Tissue Inflammation and Drug-Eluting Stent-Induced Coronary Hyperconstricting Responses in Pigs: (18)F-Fluorodeoxyglucose Positron Emission Tomography Imaging Study. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1757–1764. [Google Scholar] [CrossRef] [Green Version]
- Van Linthout, S.; Tschope, C. Inflammation-Cause or Consequence of Heart Failure or Both? Curr. Heart Fail Rep. 2017, 14, 251–265. [Google Scholar] [CrossRef] [Green Version]
- Northcott, J.M.; Yeganeh, A.; Taylor, C.G.; Zahradka, P.; Wigle, J.T. Adipokines and the cardiovascular system: Mechanisms mediating health and disease. Can. J. Physiol. Pharmacol. 2012, 90, 1029–1059. [Google Scholar] [CrossRef]
- Van Linthout, S.; Miteva, K.; Tschope, C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc. Res. 2014, 102, 258–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frantz, S.; Falcao-Pires, I.; Balligand, J.L.; Bauersachs, J.; Brutsaert, D.; Ciccarelli, M.; Dawson, D.; De Windt, L.J.; Giacca, M.; Hamdani, N.; et al. The innate immune system in chronic cardiomyopathy: A European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur. J. Heart Fail 2018, 20, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Waddingham, M.T.; Sonobe, T.; Tsuchimochi, H.; Edgley, A.J.; Sukumaran, V.; Chen, Y.C.; Hansra, S.S.; Schwenke, D.O.; Umetani, K.; Aoyama, K.; et al. Diastolic dysfunction is initiated by cardiomyocyte impairment ahead of endothelial dysfunction due to increased oxidative stress and inflammation in an experimental prediabetes model. J. Mol. Cell. Cardiol. 2019, 137, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschope, C.; Leite-Moreira, A.F.; Musters, R.J.P.; Niessen, H.W.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure With Preserved Ejection Fraction. JACC Heart Fail 2016, 4, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.T.; Wu, C.K.; Lee, J.K.; Chang, S.N.; Kuo, Y.M.; Wang, Y.C.; Lai, L.-P.; Chiang, F.-T.; Hwang, J.-J.; Lin, J.-L.; et al. TNF-alpha down-regulates sarcoplasmic reticulum Ca(2)(+) ATPase expression and leads to left ventricular diastolic dysfunction through binding of NF-kappaB to promoter response element. Cardiovasc. Res. 2015, 105, 318–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurantz, D.; Cowling, R.T.; Varki, N.; Frikovsky, E.; Moore, C.D.; Greenberg, B.H. IL-1beta and TNF-alpha upregulate angiotensin II type 1 (AT1) receptors on cardiac fibroblasts and are associated with increased AT1 density in the post-MI heart. J. Mol. Cell. Cardiol. 2005, 38, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Lindner, D.; Zietsch, C.; Tank, J.; Sossalla, S.; Fluschnik, N.; Hinrichs, S.; Maier, L.; Poller, W.; Blankenberg, S.; Schultheiss, H.-P.; et al. Cardiac fibroblasts support cardiac inflammation in heart failure. Basic Res. Cardiol. 2014, 109, 428. [Google Scholar] [CrossRef]
- Kubota, T.; McTiernan, C.F.; Frye, C.S.; Slawson, S.E.; Lemster, B.H.; Koretsky, A.P.; Demetris, A.J.; Feldman, A.M. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ. Res. 1997, 81, 627–635. [Google Scholar] [CrossRef]
- Zimmer, A.; Bagchi, A.K.; Vinayak, K.; Bello-Klein, A.; Singal, P.K. Innate immune response in the pathogenesis of heart failure in survivors of myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H435–H45. [Google Scholar] [CrossRef]
- Ramos, G.; Frantz, S. Myocardial Metabolism Under Control of a Cytokine Receptor. J. Am. Heart Assoc. 2017, 6, e006291. [Google Scholar] [CrossRef]
- Wodsedalek, D.J.; Paddock, S.J.; Wan, T.C.; Auchampach, J.A.; Kenarsary, A.; Tsaih, S.W.; Flister, M.J.; O’Meara, C.C. IL-13 promotes in vivo neonatal cardiomyocyte cell cycle activity and heart regeneration. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H24–H34. [Google Scholar] [CrossRef] [PubMed]
- Riddell, A.; McBride, M.; Braun, T.; Nicklin, S.A.; Cameron, E.; Loughrey, C.M.; Martin, T.P. RUNX1: An emerging therapeutic target for cardiovascular disease. Cardiovasc. Res. 2020, 116, 1410–1423. [Google Scholar] [CrossRef] [PubMed]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef] [PubMed]
- Lorenzatti, A.; Servato, M.L. Role of Anti-inflammatory Interventions in Coronary Artery Disease: Understanding the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS). Eur. Cardiol. 2018, 13, 38–41. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upregulated Subgroup (27 Proteins, 18 Secreted Proteins) | Downregulated Subgroup (19 Proteins, 8 Secreted Proteins) |
|---|---|
| Tumor necrosis factor Mitogen-activated protein kinase kinase kinase 8 Scavenger receptor cysteine-rich type 1 protein M130 | Tyrosine-protein kinase ABL2 5-aminolevulinate synthase |
| C-C motif chemokine 2 C-C chemokine receptor type 2 | erythroid-specific, mitochondrial Amphiphysin |
| Arachidonate 5-lipoxygenase-activating protein C-C motif chemokine 5 | Aminopeptidase N |
| Nitric oxide synthase, endothelial | Cathepsin K |
| Neutrophil cytosol factor 2 Histone-lysine N-methyltransferase PRDM16 | Dynamin-1 |
| C-C motif chemokine 18 | Gelsolin |
| Cathepsin E Leucine-rich repeat transmembrane protein FLRT3 Interleukin-7 receptor subunit alpha | Macrophage scavenger receptor types I and II Ubiquitin carboxyl-terminal hydrolase isozyme L1 |
| C-C motif chemokine 13 T-cell surface glycoprotein CD3 zeta chain | Butyrophilin-like protein 9 Zinc finger and BTB domain-containing protein 16 |
| HLA class I histocompatibility antigen protein P5 Toll-like receptor 2 | Galectin-3 |
| Interleukin-18 | Secreted frizzled-related protein 2 |
| E3 SUMO-protein ligase EGR2 | Hormone-sensitive lipase |
| Early activation antigen CD69 L-selectin | Prelamin-A/C |
| Alpha-1-antichymotrypsin | Actin, gamma-enteric smooth muscle |
| Complement C3 | Collagen alpha-1(I) chain |
| Coronin-1A Intercellular adhesion molecule 3 | Acyl-CoA-binding protein |
| Integrin beta-2 | Fructose-bisphosphate aldolase C |
| Pathway Name | Entities Found | Entities Total | Entities Ratio | Entities p Value | Entities FDR |
|---|---|---|---|---|---|
| Upregulated Proteins | |||||
| Interleukin-10 signaling | 5 | 45 | 0.004 | 8.66 × 10−8 | 1.69 × 10−5 |
| Signaling by Interleukins | 9 | 456 | 0.04 | 8.91 × 10−7 | 5.9 × 10−5 |
| Immune System | 18 | 2398 | 0.21 | 9.07 × 10−7 | 5.9 × 10−5 |
| Chemokine receptors bind chemokines | 4 | 57 | 0.005 | 1.15 × 10−5 | 5.54 × 10−4 |
| Innate Immune System | 11 | 1187 | 0.104 | 6.06 × 10−5 | 2.36 × 10−3 |
| Peptide ligand-binding receptors | 5 | 198 | 0.017 | 1.1 × 10−4 | 3.53 × 10−3 |
| Interleukin-4 and Interleukin-13 signaling | 4 | 111 | 0.01 | 1.52 × 10−4 | 4.09 × 10−3 |
| Interleukin-18 signaling | 2 | 9 | 0.001 | 2.32 × 10−4 | 5.56 × 10−3 |
| Adaptive Immune System | 9 | 944 | 0.083 | 2.9 × 10−4 | 6.09 × 10−3 |
| Cytokine Signaling in Immune system | 9 | 981 | 0.086 | 3.86 × 10−4 | 7.34 × 10−3 |
| Downregulated Proteins | |||||
| Depolymerisation of the Nuclear | 2 | 16 | 0.001 | 4.05 × 10−4 | 2.65 × 10−2 |
| Scavenging by Class A Receptors | 2 | 19 | 0.002 | 5.7 × 10−4 | 2.65 × 10−2 |
| Initiation of Nuclear Envelope (NE) Reformation | 2 | 20 | 0.002 | 6.3 × 10−4 | 2.65 × 10−2 |
| Apoptotic cleavage of cellular proteins | 2 | 38 | 0.003 | 2.23 × 10−3 | 6.91 × 10−2 |
| Apoptotic execution phase | 2 | 52 | 0.005 | 4.11 × 10−3 | 8.17 × 10−2 |
| Nuclear Envelope Breakdown | 2 | 58 | 0.005 | 5.08 × 10−3 | 8.17 × 10−2 |
| Breakdown of the nuclear lamina | 1 | 3 | 0 | 5.5 × 10−3 | 8.17 × 10−2 |
| Collagen degration | 2 | 64 | 0.006 | 6.14 × 10-3 | 8.17 × 10−2 |
| Nuclear Envelope (NE) Reassembly | 2 | 78 | 0.007 | 8.99 × 10−3 | 8.17 × 10−2 |
| RUNX2 regulates genes involved in differentiation of myeloid cells | 1 | 5 | 0 | 9.15 × 10−3 | 8.17 × 10−2 |
| Pathway Name | Entities Found | Entities Total | Entities Ratio | Entities p Value | Entities FDR |
|---|---|---|---|---|---|
| Upregulated Proteins | |||||
| Interleukin-10 signaling | 4 | 45 | 0.004 | 8.91 × 10−7 | 1.08 × 10−4 |
| Signaling by Interleukins | 7 | 456 | 0.04 | 5.33 × 10−6 | 3.2 × 10−4 |
| Immune System | 13 | 2398 | 0.21 | 1.14 × 10−5 | 4.58 × 10−4 |
| Interleukin-4 and Interleukin-13 signaling | 4 | 111 | 0.01 | 3.08 × 10−5 | 9.23 × 10−4 |
| Interleukin-18 signaling | 2 | 9 | 0.001 | 1.05 × 10−4 | 2.27 × 10−3 |
| Chemokine receptors bind chemokines | 3 | 57 | 0.005 | 1.13 × 10−4 | 2.27 × 10−3 |
| Peptide ligand-binding receptors | 4 | 198 | 0.017 | 2.84 × 10−4 | 4.83 × 10−3 |
| Cytokine Signaling in Immune system | 7 | 981 | 0.086 | 6.86 × 10−4 | 9.59 × 10−3 |
| Purinergic signaling in Leishmaniasis infection | 2 | 25 | 0.002 | 8.00 × 10−4 | 9.59 × 10−3 |
| Cell recruitment (proinflammatory response) | 2 | 25 | 0.002 | 8.00 × 10−4 | 9.59 × 10−3 |
| Downregulated Proteins | |||||
| Binding and Uptake of Ligands by Scavenger Receptors | 2 | 19 | 0.002 | 7.7 × 10−5 | 4.85 × 10−3 |
| RUNX2 regulates genes involved in differentiation of myeloid cells | 2 | 64 | 0.006 | 8.6 × 10−4 | 2.58 × 10−2 |
| Neutrophil degranulation | 2 | 121 | 0.011 | 3.01 × 10−4 | 2.58 × 10−2 |
| Degradation of the extracellular matrix | 2 | 129 | 0.011 | 3.41 × 10−3 | 2.58 × 10−2 |
| RUNX1 regulates transcription of genes involved in differentiation of myeloid cells | 1 | 5 | 0 | 3.5 × 10−3 | 2.58 × 10−2 |
| RUNX1 regulates transcription of genes involved in differentiation of keratinocytes | 3 | 480 | 0.042 | 3.55 × 10−3 | 2.58 × 10−2 |
| Innate Immune System | 2 | 140 | 0.012 | 4.01 × 10−3 | 2.58 × 10−2 |
| GP1b-IX-V activation signalling | 1 | 8 | 0.001 | 5.59 × 10−3 | 2.58 × 10−2 |
| Caspase-mediated cleavage of cytoskeletal proteins | 1 | 8 | 0.001 | 5.59 × 10−3 | 2.58 × 10−2 |
| Transcriptional regulation by RUNX1 | 4 | 1187 | 0.104 | 5.79 × 10−3 | 2.58 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conceição, G.; Martins, D.; M. Miranda, I.; Leite-Moreira, A.F.; Vitorino, R.; Falcão-Pires, I. Unraveling the Role of Epicardial Adipose Tissue in Coronary Artery Disease: Partners in Crime? Int. J. Mol. Sci. 2020, 21, 8866. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228866
Conceição G, Martins D, M. Miranda I, Leite-Moreira AF, Vitorino R, Falcão-Pires I. Unraveling the Role of Epicardial Adipose Tissue in Coronary Artery Disease: Partners in Crime? International Journal of Molecular Sciences. 2020; 21(22):8866. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228866
Chicago/Turabian StyleConceição, Glória, Diana Martins, Isabel M. Miranda, Adelino F. Leite-Moreira, Rui Vitorino, and Inês Falcão-Pires. 2020. "Unraveling the Role of Epicardial Adipose Tissue in Coronary Artery Disease: Partners in Crime?" International Journal of Molecular Sciences 21, no. 22: 8866. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228866