Fragments of gD Protein as Inhibitors of BTLA/HVEM Complex Formation - Design, Synthesis, and Cellular Studies

 , , , , , , and
, , , , , , and
Abstract
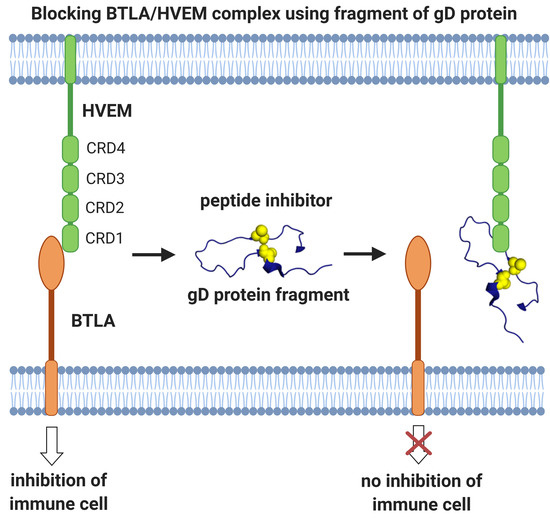
:
1. Introduction
2. Results
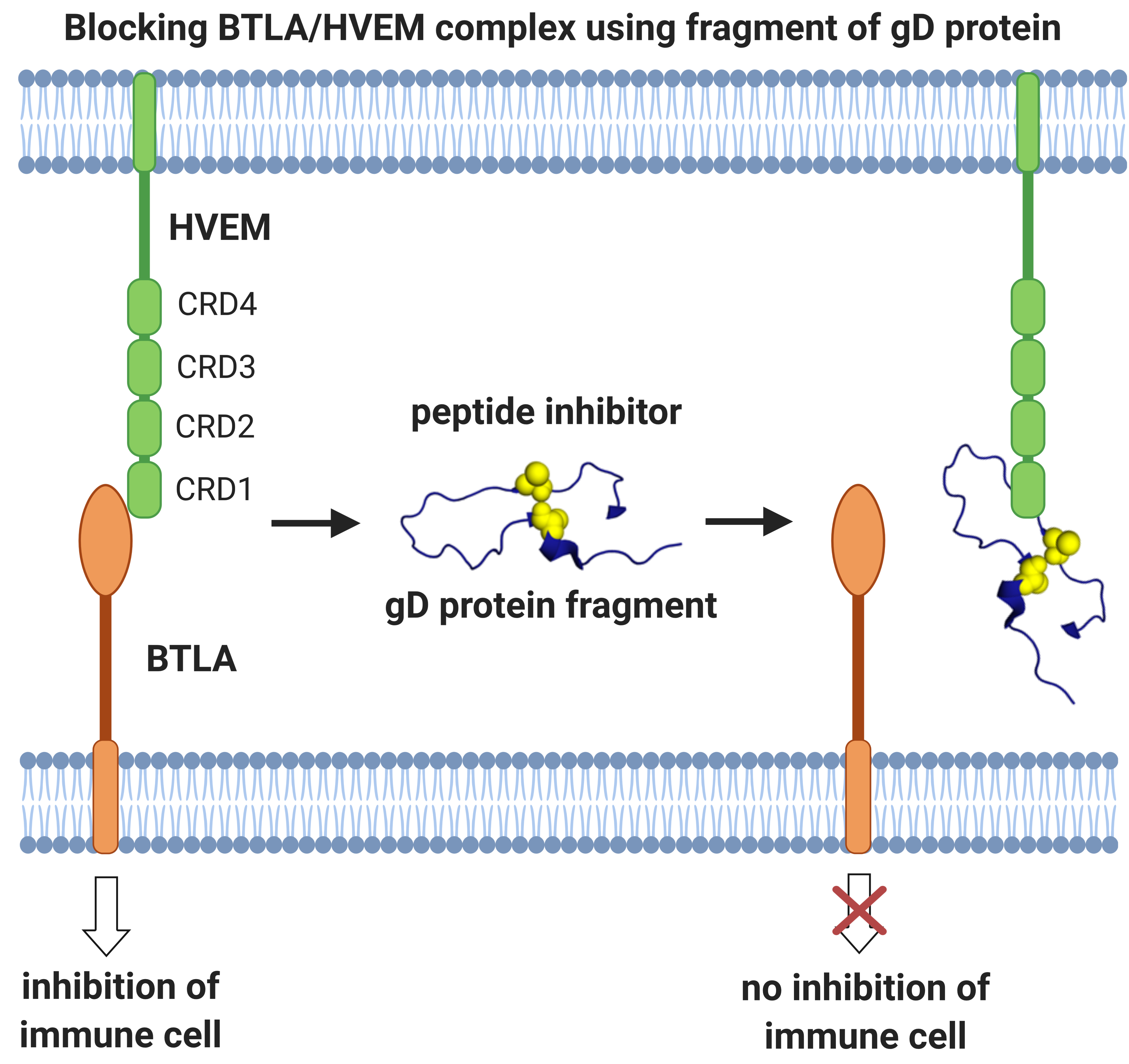
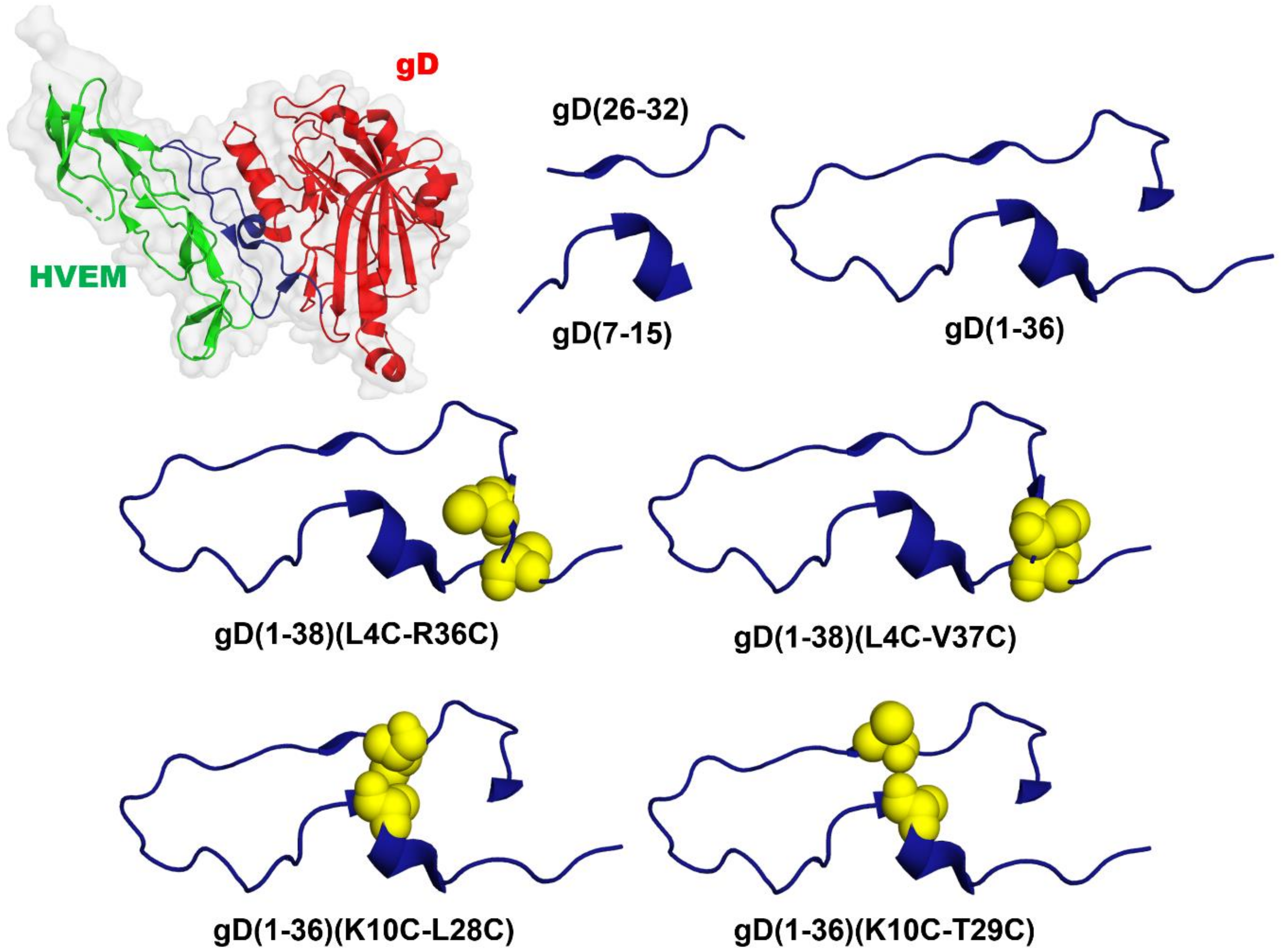
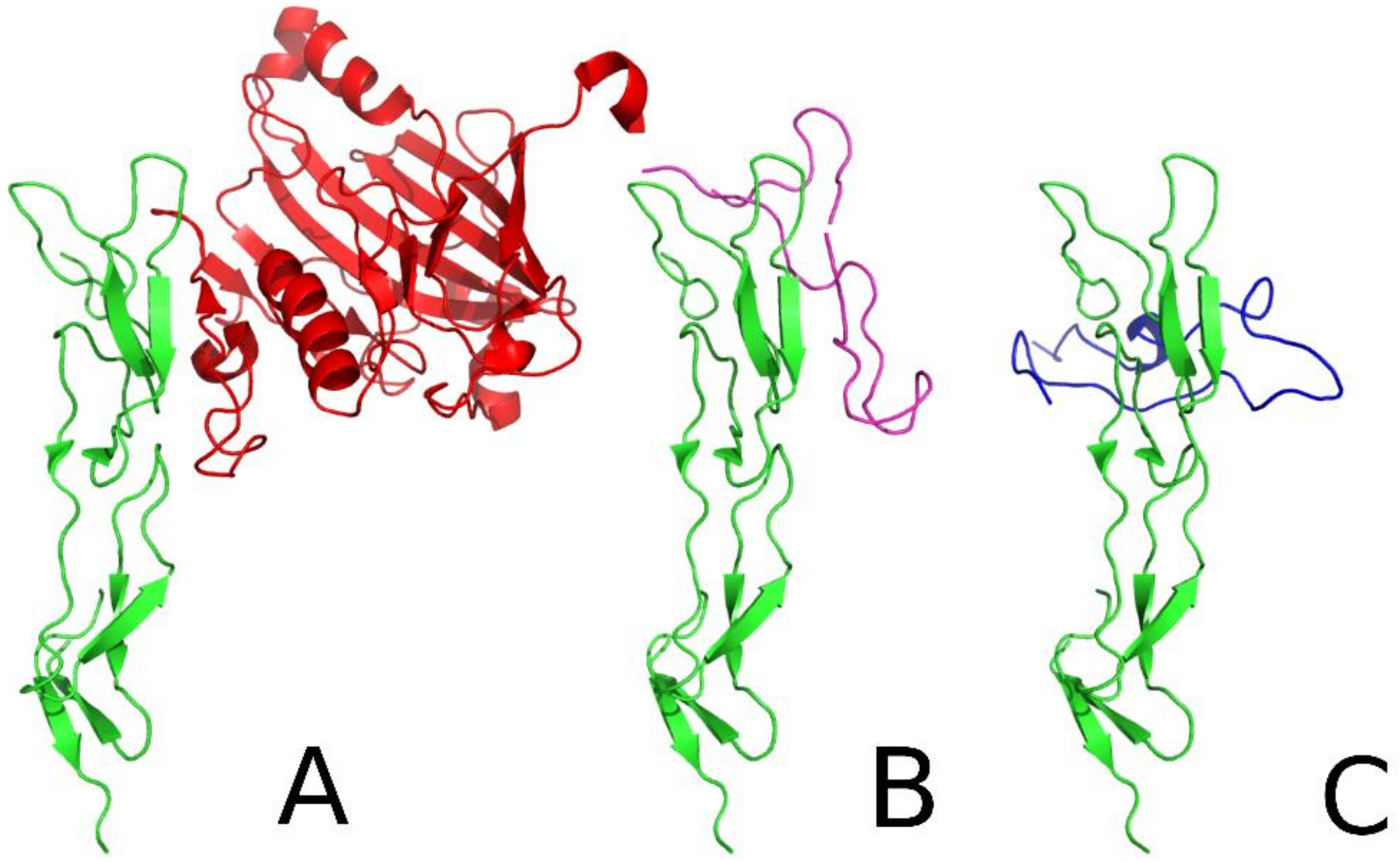
2.1. Peptide Design
2.2. Affinity Studies
2.3. ELISA Tests for BTLA/HVEM and HVEM/LIGHT Complexes
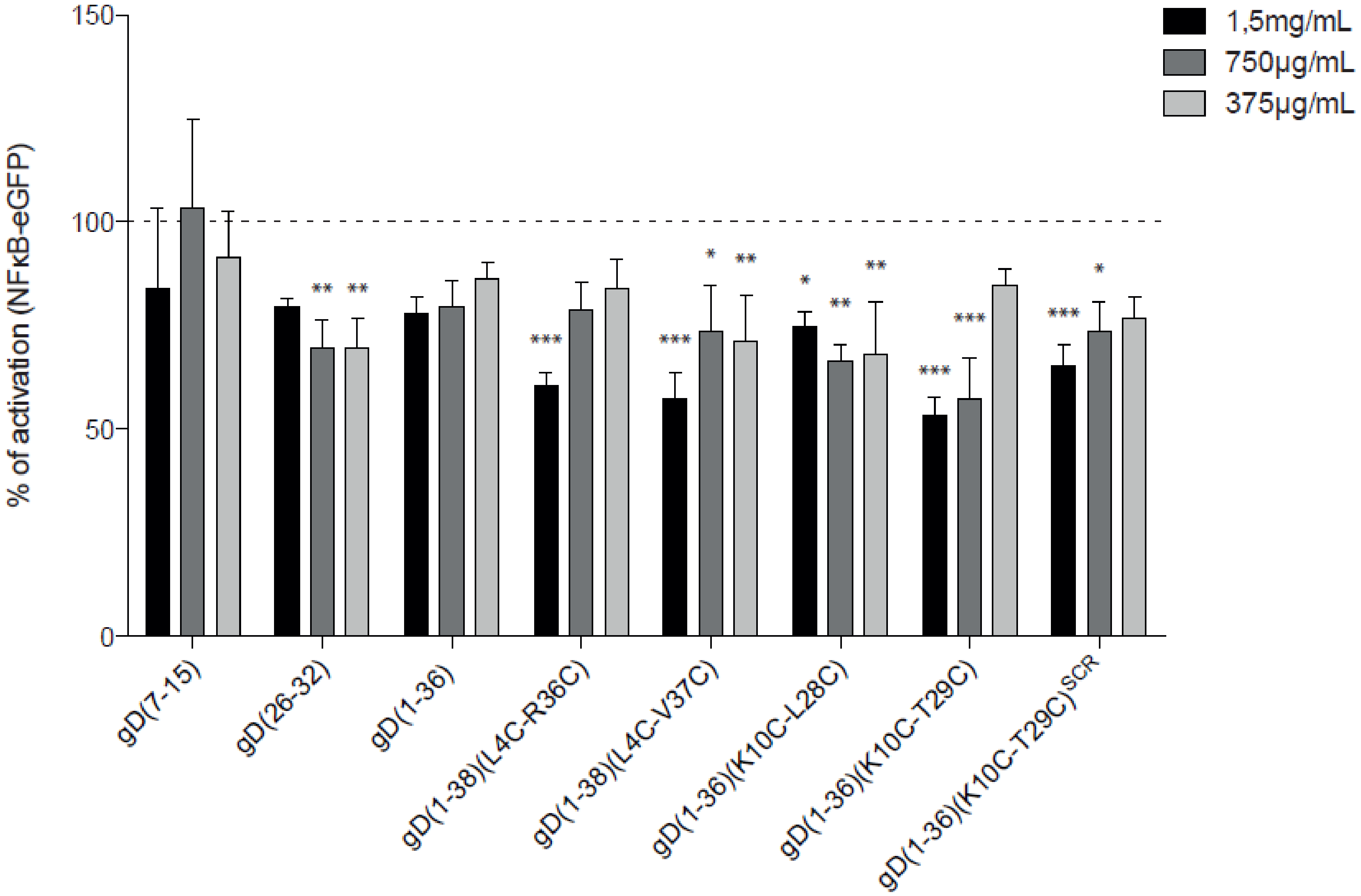
2.4. Evaluation of the Inhibitory Properties of gD Peptides in a Cellular Reporter System
2.5. Stability of the Peptides in PBS, Cell Culture Medium, and Plasma
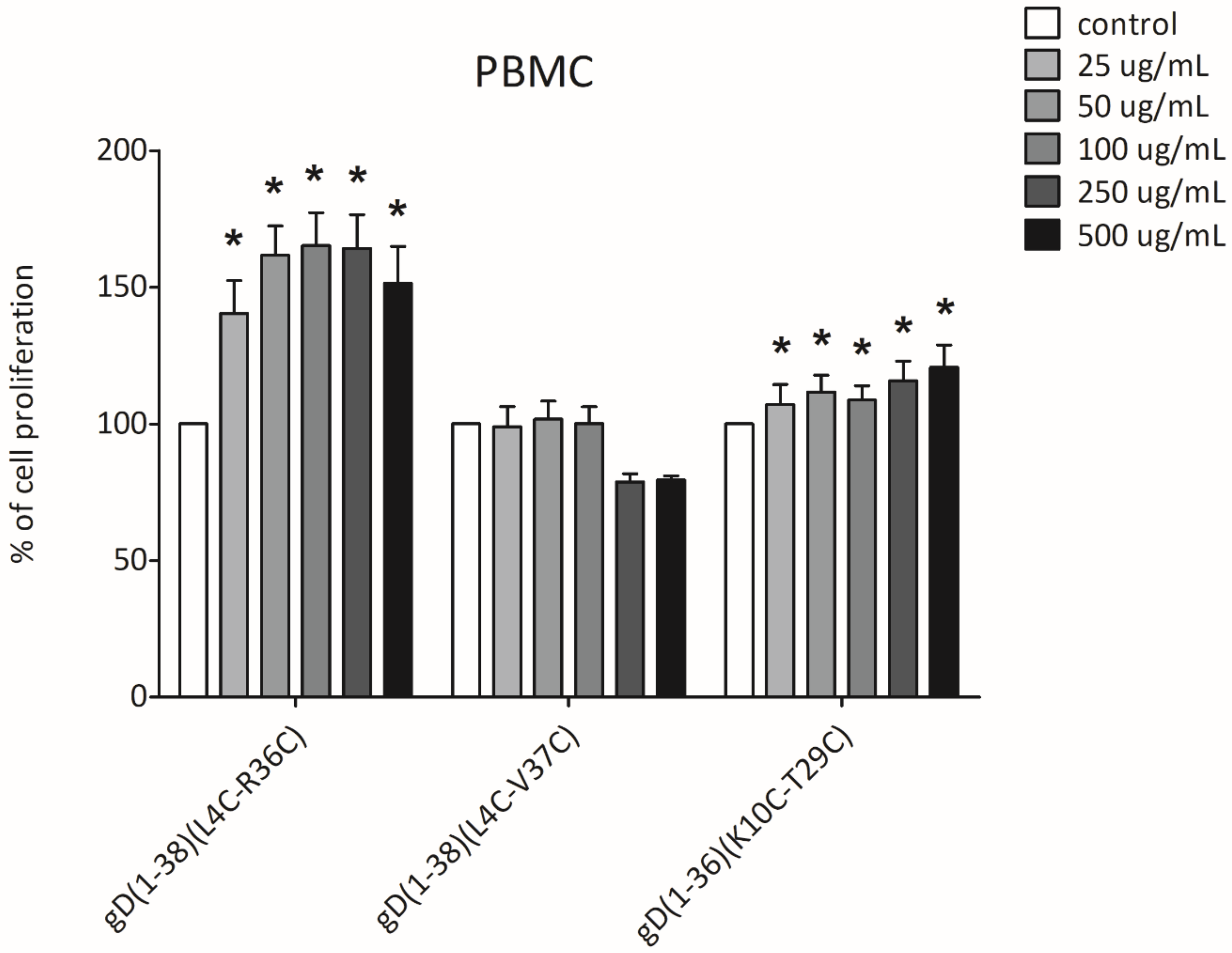
2.6. XTT Cell Proliferation Assay
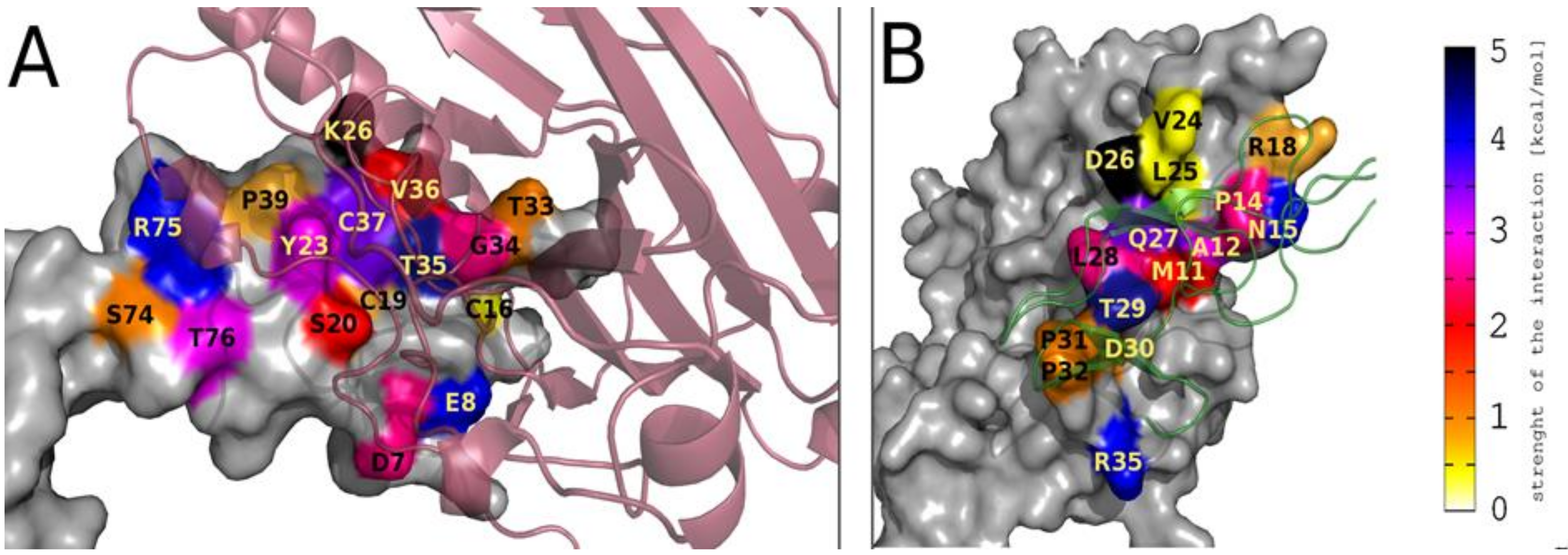
2.7. Flexibility Studies and Docking Simulations of Leading Peptides
3. Discussion
4. Materials and Methods
4.1. Sources of Proteins
4.2. MD Simulation Protocol
4.3. Free Energy Calculations
4.4. Post-Processing of the MD Trajectory
4.5. Peptides Synthesis, Purification and Disulfide Bond Formation
4.6. Affinity Tests
4.7. ELISA
4.8. Stability of Peptides in PBS
4.9. Stability of Peptides in the Medium and Human Plasma
4.10. Cell Culture, Antibodies and Flow Cytometry
4.11. Cell-Based Reporter Assays
4.12. UNRES Docking Procedure
4.13. XTT Cell Proliferation Assay
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BTLA | B- and T-lymphocyte attenuator |
| CD80/CD86 | cluster of differentiation 80/86 |
| CD160 | cluster of differentiation 160 |
| CRD | cysteine rich domain |
| CTLA-4 | cytotoxic T lymphocyte antigen-4 |
| DTT | dithiothreitol |
| ELISA | enzyme-linked immunosorbent assay |
| Fc | fragment crystallizable |
| FCS | fetal calf serum |
| FDA | U.S. Food and Drug Administration |
| gD | glycoprotein D |
| HVEM | herpes virus entry mediator |
| HSV | 1 - herpes simplex virus type 1 |
| HSV-2 | herpes simplex virus type 2 |
| ICP | immune checkpoint protein |
| Ig | Superfamily immunoglobulin-like superfamily |
| LIGHT | homologous to lymphotoxins |
| LTα | lymphotoxin α |
| MD | molecular dynamics |
| MM/GBSA | molecular mechanics Generalized Born surface area |
| MREMD | multiplexed replica exchange molecular dynamics |
| PBMC | peripheral blood mononuclear cell |
| PBS | phosphate-buffered saline |
| PD-1 | programmed cell death 1 |
| PDB | protein data bank |
| PD-L1 | programmed cell death-ligand 1 |
| RP-HPLC | reversed phase-high performance liquid chromatography |
| TCS | T cell stimulator cells |
| TMB | 3,3’,5,5’-tetramethylbenzidine |
| TNF | tumor necrosis factor |
| UNRES | UNited RESidue |
| WHAM | weighted histogram analysis method |
| XTT | 2,3-Bis-(2-Methoxy-4-Nitro-5-Sulfophenyl)-2H-Tetrazolium-5-Carboxanilide |
References
- Sedy, J.R.; Gavrieli, M.; Potter, K.G.; Hurchla, M.A.; Lindsley, R.C.; Hildner, K.; Scheu, S.; Pfeffer, K.; Ware, C.F.; Murphy, T.L.; et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol. 2005, 6, 90–98. [Google Scholar] [CrossRef]
- Watanabe, N.; Gavrieli, M.; Sedy, J.R.; Yang, J.; Fallarino, F.; Loftin, S.K.; Hurchla, M.A.; Zimmerman, N.; Sim, J.; Zang, X.; et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat. Immunol. 2003, 4, 670–679. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Lan, X.; Li, S.; Gao, H.; Nanding, A.; Quan, L.; Yang, C.; Ding, S.; Xue, Y. Increased BTLA and HVEM in gastric cancer are associated with progression and poor prognosis. Onco. Targets. Ther. 2017, 10, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevalier, M.F.; Bohner, P.; Pieraerts, C.; Lhermitte, B.; Gourmaud, J.; Nobile, A.; Rotman, S.; Cesson, V.; Martin, V.; Legris, A.S.; et al. Immunoregulation of Dendritic Cell Subsets by Inhibitory Receptors in Urothelial Cancer. Eur. Urol. 2017, 71, 854–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, J.; He, M.; Zhang, G.L.; Zhao, Q. Distinct Changes of BTLA and HVEM Expressions in Circulating CD4+ and CD8+ T Cells in Hepatocellular Carcinoma Patients. J. Immunol. Res. 2018, 2018, 4561571. [Google Scholar] [CrossRef] [Green Version]
- Derré, L.; Rivals, J.P.; Jandus, C.; Pastor, S.; Rimoldi, D.; Romero, P.; Michielin, O.; Olive, D.; Speiser, D.E. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J. Clin. Invest. 2010, 120, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Safety, Tolerability and Pharmacokinetics of a Monoclonal Antibody Specific to B-and T-Lymphocyte Attenuator (BTLA) for Injection in Subjects With Advanced Malignancies - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04137900?cond=BTLA&draw=2&rank=2 (accessed on 18 September 2020).
- Safety, Tolerability and Pharmacokinetics of a Recombinant Humanized mAb Specific to B-and T-Lymphocyte Attenuator (BTLA) for Injection in Subjects With Advanced Malignancies - Full Text View - ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04278859?cond=BTLA&draw=2&rank=1 (accessed on 18 September 2020).
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Compaan, D.M.; Gonzalez, L.C.; Tom, I.; Loyet, K.M.; Eaton, D.; Hymowitz, S.G. Attenuating lymphocyte activity: The crystal structure of the BTLA-HVEM complex. J. Biol. Chem. 2005, 280, 39553–39561. [Google Scholar] [CrossRef] [Green Version]
- Mauri, D.N.; Ebner, R.; Montgomery, R.I.; Kochel, K.D.; Cheung, T.C.; Yu, G.L.; Ruben, S.; Murphy, M.; Eisenberg, R.J.; Cohen, G.H.; et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin α are ligands for herpesvirus entry mediator. Immunity 1998, 8, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Cai, G.; Anumanthan, A.; Brown, J.A.; Greenfield, E.A.; Zhu, B.; Freeman, G.J. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat. Immunol. 2008, 9, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Granger, S.W.; Ware, C.F. Turning on LIGHT. J. Clin. Invest. 2001, 108, 1741–1742. [Google Scholar] [CrossRef]
- Wang, Y.; Subudhi, S.K.; Anders, R.A.; Lo, J.; Sun, Y.; Blink, S.; Wang, Y.; Wang, J.; Liu, X.; Mink, K.; et al. The role of herpesvirus entry mediator as a negative regulator of T cell-mediated responses. J. Clin. Invest. 2005, 115, 711–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, R.I.; Warner, M.S.; Lum, B.J.; Spear, P.G. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 1996, 87, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Kwon, B.S.; Tan, K.B.; Ni, J.; Kwi-Ok-Oh; Lee, Z.H.; Kim, K.K.; Kim, Y.J.; Wang, S.; Gentz, R.; Yu, G.L.; et al. A newly identified member of the tumor necrosis factor receptor superfamily with a wide tissue distribution and involvement in lymphocyte activation. J. Biol. Chem. 1997, 272, 14272–14276. [Google Scholar] [CrossRef] [Green Version]
- Cheung, T.C.; Humphreys, I.R.; Potter, K.G.; Norris, P.S.; Shumway, H.M.; Tran, B.R.; Patterson, G.; Jean-Jacques, R.; Yoon, M.; Spear, P.G.; et al. Evolutionarily divergent herpesviruses modulate T cell activation by targeting the herpesvirus entry mediator cosignaling pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 13218–13223. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Garrett, S.C.; Fedorov, E.V.; Ramagopal, U.A.; Garforth, S.J.; Bonanno, J.B.; Almo, S.C.; Liu, W.; Garrett, S.C. Structural Basis of CD160: HVEM Recognition Article Structural Basis of CD160: HVEM Recognition. Structure 2019, 27, 1286–1295. [Google Scholar] [CrossRef]
- Whitbeck, J.C.; Peng, C.; Lou, H.; Xu, R.; Willis, S.H.; Ponce de Leon, M.; Peng, T.; Nicola, A.V.; Montgomery, R.I.; Warner, M.S.; et al. Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a member of the tumor necrosis factor receptor superfamily and a mediator of HSV entry. J. Virol. 1997, 71, 6083–6093. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, R.J.; Cerini, C.P.; Heilman, C.J.; Joseph, A.D.; Dietzschold, B.; Golub, E.; Long, D.; Ponce de Leon, M.; Cohen, G.H. Synthetic glycoprotein D-related peptides protect mice against herpes simplex virus challenge. J. Virol. 1985, 56, 1014–1017. [Google Scholar] [CrossRef] [Green Version]
- Lasaro, M.O.; Ertl, H.C.J. Potentiating vaccine immunogenicity by manipulating the HVEM/BTLA pathway and other co-stimulatory and co-inhibitory signals of the immune system. Hum. Vaccin. 2009, 5, 6–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarrias, M.R.; Whitbeck, J.C.; Rooney, I.; Ware, C.F.; Eisenberg, R.J.; Cohen, G.H.; Lambris, J.D. The three HveA receptor ligands, gD, LT-α and LIGHT bind to distinct sites on HveA. Mol. Immunol. 2000, 37, 665–673. [Google Scholar] [CrossRef]
- Stiles, K.M.; Whitbeck, J.C.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C. Herpes Simplex Virus Glycoprotein D Interferes with Binding of Herpesvirus Entry Mediator to Its Ligands through Downregulation and Direct Competition. J. Virol. 2010, 84, 11646–11660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carfí, A.; Willis, S.H.; Whitbeck, J.C.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Wiley, D.C. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell 2001, 8, 169–179. [Google Scholar] [CrossRef]
- Connolly, S.A.; Landsburg, D.J.; Carfi, A.; Wiley, D.C.; Cohen, G.H.; Eisenberg, R.J. Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD-HveA/HVEM binding interface. J. Virol. 2003, 77, 8127–8140. [Google Scholar] [CrossRef] [Green Version]
- Lazear, E.; Whitbeck, J.C.; Zuo, Y.; Carfí, A.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C. Induction of conformational changes at the N-terminus of herpes simplex virus glycoprotein D upon binding to HVEM and nectin-1. Virology 2014, 448, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Krummenacher, C.; Supekar, V.M.; Whitbeck, J.C.; Lazear, E.; Connolly, S.A.; Eisenberg, R.J.; Cohen, G.H.; Wiley, D.C.; Carfí, A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 2005, 24, 4144–4153. [Google Scholar] [CrossRef]
- Stump, J.D.; Sticht, H. Mutations in herpes simplex virus gD protein affect receptor binding by different molecular mechanisms. J. Mol. Model. 2014, 20. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 14. Available online: https://ambermd.org/doc12/Amber14.pdf (accessed on 18 January 2020).
- Onufriev, A.; Bashford, D.; Case, D.A. Modification of the Generalized Born Model Suitable for Macromolecules. J. Phys. Chem. B 2002, 104, 3712–3720. [Google Scholar] [CrossRef] [Green Version]
- Spodzieja, M.; Lach, S.; Iwaszkiewicz, J.; Cesson, V.; Kalejta, K.; Olive, D.; Michielin, O.; Speiser, D.E.; Zoete, V.; Derré, L.; et al. Design of short peptides to block BTLA/HVEM interactions for promoting anticancer T-cell responses. PLoS ONE 2017, 12, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Jutz, S.; Hennig, A.; Paster, W.; Asrak, Ö.; Dijanovic, D.; Kellner, F.; Pickl, W.F.; Huppa, J.B.; Leitner, J.; Steinberger, P. A cellular platform for the evaluation of immune checkpoint molecules. Oncotarget 2017, 8, 64892–64906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitner, J.; Kuschei, W.; Grabmeier-Pfistershammer, K.; Woitek, R.; Kriehuber, E.; Majdic, O.; Zlabinger, G.; Pickl, W.F.; Steinberger, P. T cell stimulator cells, an efficient and versatile cellular system to assess the role of costimulatory ligands in the activation of human T cells. J. Immunol. Methods 2010, 362, 131–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zapadka, K.L.; Becher, F.J.; Gomes dos Santos, A.L.; Jackson, S.E. Factors affecting the physical stability (aggregation) of peptide therapeutics. Interface Focus 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boöttger, R.; Hoffmann, R.; Knappe, D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS ONE 2017, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Globus, O.; Evron, T.; Caspi, M.; Siman-Tov, R.; Rosin-Arbesfeld, R. High-Temperature Requirement A1 (Htra1) - A Novel Regulator of Canonical Wnt Signaling. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Lee, M.O.; Moon, B.H.; Shim, S.H.; Fornace, A.J.; Cha, H.J. Senescent growth arrest in mesenchymal stem cells is bypassed by Wip1-mediated downregulation of intrinsic stress signaling pathways. Stem Cells 2009, 27, 1963–1975. [Google Scholar] [CrossRef]
- Boohaker, R.J.; Sambandam, V.; Segura, I.; Miller, J.; Suto, M.; Xu, B. Rational design and development of a peptide inhibitor for the PD-1/PD-L1 interaction. Cancer Lett. 2018, 434, 11–21. [Google Scholar] [CrossRef]
- Abbas, A.B.; Lin, B.; Liu, C.; Morshed, A.; Hu, J.; Xu, H. Design and synthesis of A PD-1 binding peptide and evaluation of its anti-tumor activity. Int. J. Mol. Sci. 2019, 20, 572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, K.; Lu, J.; Yin, X.; Xu, H.; Li, L.; Ma, B. Structure-based derivation and intramolecular cyclization of peptide inhibitors from PD-1/PD-L1 complex interface as immune checkpoint blockade for breast cancer immunotherapy. Biophys. Chem. 2019, 253. [Google Scholar] [CrossRef]
- Podlesnykh, S.V.; Shanshin, D.V.; Kolosova, E.A.; Murashkin, D.E.; Shaprova, O.N.; Shcherbakov, D.N.; Chapoval, A.I. Development of Search Strategy for Peptide Inhibitors of Immune Checkpoints. Russ. J. Bioorganic Chem. 2018, 44, 150–157. [Google Scholar] [CrossRef]
- Spodzieja, M.; Kuncewicz, K.; Sieradzan, A.; Karczyńska, A.; Iwaszkiewicz, J.; Cesson, V.; Węgrzyn, K.; Zhukov, I.; Maszota-Zieleniak, M.; Michielin, O.; et al. Disulfide-linked peptides for blocking BTLA/HVEM binding. Int. J. Mol. Sci. 2020, 21, 636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ware, C.F.; VanArsdale, T.L.; Crowe, P.D.; Browning, J.L. The ligands and receptors of the lymphotoxin system. Curr. Top. Microbiol. Immunol. 1995, 198, 175–218. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.; Spear, P.G. Random mutagenesis of the gene encoding a viral ligand for multiple cell entry receptors to obtain viral mutants altered for receptor usage. Proc. Natl. Acad. Sci. USA 2004, 101, 17252–17257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Tsui, V. Theory and Applications of the Generalized Born Solvation Model in Macromolecular Simulations. Biopolymers 2001, 275–291. [Google Scholar]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- FIELDS, G.B.; NOBLE, R.L. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int. J. Pept. Protein Res. 1990, 35, 161–214. [Google Scholar] [CrossRef]
- Jutz, S.; Leitner, J.; Schmetterer, K.; Doel-Perez, I.; Majdic, O.; Grabmeier-Pfistershammer, K.; Paster, W.; Huppa, J.B.; Steinberger, P. Assessment of costimulation and coinhibition in a triple parameter T cell reporter line: Simultaneous measurement of NF-κB, NFAT and AP-1. J. Immunol. Methods 2016, 430, 10–20. [Google Scholar] [CrossRef] [Green Version]
- Battin, C.; Hennig, A.; Mayrhofer, P.; Kunert, R.; Zlabinger, G.J.; Steinberger, P.; Paster, W. A human monocytic NF-κB fluorescent reporter cell line for detection of microbial contaminants in biological samples. PLoS ONE 2017, 12, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef] [Green Version]
- Liwo, A.; Sieradzan, A.K.; Lipska, A.G.; Czaplewski, C.; Joung, I.; Zmudzińska, W.; Hałabis, A.; Ołdziej, S. A general method for the derivation of the functional forms of the effective energy terms in coarse-grained energy functions of polymers. III. Determination of scale-consistent backbone-local and correlation potentials in the UNRES force field and force-f. J. Chem. Phys. 2019, 150. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Rosenberg, J.M.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Deptuła, M.; Karpowicz, P.; Wardowska, A.; Sass, P.; Sosnowski, P.; Mieczkowska, A.; Filipowicz, N.; Dzierżyńska, M.; Sawicka, J.; Nowicka, E.; et al. Development of a Peptide Derived from Platelet-Derived Growth Factor (PDGF-BB) into a Potential Drug Candidate for the Treatment of Wounds. Adv. Wound Care 2019, 9, 12. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of the Energy Decomposition | Amino Acid Residues of gD Involved in Important gD/HVEM Interactions | Amino Acid Residues of HVEM Involved in Important gD/HVEM Interactions |
|---|---|---|
| Per-residue | M11, A12, P14, N15, V24, Q27, L28, T29, P31, P32, R35 | P17, Y23, G34, T35, V36, C37, P39, R75, T76 |
| Pairwise per-residue | M11, A12, P14, N15, R18, V24, L25, D26, Q27, L28, T29, D30, P31, P32, R35 | D7, E8, C16, P17, C19, S20, Y23, K26, T33, G34, T35, V36, C37, P39, S74, R75, T76 |
| Peptide Name | Amino Acid Sequence |
|---|---|
| gD(7-15) | Ac-ASLKMADPN-NH2 |
| gD(26-32) | Ac-DQLTDPP-NH2 |
| gD(1-36) | Ac-KYALVDASLKMADPNRFRGKDLPVLDQLTDPPGVRR-NH2 |
| gD(1-38)(L4C-R36C) |  |
| gD(1-38)(L4C-V37C) |  |
| gD(1-36)(K10C-L28C) |  |
| gD(1-36)(K10C-T29C) |  |
| Peptide | [M+H]+Calc. | Supernatant [M+H]+ | Last Wash | Elution [M+H]+ |
|---|---|---|---|---|
| gD(7-15) | 987.12 | 987.48 | - | - |
| gD(26-32) | 825.86 | 848.32 [M+Na]+ | - | - |
| gD(1-36) | 4094.72 | 4094.88 | - | 4094.88 |
| gD(1-38)(L4C-R36C) | 4291.97 | 4291.37 | - | 4292.61 |
| gD(1-38)(L4C-V37C) | 4349.03 | 4349.20 | - | 4349.20 |
| gD(1-36)(K10C-L28C) | 4057.68 | 4057.11 | - | 4057.11 |
| gD(1-36)(K10C-T29C) | 4069.73 | 4070.37 | - | 4068.45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuncewicz, K.; Battin, C.; Sieradzan, A.; Karczyńska, A.; Orlikowska, M.; Wardowska, A.; Pikuła, M.; Steinberger, P.; Rodziewicz-Motowidło, S.; Spodzieja, M. Fragments of gD Protein as Inhibitors of BTLA/HVEM Complex Formation - Design, Synthesis, and Cellular Studies. Int. J. Mol. Sci. 2020, 21, 8876. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228876
Kuncewicz K, Battin C, Sieradzan A, Karczyńska A, Orlikowska M, Wardowska A, Pikuła M, Steinberger P, Rodziewicz-Motowidło S, Spodzieja M. Fragments of gD Protein as Inhibitors of BTLA/HVEM Complex Formation - Design, Synthesis, and Cellular Studies. International Journal of Molecular Sciences. 2020; 21(22):8876. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228876
Chicago/Turabian StyleKuncewicz, Katarzyna, Claire Battin, Adam Sieradzan, Agnieszka Karczyńska, Marta Orlikowska, Anna Wardowska, Michał Pikuła, Peter Steinberger, Sylwia Rodziewicz-Motowidło, and Marta Spodzieja. 2020. "Fragments of gD Protein as Inhibitors of BTLA/HVEM Complex Formation - Design, Synthesis, and Cellular Studies" International Journal of Molecular Sciences 21, no. 22: 8876. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21228876