A Computational Framework Based on Ensemble Deep Neural Networks for Essential Genes Identification

 ,
, 
 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Hyperparameters Optimization
2.2. Effectiveness of Ensemble Model
2.3. Comparison with Other State-of-the-Art DNA Sequencing Features
2.4. Imbalance Solving
2.5. Comparison to the Existing Predictors in Identifying Essential Genes
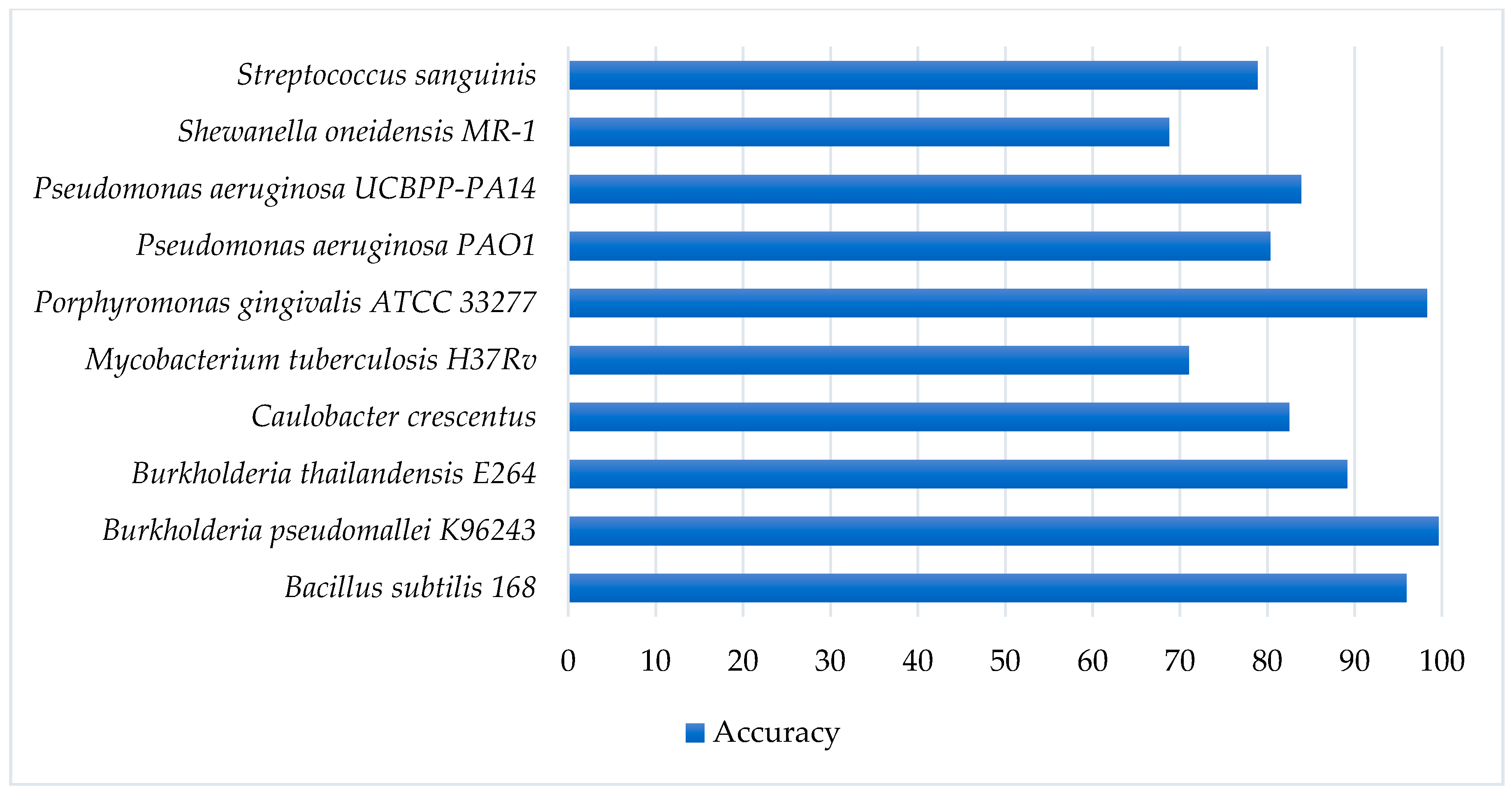
2.6. Validation of the Proposed Model on Cross-Species Datasets
3. Materials and Methods
- A benchmark dataset that was verified and used in previous publication with high confidence was retrieved.
- The genes were extracted by using an NLP model that aimed to integrate the hidden information of gene sequences.
- Different machine learning and deep learning-based methods were developed to learn and analyze the extracted features.
3.1. Benchmark Dataset
3.2. Feature Engineering
- (1)
- We split each DNA sequence into a “sentence” comprising biological sub-words (token) in an overlapping manner. A detail explanation of generating different n-gram levels for DNA sequences is shown in Supplementary Figure S1.
- (2)
- We trained the language model to generate word embeddings for each word. No matter the sequence length, the NLP model could generate the word embeddings with the same vector sizes. Thus, we did not apply any padding approach on DNA sequences.
- (3)
- We concatenated these word embeddings to become a vector that represented for whole DNA sequence.
- (4)
- Consequently, these word vectors were fed into our ensemble neural network to evaluate the potential of the network in learning such kinds of features.
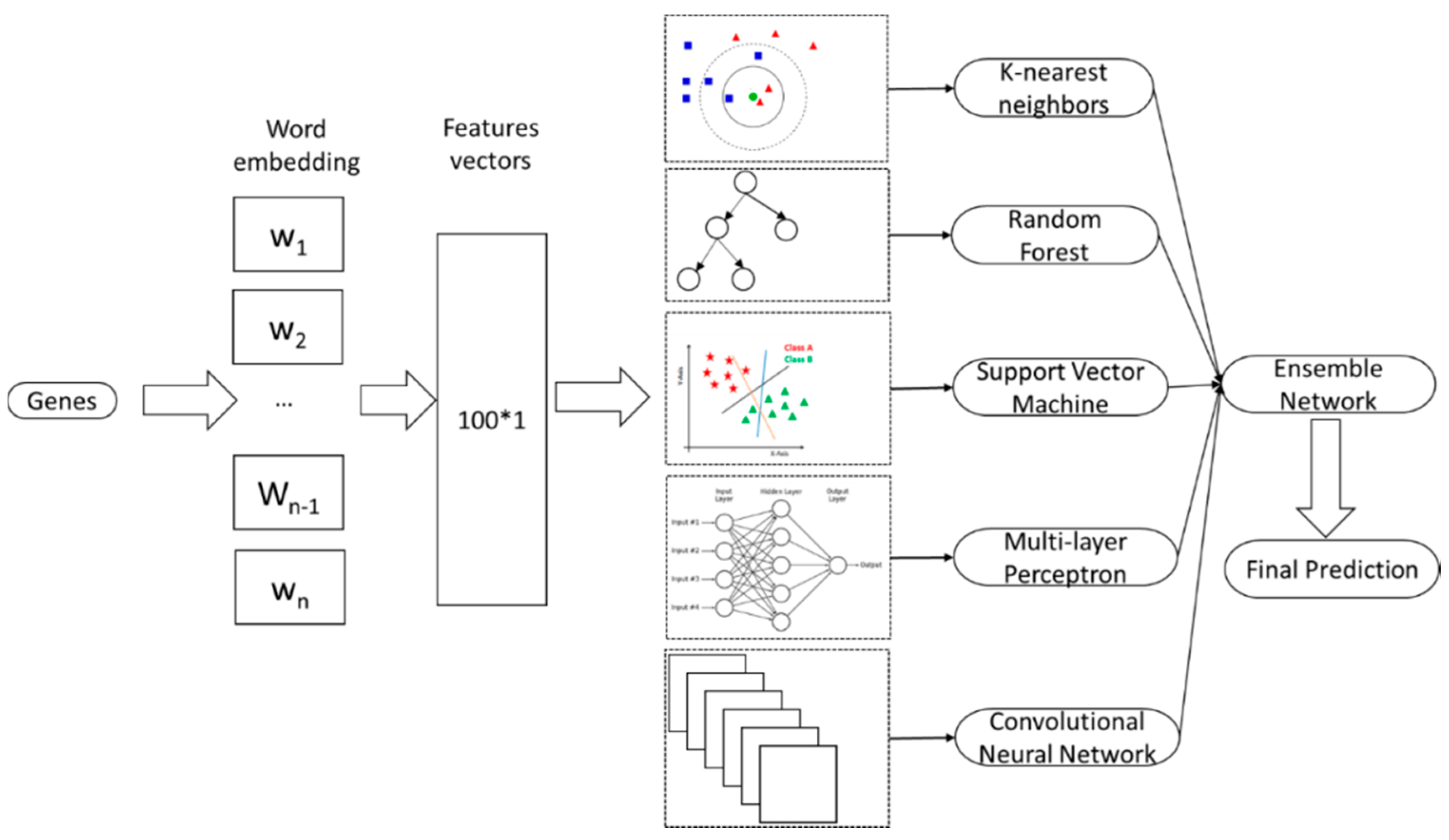
3.3. Ensemble Neural Network
3.3.1. k-Nearest Neighbors
3.3.2. Random Forest
3.3.3. Support Vector Machine
3.3.4. Multi-Layer Perceptron
3.3.5. Convolutional Neural Network
3.3.6. Ensemble Neural Network
3.4. Validation Method and Performance Metrics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- O’Neill, R.S.; Clark, D.V. The Drosophila melanogaster septin gene Sep2 has a redundant function with the retrogene Sep5 in imaginal cell proliferation but is essential for oogenesis. Genome 2013, 56, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Juhas, M.; Eberl, L.; Glass, J.I. Essence of life: Essential genes of minimal genomes. Trends Cell Biol. 2011, 21, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V. How many genes can make a cell: The minimal-gene-set concept. Annu. Rev. Genom. Hum. Genet. 2000, 1, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Juhas, M.; Reuß, D.R.; Zhu, B.; Commichau, F.M. Bacillus subtilis and Escherichia coli essential genes and minimal cell factories after one decade of genome engineering. Microbiology 2014, 160, 2341–2351. [Google Scholar] [CrossRef] [Green Version]
- Itaya, M. An estimation of minimal genome size required for life. FEBS Lett. 1995, 362, 257–260. [Google Scholar] [CrossRef] [Green Version]
- Dickerson, J.E.; Zhu, A.; Robertson, D.L.; Hentges, K.E. Defining the role of essential genes in human disease. PLoS ONE 2011, 6, e27368. [Google Scholar] [CrossRef] [Green Version]
- Giaever, G.; Chu, A.M.; Ni, L.; Connelly, C.; Riles, L.; Véronneau, S.; Dow, S.; Lucau-Danila, A.; Anderson, K.; André, B. Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002, 418, 387–391. [Google Scholar] [CrossRef]
- Roemer, T.; Jiang, B.; Davison, J.; Ketela, T.; Veillette, K.; Breton, A.; Tandia, F.; Linteau, A.; Sillaots, S.; Marta, C. Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol. Microbiol. 2003, 50, 167–181. [Google Scholar] [CrossRef]
- Veeranagouda, Y.; Husain, F.; Tenorio, E.L.; Wexler, H.M. Identification of genes required for the survival of B. fragilis using massive parallel sequencing of a saturated transposon mutant library. BMC Genom. 2014, 15, 429. [Google Scholar] [CrossRef] [Green Version]
- Cullen, L.M.; Arndt, G.M. Genome-wide screening for gene function using RNAi in mammalian cells. Immunol. Cell Biol. 2005, 83, 217–223. [Google Scholar] [CrossRef]
- Rancati, G.; Moffat, J.; Typas, A.; Pavelka, N. Emerging and evolving concepts in gene essentiality. Nat. Rev. Genet. 2018, 19, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Grazziotin, A.L.; Vidal, N.M.; Venancio, T.M. Uncovering major genomic features of essential genes in Bacteria and a methanogenic Archaea. FEBS J. 2015, 282, 3395–3411. [Google Scholar] [CrossRef] [PubMed]
- Mobegi, F.M.; Zomer, A.; De Jonge, M.I.; Van Hijum, S.A. Advances and perspectives in computational prediction of microbial gene essentiality. Brief. Funct. Genom. 2017, 16, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Acencio, M.L.; Lemke, N. Towards the prediction of essential genes by integration of network topology, cellular localization and biological process information. BMC Bioinform. 2009, 10, 290. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, A.M.; Snitkin, E.S.; Parker, S.C.; DeLisi, C.; Kasif, S. Towards the identification of essential genes using targeted genome sequencing and comparative analysis. BMC Genom. 2006, 7, 265. [Google Scholar] [CrossRef] [Green Version]
- Plaimas, K.; Eils, R.; König, R. Identifying essential genes in bacterial metabolic networks with machine learning methods. BMC Syst. Biol. 2010, 4, 56. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Ning, L.-W.; Ye, Y.-N.; Guo, F.-B. Geptop: A gene essentiality prediction tool for sequenced bacterial genomes based on orthology and phylogeny. PLoS ONE 2013, 8, e72343. [Google Scholar] [CrossRef]
- Song, K.; Tong, T.; Wu, F. Predicting essential genes in prokaryotic genomes using a linear method: ZUPLS. Integr. Biol. 2014, 6, 460–469. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Xu, Z.; Wu, W.; Zhao, L.; Li, X.; Liu, Y.; Tao, S. Training set selection for the prediction of essential genes. PLoS ONE 2014, 9, e86805. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Deng, L.; Su, S.; Zhang, M.; Lin, X.; Wei, L.; Minai, A.A.; Hassett, D.J.; Lu, L.J. Investigating the predictability of essential genes across distantly related organisms using an integrative approach. Nucleic Acids Res. 2010, 39, 795–807. [Google Scholar] [CrossRef]
- Deng, J. An Integrated Machine-Learning Model to Predict Prokaryotic Essential Genes. In Gene Essentiality: Methods and Protocols; Lu, L.J., Ed.; Springer: New York, NY, USA, 2015; pp. 137–151. [Google Scholar]
- Chen, Y.; Xu, D. Understanding protein dispensability through machine-learning analysis of high-throughput data. Bioinformatics 2005, 21, 575–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seringhaus, M.; Paccanaro, A.; Borneman, A.; Snyder, M.; Gerstein, M. Predicting essential genes in fungal genomes. Genome Res. 2006, 16, 1126–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Xu, Y.; Xu, J.; Ball, R.L.; Liang, H. Predicting the lethal phenotype of the knockout mouse by integrating comprehensive genomic data. Bioinformatics 2012, 28, 1246–1252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Acencio, M.L.; Lemke, N. Predicting essential genes and proteins based on machine learning and network topological features: A comprehensive review. Front. Physiol. 2016, 7, 75. [Google Scholar]
- Lloyd, J.P.; Seddon, A.E.; Moghe, G.D.; Simenc, M.C.; Shiu, S.-H. Characteristics of plant essential genes allow for within-and between-species prediction of lethal mutant phenotypes. Plant. Cell 2015, 27, 2133–2147. [Google Scholar] [CrossRef]
- Zhang, X.; Xiao, W.; Xiao, W. DeepHE: Accurately predicting human essential genes based on deep learning. PLoS Comput. Biol. 2020, 16, e1008229. [Google Scholar] [CrossRef]
- Hua, H.-L.; Zhang, F.-Z.; Labena, A.A.; Dong, C.; Jin, Y.-T.; Guo, F.-B. An approach for predicting essential genes using multiple homology mapping and machine learning algorithms. Biomed. Res. Int. 2016, 2016, 7639397. [Google Scholar] [CrossRef]
- Liu, X.; Wang, B.-J.; Xu, L.; Tang, H.-L.; Xu, G.-Q. Selection of key sequence-based features for prediction of essential genes in 31 diverse bacterial species. PLoS ONE 2017, 12, e0174638. [Google Scholar] [CrossRef] [Green Version]
- Acevedo-Rocha, C.G.; Fang, G.; Schmidt, M.; Ussery, D.W.; Danchin, A. From essential to persistent genes: A functional approach to constructing synthetic life. Trends Genet. 2013, 29, 273–279. [Google Scholar] [CrossRef] [Green Version]
- Le, N.Q.K.; Yapp, E.K.Y.; Nagasundaram, N.; Yeh, H.-Y. Classifying Promoters by Interpreting the Hidden Information of DNA Sequences via Deep Learning and Combination of Continuous FastText N-Grams. Front. Bioeng. Biotechnol. 2019, 7, 305. [Google Scholar] [CrossRef] [Green Version]
- Le, N.Q.K. iN6-methylat (5-step): Identifying DNA N6-methyladenine sites in rice genome using continuous bag of nucleobases via Chou’s 5-step rule. Mol. Genet. Genom. 2019, 294, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Do, D.T.; Le, N.Q.K. Using extreme gradient boosting to identify origin of replication in Saccharomyces cerevisiae via hybrid features. Genomics 2020, 112, 2445–2451. [Google Scholar] [CrossRef] [PubMed]
- Le, N.Q.K.; Huynh, T.-T.; Yapp, E.K.Y.; Yeh, H.-Y. Identification of clathrin proteins by incorporating hyperparameter optimization in deep learning and PSSM profiles. Comput. Methods Programs Biomed. 2019, 177, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, Y.; Liao, Q.; Liu, B. iEsGene-ZCPseKNC: Identify Essential Genes Based on Z Curve Pseudo $k$ -Tuple Nucleotide Composition. IEEE Access 2019, 7, 165241–165247. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Aromolaran, O.; Beder, T.; Oswald, M.; Oyelade, J.; Adebiyi, E.; Koenig, R. Essential gene prediction in Drosophila melanogaster using machine learning approaches based on sequence and functional features. Comput. Struct. Biotechnol. J. 2020, 18, 612–621. [Google Scholar] [CrossRef]
- Campos, T.L.; Korhonen, P.K.; Hofmann, A.; Gasser, R.B.; Young, N.D. Combined use of feature engineering and machine-learning to predict essential genes in Drosophila melanogaster. NAR Genom. Bioinform. 2020, 2, lqaa051. [Google Scholar] [CrossRef]
- Guo, F.-B.; Dong, C.; Hua, H.-L.; Liu, S.; Luo, H.; Zhang, H.-W.; Jin, Y.-T.; Zhang, K.-Y. Accurate prediction of human essential genes using only nucleotide composition and association information. Bioinformatics 2017, 33, 1758–1764. [Google Scholar] [CrossRef] [Green Version]
- Nigatu, D.; Sobetzko, P.; Yousef, M.; Henkel, W. Sequence-based information-theoretic features for gene essentiality prediction. BMC Bioinform. 2017, 18, 473. [Google Scholar] [CrossRef] [Green Version]
- Tian, D.; Wenlock, S.; Kabir, M.; Tzotzos, G.; Doig, A.J.; Hentges, K.E. Identifying mouse developmental essential genes using machine learning. Dis. Models Mech. 2018, 11, dmm034546. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Guo, Z.; Liu, X. Prediction of essential genes in prokaryote based on artificial neural network. Genes Genom. 2020, 42, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhang, F.Z.; Xue, K.; Gao, Y.Z.; Guo, F.B. Identifying Bacterial Essential Genes Based on a Feature-Integrated Method. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 1274–1279. [Google Scholar] [CrossRef] [PubMed]
- Azhagesan, K.; Ravindran, B.; Raman, K. Network-based features enable prediction of essential genes across diverse organisms. PLoS ONE 2018, 13, e0208722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, M.A.; Lonardi, S. DeeplyEssential: A deep neural network for predicting essential genes in microbes. BMC Bioinform. 2020, 21, 367. [Google Scholar] [CrossRef]
- Zhang, R.; Ou, H.Y.; Zhang, C.T. DEG: A database of essential genes. Nucleic Acids Res. 2004, 32, D271–D272. [Google Scholar] [CrossRef]
- Bojanowski, P.; Grave, E.; Joulin, A.; Mikolov, T. Enriching word vectors with subword information. Trans. Assoc. Comput. Linguist. 2017, 5, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Do, D.T.; Le, T.Q.T.; Le, N.Q.K. Using deep neural networks and biological subwords to detect protein S-sulfenylation sites. Brief. Bioinform. 2020, bbaa128. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, C.J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. (TIST) 2011, 2, 1–27. [Google Scholar] [CrossRef]
- Cybenko, G. Approximation by superpositions of a sigmoidal function. Math. Control. Signals Syst. 1989, 2, 303–314. [Google Scholar] [CrossRef]
- Nair, V.; Hinton, G.E. Rectified linear units improve restricted boltzmann machines. In Proceedings of the 27th International Conference on International Conference on Machine Learning, Omnipress, Haifa, Israel, 21–24 June 2010; pp. 807–814. [Google Scholar]
- Le, N.Q.K.; Yapp, E.K.Y.; Nagasundaram, N.; Chua, M.C.H.; Yeh, H.-Y. Computational identification of vesicular transport proteins from sequences using deep gated recurrent units architecture. Comput. Struct. Biotechnol. J. 2019, 17, 1245–1254. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Classifier | Optimal Parameters |
|---|---|
| kNN | k = 10 |
| RF | n_estimators = 500, n_features = 20 |
| SVM | c = 23768, g = 0.001953125 |
| MLP | 100–50 nodes, dropout = 0.5, optimizer = adam, learning rate = 0.001 |
| CNN | n_filters = 64, dropout = 0.1, optimizer = adadelta |
| Classifier | Sens (%) | Spec (%) | Acc (%) | MCC | AUC | Time (s) |
|---|---|---|---|---|---|---|
| kNN | 43.5 | 87.6 | 73.2 | 0.348 | 0.747 | 0.27 |
| RF | 46.9 | 86.6 | 73.6 | 0.367 | 0.762 | 12.85 |
| SVM | 35.9 | 92.3 | 74 | 0.353 | 0.775 | 3.37 |
| MLP | 43.5 | 89.9 | 74.8 | 0.385 | 0.775 | 94.32 |
| CNN | 42.3 | 90.4 | 74.7 | 0.381 | 0.775 | 105.18 |
| Ensemble | 50.5 | 90.2 | 77.3 | 0.452 | 0.814 | 208.12 |
| Features | Sens (%) | Spec (%) | Acc (%) | MCC | AUC |
|---|---|---|---|---|---|
| k-mer | 35.9 | 90.2 | 72.4 | 0.316 | 0.698 |
| PseDNC | 36.5 | 91 | 73.2 | 0.337 | 0.637 |
| PseTNC | 31.7 | 93.4 | 73.3 | 0.331 | 0.625 |
| PCPseDNC | 37.6 | 89.5 | 72.6 | 0.322 | 0.704 |
| PCPseTNC | 33.4 | 93 | 73.5 | 0.341 | 0.72 |
| fastText | 50.5 | 90.2 | 77.3 | 0.452 | 0.814 |
| Predictors | Feature | Sens | Spec | Acc | MCC | |
|---|---|---|---|---|---|---|
| Original | Aromolaran [37] | Auto covariance, pseudo nucleotide composition, k-mer | 40.8 | 90.7 | 74.4 | 0.371 |
| Campos et al. [38] | Nucleotide composition, correlation features | 38.5 | 93 | 75.2 | 0.39 | |
| Liu et al. [29] | Sequence-based features and LASSO | 41.3 | 89.7 | 73.8 | 0.361 | |
| Tian et al. [41] | Hybrid features | 36.5 | 93.9 | 75.2 | 0.389 | |
| Deng et al. [20] | Intrinsic and context-dependent genomic features | 34 | 93.5 | 74.1 | 0.355 | |
| Xu et al. [42] | Hybrid features and WPCA | 42.7 | 86.9 | 72.6 | 0.331 | |
| Nigatu et al. [40] | Information theoretic features | 38.8 | 92.1 | 74.8 | 0.377 | |
| Lin et al. [43] | Hybrid features | 35.6 | 92 | 73.5 | 0.345 | |
| Pheg [39] | Nucleotide composition | 35 | 94.4 | 75.1 | 0.383 | |
| iEsGene-ZCPseKNC [35] | Nucleotide composition | 44.6 | 89 | 74.5 | 0.38 | |
| Ours | NLP-based features | 50.5 | 90.2 | 77.3 | 0.452 | |
| SMOTE | Aromolaran [37] | Auto covariance, pseudo nucleotide composition, k-mer | 55.3 | 82.2 | 73.5 | 0.384 |
| Campos et al. [38] | Nucleotide composition, correlation features | 52.9 | 85 | 74.5 | 0.399 | |
| Liu et al. [29] | Sequence-based features and LASSO | 45.2 | 89.2 | 74.8 | 0.389 | |
| Tian et al. [41] | Hybrid features | 50.5 | 85.5 | 74.1 | 0.384 | |
| Deng et al. [20] | Intrinsic and context-dependent genomic features | 45.2 | 85.9 | 72.6 | 0.341 | |
| Xu et al. [42] | Hybrid features and WPCA | 54.8 | 82.7 | 73.6 | 0.386 | |
| Nigatu et al. [40] | Information theoretic features | 46.6 | 86.9 | 73.8 | 0.368 | |
| Lin et al. [43] | Hybrid features | 44.2 | 87.8 | 73.5 | 0.359 | |
| Pheg [39] | Nucleotide composition | 53.8 | 86.4 | 75.7 | 0.426 | |
| iEsGene-ZCPseKNC [35] | Nucleotide composition | 63.7 | 77 | 72.6 | 0.396 | |
| Ours | NLP-based features | 60.2 | 84.6 | 76.3 | 0.449 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, N.Q.K.; Do, D.T.; Hung, T.N.K.; Lam, L.H.T.; Huynh, T.-T.; Nguyen, N.T.K. A Computational Framework Based on Ensemble Deep Neural Networks for Essential Genes Identification. Int. J. Mol. Sci. 2020, 21, 9070. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239070
Le NQK, Do DT, Hung TNK, Lam LHT, Huynh T-T, Nguyen NTK. A Computational Framework Based on Ensemble Deep Neural Networks for Essential Genes Identification. International Journal of Molecular Sciences. 2020; 21(23):9070. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239070
Chicago/Turabian StyleLe, Nguyen Quoc Khanh, Duyen Thi Do, Truong Nguyen Khanh Hung, Luu Ho Thanh Lam, Tuan-Tu Huynh, and Ngan Thi Kim Nguyen. 2020. "A Computational Framework Based on Ensemble Deep Neural Networks for Essential Genes Identification" International Journal of Molecular Sciences 21, no. 23: 9070. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239070