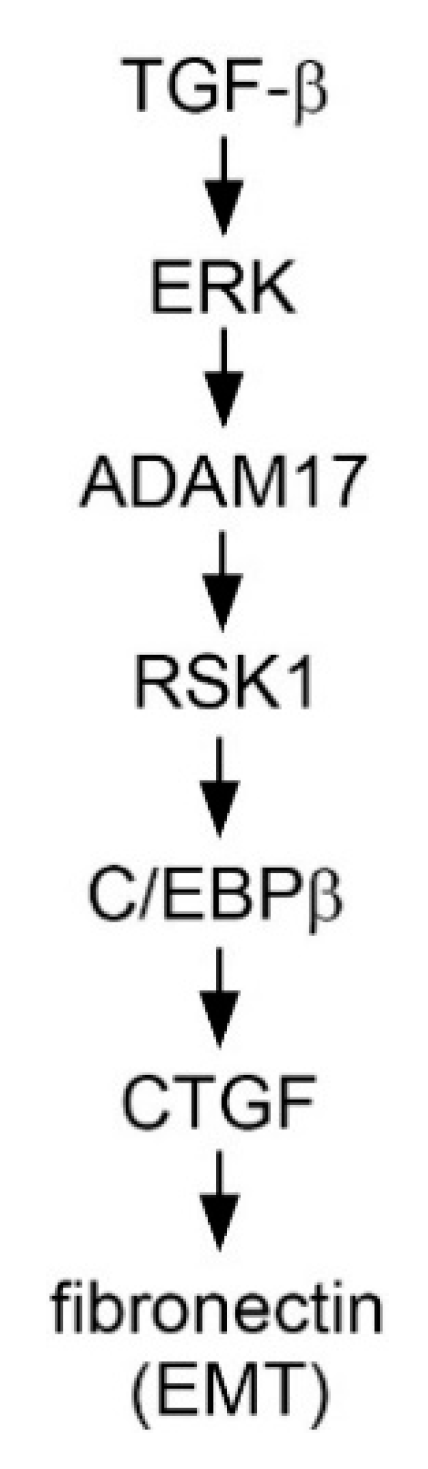
TGF-β Induced CTGF Expression in Human Lung Epithelial Cells through ERK, ADAM17, RSK1, and C/EBPβ Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
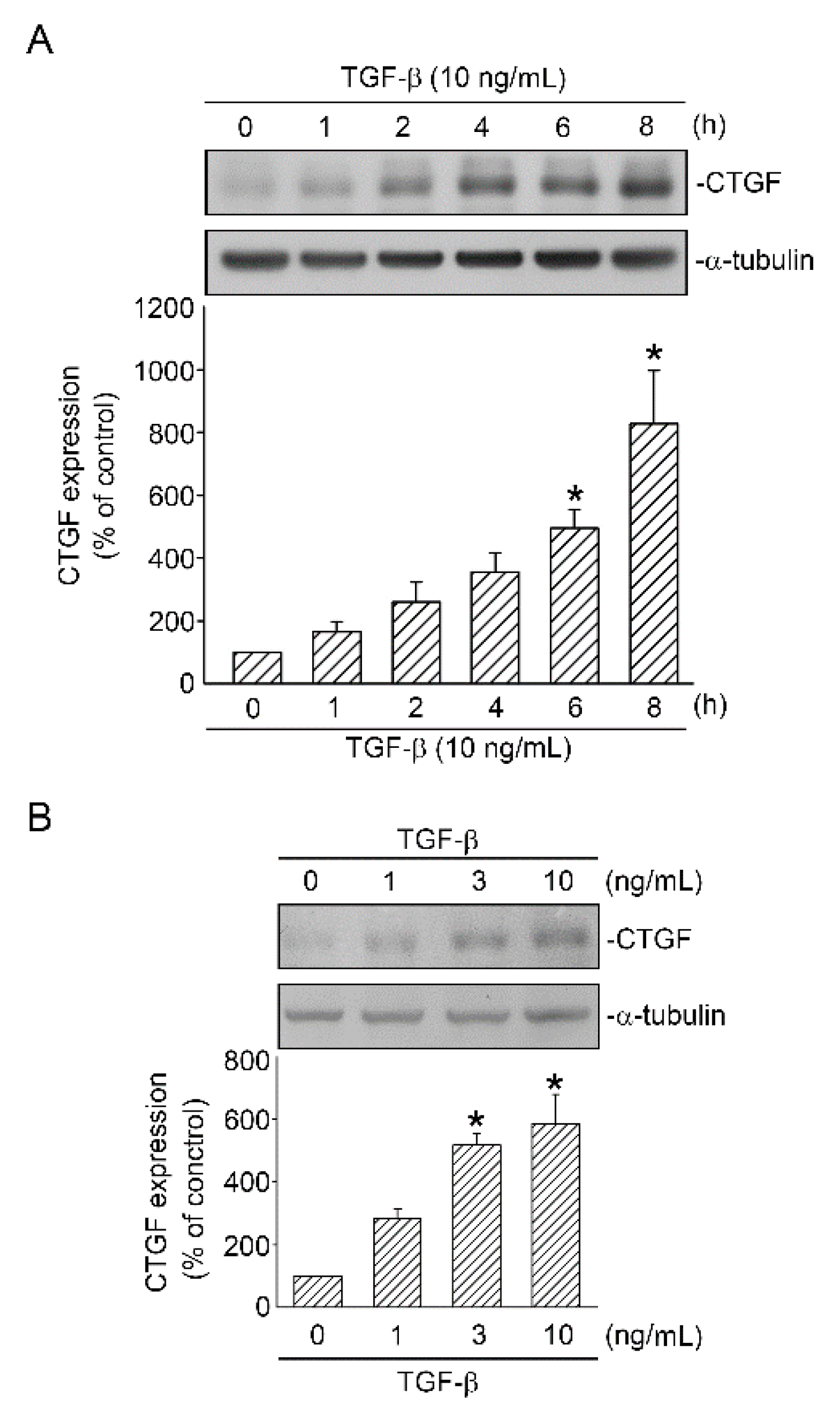
2.1. TGF-β Induced CTGF Expression in Human Lung Epithelial Cells
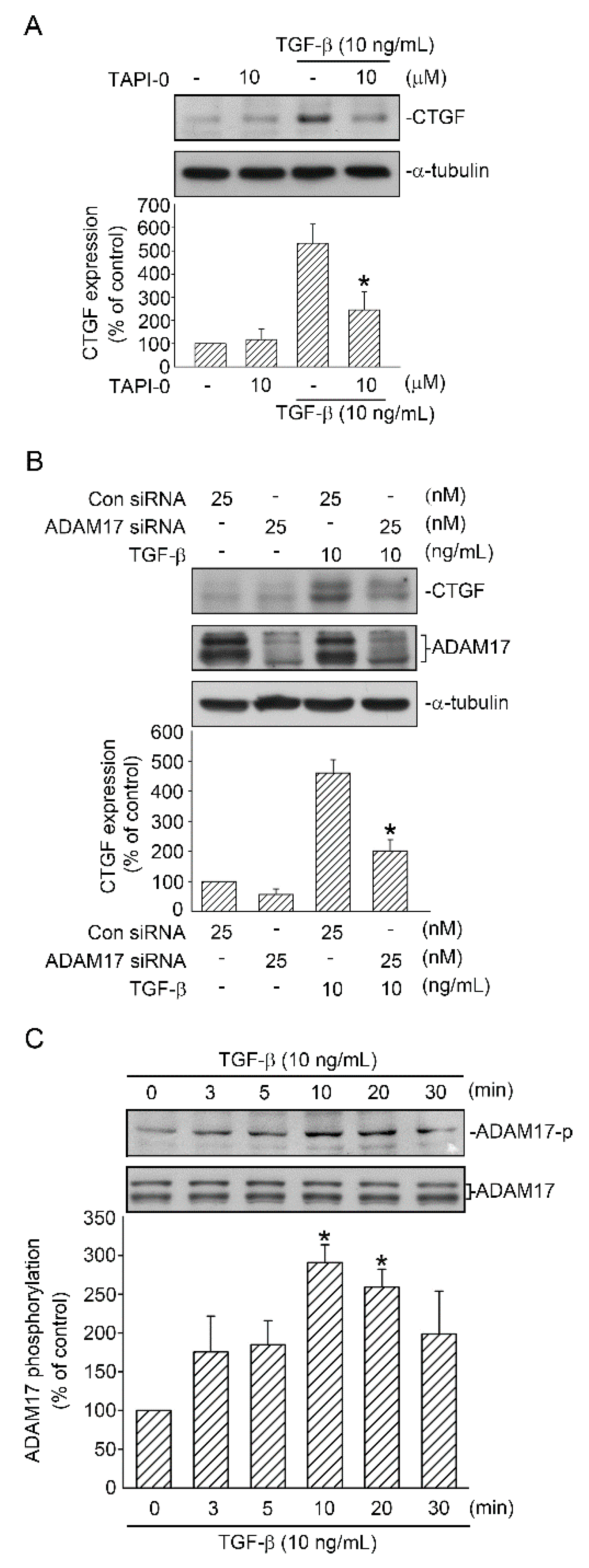
2.2. Involvement of ADAM17 Activation in TGF-β Induced CTGF Expression in A549 Cells
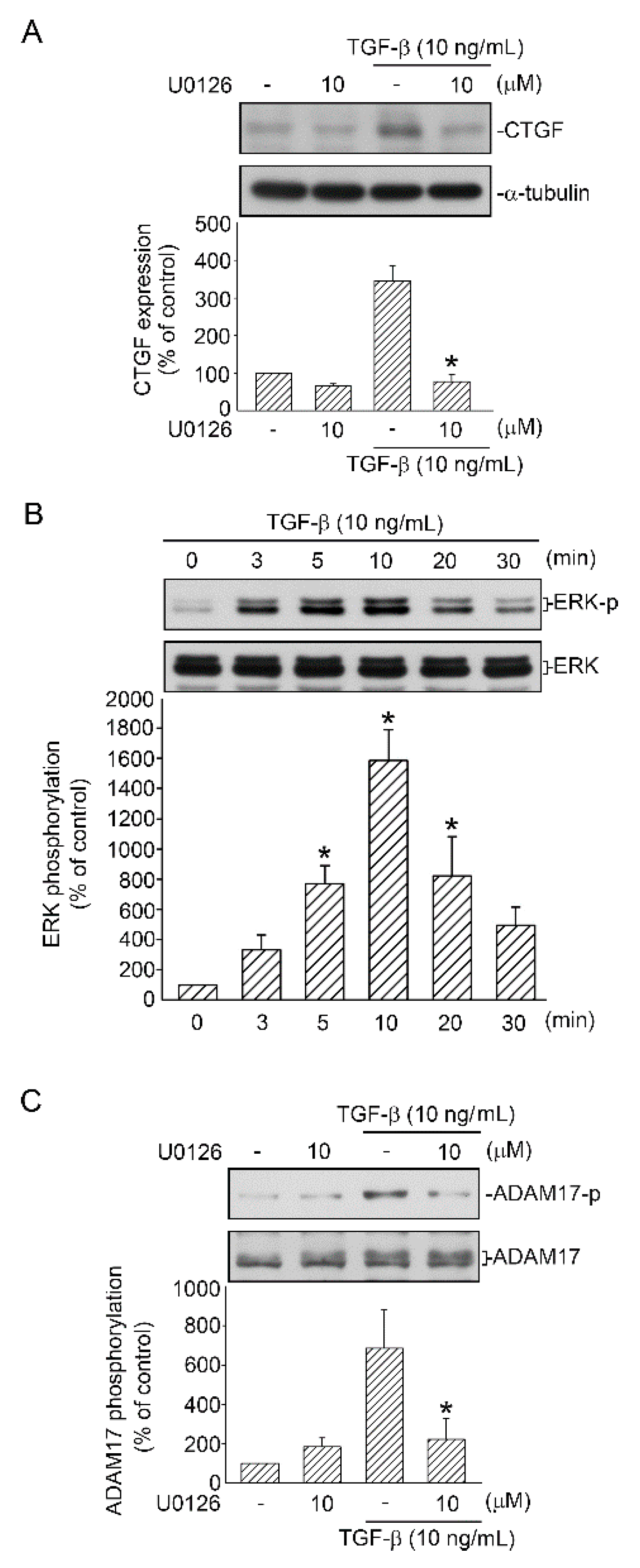
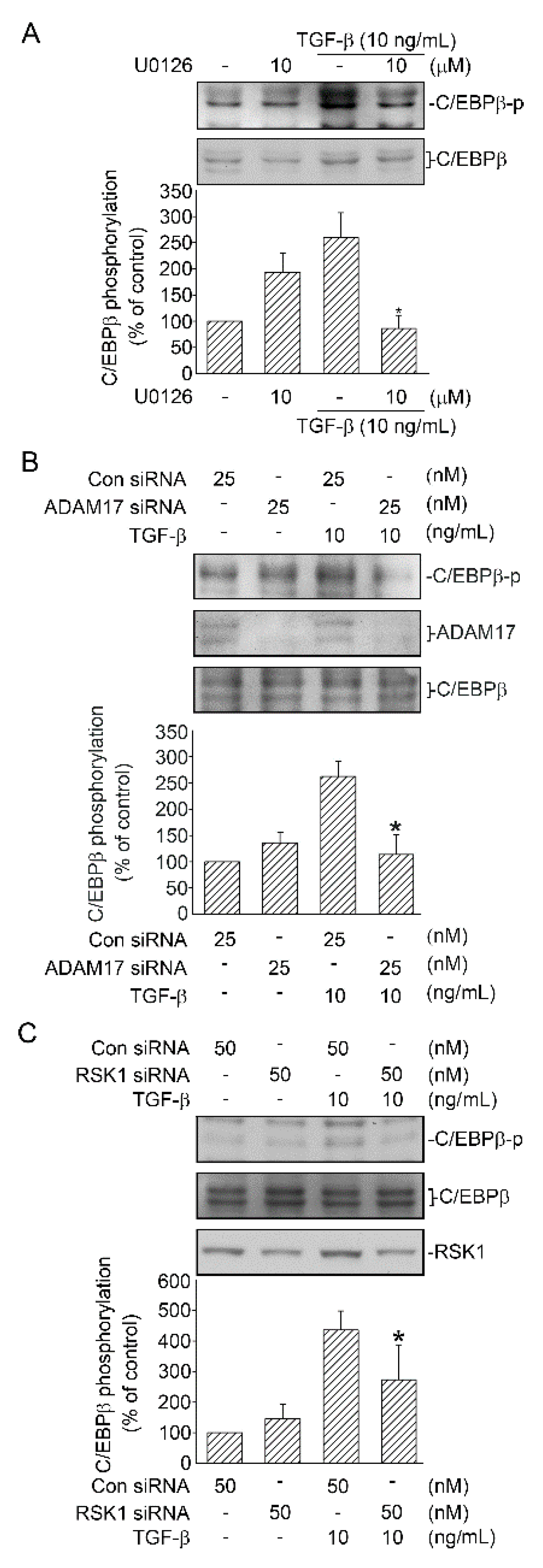
2.3. Mediating Effects of ERK on TGF-β-Induced CTGF Expression and ADAM17 Phosphorylation
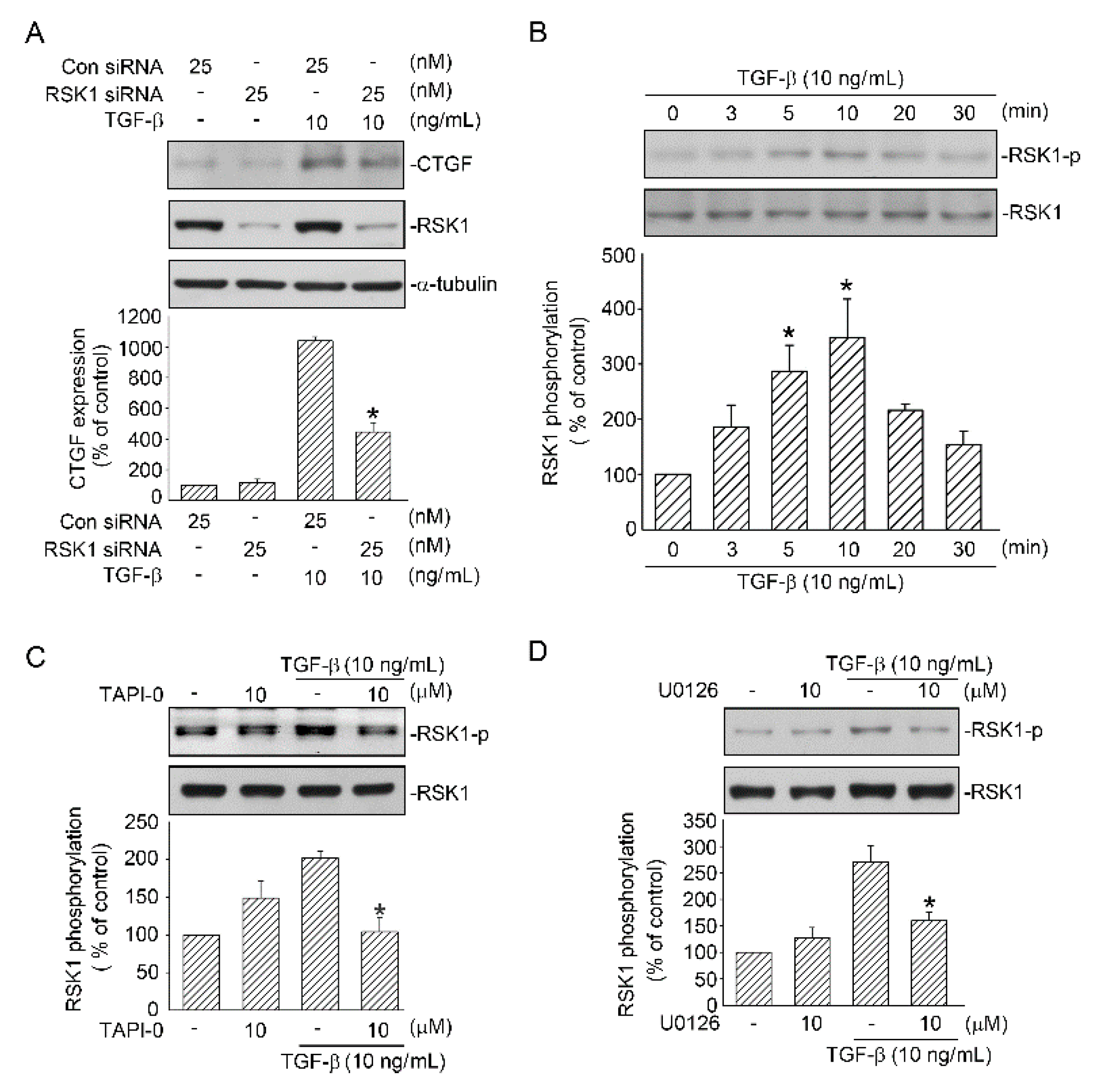
2.4. RSK1-Mediated TGF-β-Induced CTGF Expression
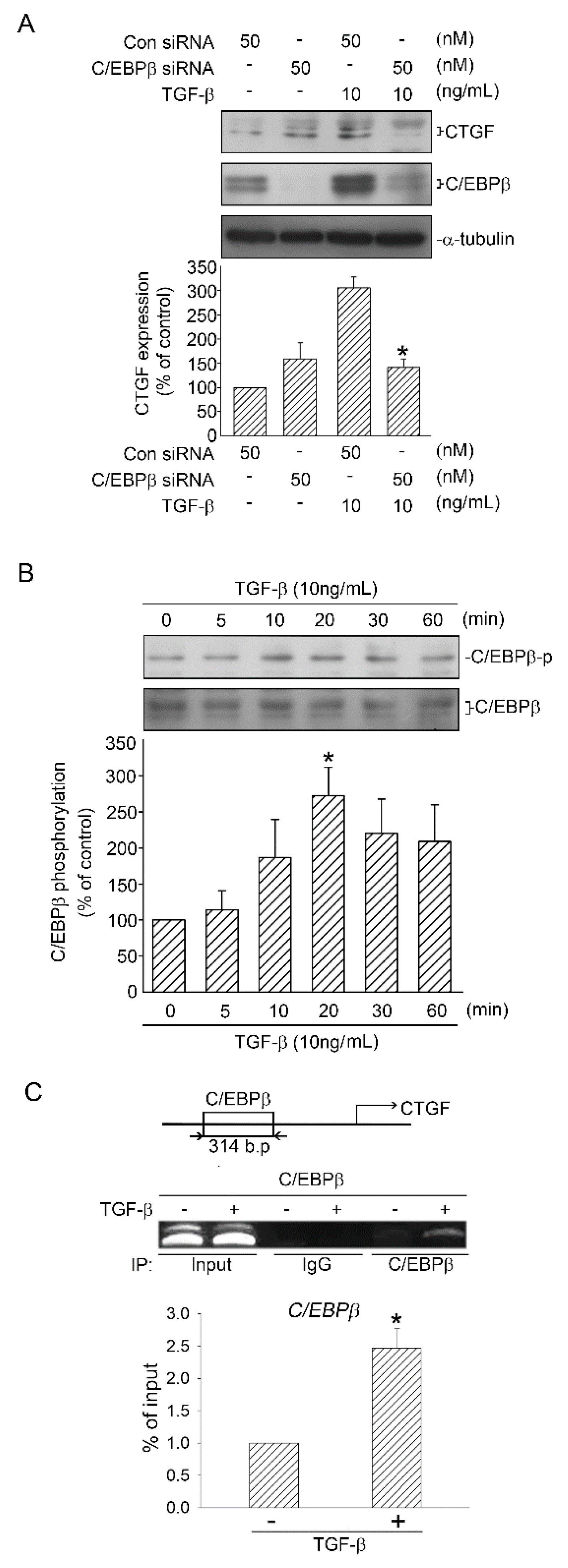
2.5. Role of C/EBPβ in TGF-β-Induced CTGF
2.6. Activation of C/EBPβ by TGF-β through ERK, ADAM17, and RSK1
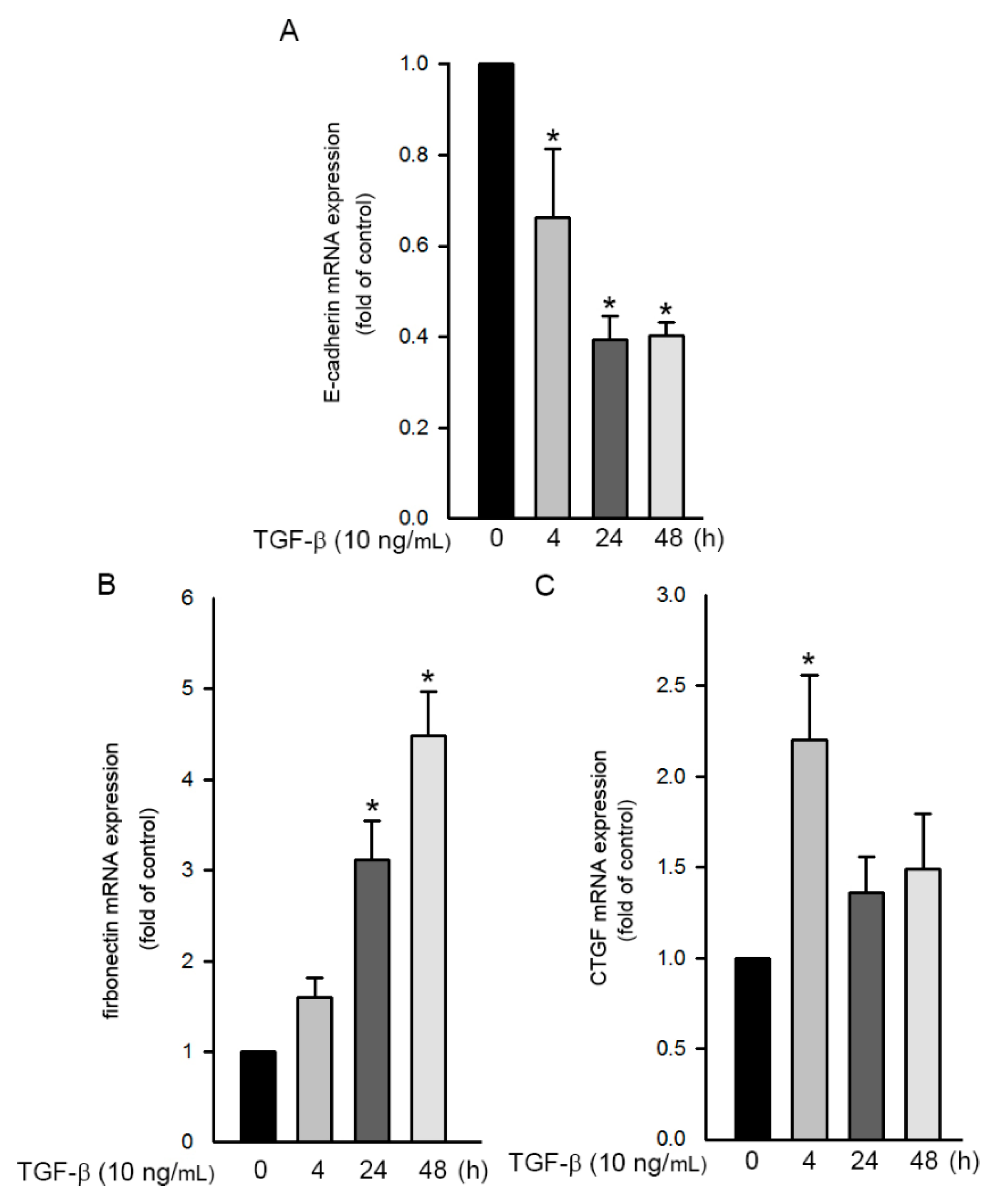
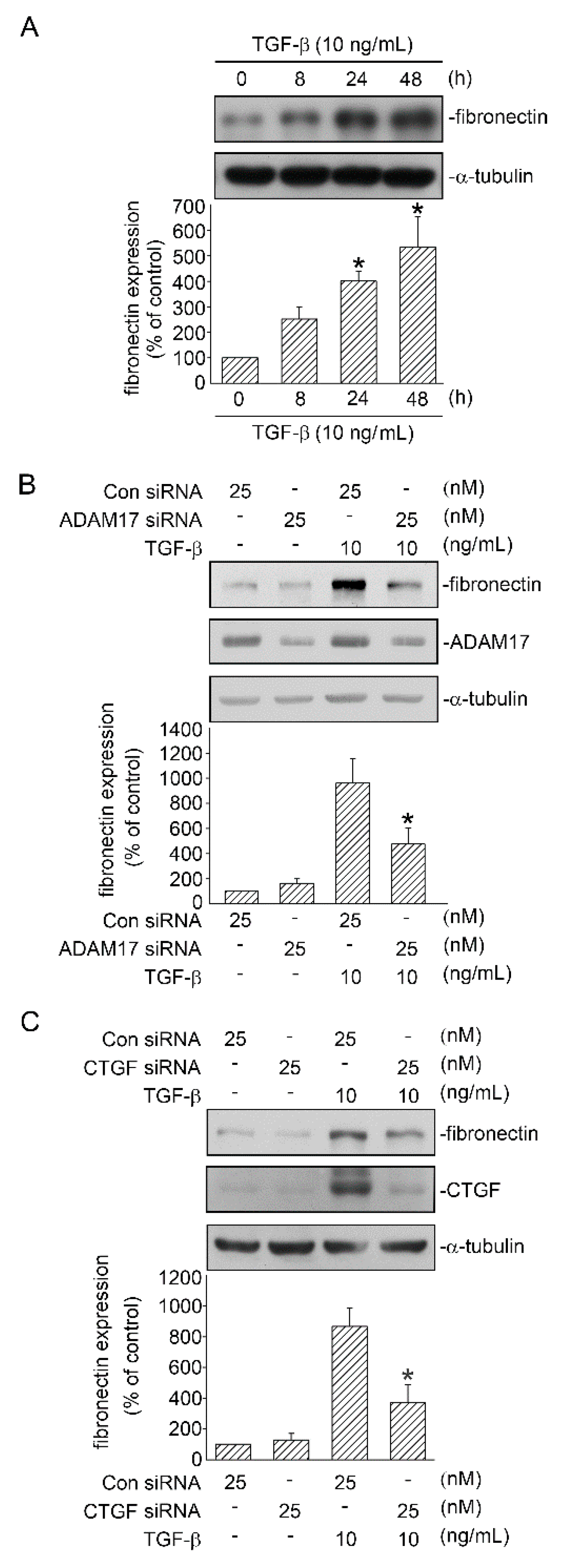
2.7. TGF-β-Induced EMT Process in Human Lung Epithelial Cells
2.8. Involvement of ADAM17 and CTGF in TGF-β-Induced FN Expression
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. siRNAs Transfection
4.4. Western Blotting
4.5. Chromatin Immunoprecipitation Assay
4.6. RNA Isolation and RT-PCR
4.7. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAM17 | A disintegrin and metalloproteinase 17 |
| ANOVA | Analysis of variance |
| C/EBPβ | CCAAT/enhancer-binding protein β |
| ChIP | Chromatin immunoprecipitation |
| CTGF | Connective tissue growth factor |
| ECM | Extracellular matrix |
| EMT | Epithelial–mesenchymal transition |
| ERK | Extracellular signal-regulated kinase |
| FN | Fibronectin |
| IPF | Idiopathic pulmonary fibrosis |
| RSK | Ribosomal S6 kinases 1 |
| siRNA | Small interfering RNA |
| TGF-β | Transforming growth factor-β |
References
- Rajasekaran, S.; Rajaguru, P.; Gandhi, P.S.S. MicroRNAs as potential targets for progressive pulmonary fibrosis. Front Pharmacol. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giarnieri, E.; Bellipanni, G.; Macaluso, M.; Mancini, R.; Holstein, A.C.; Milanese, C.; Giovagnoli, M.R.; Giordano, A.; Russo, G. Review: Cell Dynamics in Malignant Pleural Effusions. J. Cell. Physiol. 2015, 230, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Liu, Y.; Zhu, B.; Ding, K.; Yao, T.P.; Chen, F.; Zhan, L.; Xu, P.; Ehrlich, M.; Liang, T.; et al. Loss of alpha-Tubulin Acetylation Is Associated with TGF-beta-induced Epithelial-Mesenchymal Transition. J. Biol. Chem. 2016, 291, 5396–5405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Lu, W.; Yu, D.; Sun, C.; Guo, J.; Li, Z.; Guan, F. Quantitative Analysis of Differential Proteome Expression in Epithelial-to-Mesenchymal Transition of Bladder Epithelial Cells Using SILAC Method. Molecules 2016, 21, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitoiu, D.; Etheridge, S.L.; Kelsell, D.P. Insights into Desmosome Biology from Inherited Human Skin Disease and Cardiocutaneous Syndromes. Cell Commun. Adhes. 2014, 21, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Jiang, J.; Lu, K.; Chen, Q.; Tao, D.; Chen, Z. Therapeutic potential of ADAM17 modulation in gastric cancer through regulation of the EGFR and TNF-alpha signalling pathways. Mol. Cell. Biochem. 2017, 426, 17–26. [Google Scholar] [CrossRef]
- Malapeira, J.; Esselens, C.; Bech-Serra, J.J.; Canals, F.; Arribas, J. ADAM17 (TACE) regulates TGFbeta signaling through the cleavage of vasorin. Oncogene 2011, 30, 1912–1922. [Google Scholar] [CrossRef] [Green Version]
- Uhal, B.D.; Nguyen, H.; Dang, M.; Gopallawa, I.; Jiang, J.; Dang, V.; Ono, S.; Morimoto, K. Abrogation of ER stress-induced apoptosis of alveolar epithelial cells by angiotensin 1-7. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L33–L41. [Google Scholar] [CrossRef] [Green Version]
- Blom, I.E.; Goldschmeding, R.; Leask, A. Gene regulation of connective tissue growth factor: New targets for antifibrotic therapy? Matrix Biol. 2002, 21, 473–482. [Google Scholar] [CrossRef]
- Moussad, E.E.; Brigstock, D.R. Connective tissue growth factor: What’s in a name? Mol. Genet. Metab. 2000, 71, 276–292. [Google Scholar] [CrossRef]
- Ihn, H. Autocrine TGF-beta signaling in the pathogenesis of systemic sclerosis. J. Dermatol. Sci. 2008, 49, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Kawara, S.; Shinozaki, M.; Hayashi, N.; Kakinuma, T.; Igarashi, A.; Takigawa, M.; Nakanishi, T.; Takehara, K. Role and interaction of connective tissue growth factor with transforming growth factor-beta in persistent fibrosis: A mouse fibrosis model. J. Cell. Physiol. 1999, 181, 153–159. [Google Scholar] [CrossRef]
- Sakai, N.; Tager, A.M. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 911–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafieian, M.; Chen, S.; Wu, S. Integrin-linked kinase mediates CTGF-induced epithelial to mesenchymal transition in alveolar type II epithelial cells. Pediatr. Res. 2015, 77, 520–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.H.; Nai, P.L.; Bien, M.Y.; Yu, C.C.; Chen, B.C. Thrombin-induced CCAAT/enhancer-binding protein beta activation and IL-8/CXCL8 expression via MEKK1, ERK, and p90 ribosomal S6 kinase 1 in lung epithelial cells. J. Immunol. 2014, 192, 338–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.G.; Lee, S.J. PI3K, RSK, and mTOR signal networks for the GST gene regulation. Toxicol Sci 2007, 96, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Chiou, M.J.; Chao, T.T.; Wu, J.L.; Kuo, C.M.; Chen, J.Y. The physiological role of CTGF/CCN2 in zebrafish notochond development and biological analysis of the proximal promoter region. Biochem. Biophys. Res. Commun. 2006, 349, 750–758. [Google Scholar] [CrossRef]
- Weng, L.; Wang, W.; Su, X.; Huang, Y.; Su, L.; Liu, M.; Sun, Y.; Yang, B.; Zhou, H. The Effect of cAMP-PKA Activation on TGF-beta1-Induced Profibrotic Signaling. Cell. Physiol. Biochem. 2015, 36, 1911–1927. [Google Scholar] [CrossRef]
- Iyer, S.N.; Gurujeyalakshmi, G.; Giri, S.N. Effects of pirfenidone on transforming growth factor-beta gene expression at the transcriptional level in bleomycin hamster model of lung fibrosis. J. Pharmacol. Exp. Ther. 1999, 291, 367–373. [Google Scholar]
- Phanish, M.K.; Winn, S.K.; Dockrell, M.E.C. Connective Tissue Growth Factor-(CTGF, CCN2)—A Marker, Mediator and Therapeutic Target for Renal Fibrosis. Nephron.. Exp. Nephrol. 2010, 114, E83–E92. [Google Scholar] [CrossRef]
- Xu, P.; Liu, J.; Sakaki-Yumoto, M.; Derynck, R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci. Signal 2012, 5, ra34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.H.; Chang, M.C.; Wu, I.H.; Huang, G.F.; Huang, W.L.; Wang, Y.L.; Lee, S.Y.; Yeh, C.Y.; Guo, M.K.; Chan, C.P.; et al. Role of ALK5/Smad2/3 and MEK1/ERK Signaling in Transforming Growth Factor Beta 1-modulated Growth, Collagen Turnover, and Differentiation of Stem Cells from Apical Papilla of Human Tooth. J. Endod. 2015, 41, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.; Pan, T.; Yang, H.; Wang, H.; Wang, Y.J.E. Cadmium induces A549 cell migration and invasion by activating ERK. Exp. Ther. Med. 2019, 18, 1793–1799. [Google Scholar] [PubMed]
- Bell, H.L.; Gooz, M. ADAM-17 Is Activated by the Mitogenic Protein Kinase ERK in a Model of Kidney Fibrosis. Am. J. Med. Sci. 2010, 339, 105–107. [Google Scholar]
- Romeo, Y.; Zhang, X.; Roux, P.P. Regulation and function of the RSK family of protein kinases. Biochem. J. 2012, 441, 553–569. [Google Scholar] [CrossRef] [Green Version]
- Pruessmeyer, J.; Ludwig, A. The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Semin. Cell Dev. Biol. 2009, 20, 164–174. [Google Scholar] [CrossRef]
- Saftig, P.; Reiss, K. The “A Disintegrin And Metalloproteases” ADAM10 and ADAM17: Novel drug targets with therapeutic potential? Eur. J. Cell Biol. 2011, 90, 527–535. [Google Scholar] [CrossRef]
- Kreuter, M.; Bonella, F.; Wijsenbeek, M.; Maher, T.M.; Spagnolo, P. Pharmacological Treatment of Idiopathic Pulmonary Fibrosis: Current Approaches, Unsolved Issues, and Future Perspectives. Biomed. Res. Int. 2015, 2015, 329481. [Google Scholar] [CrossRef] [Green Version]
- Ohkouchi, S.; Ono, M.; Kobayashi, M.; Hirano, T.; Tojo, Y.; Hisata, S.; Ichinose, M.; Irokawa, T.; Ogawa, H.; Kurosawa, H. Myriad Functions of Stanniocalcin-1 (STC1) Cover Multiple Therapeutic Targets in the Complicated Pathogenesis of Idiopathic Pulmonary Fibrosis (IPF). Clin. Med. Insights Circ. Respir. Pulm. Med. 2015, 9, 91–96. [Google Scholar] [CrossRef]
- Chen, Y.L.; Zhang, X.; Bai, J.; Gai, L.; Ye, X.L.; Zhang, L.; Xu, Q.; Zhang, Y.X.; Xu, L.; Li, H.P.; et al. Sorafenib ameliorates bleomycin-induced pulmonary fibrosis: Potential roles in the inhibition of epithelial-mesenchymal transition and fibroblast activation. Cell Death Dis. 2013, 4, e665. [Google Scholar] [CrossRef] [Green Version]
- Sonnylal, S.; Shi-Wen, X.; Leoni, P.; Naff, K.; Van Pelt, C.S.; Nakamura, H.; Leask, A.; Abraham, D.; Bou-Gharios, G.; de Crombrugghe, B. Selective expression of connective tissue growth factor in fibroblasts in vivo promotes systemic tissue fibrosis. Arthritis Rheum. 2010, 62, 1523–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnylal, S.; Xu, S.; Jones, H.; Tam, A.; Sreeram, V.R.; Ponticos, M.; Norman, J.; Agrawal, P.; Abraham, D.; de Crombrugghe, B. Connective tissue growth factor causes EMT-like cell fate changes in vivo and in vitro. J. Cell Sci. 2013, 126, 2164–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, A.; Okura, T.; Hamada, C.; Miyoshi, S.; Katayama, H.; Higaki, J.; Ito, R. Cell stress induces upregulation of osteopontin via the ERK pathway in type II alveolar epithelial cells. PLoS ONE 2014, 9, e100106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe-Takano, H.; Takano, K.; Hatano, M.; Tokuhisa, T.; Endo, T. DA-Raf-Mediated Suppression of the Ras--ERK Pathway Is Essential for TGF-beta1-Induced Epithelial-Mesenchymal Transition in Alveolar Epithelial Type 2 Cells. PLoS ONE 2015, 10, e0127888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Wang, Y.; Xue, J.; Ma, Q.; Zhang, J.; Chen, Y.F.; Shang, Z.Z.; Li, Q.Q.; Zhang, S.L.; Zhao, L. Effect of Corilagin on the miR-21/smad7/ERK signaling pathway in a schistosomiasis-induced hepatic fibrosis mouse model. Parasitol. Int. 2016, 65, 308–315. [Google Scholar] [CrossRef]
- Gogl, G.; Alexa, A.; Kiss, B.; Katona, G.; Kovacs, M.; Bodor, A.; Remenyi, A.; Nyitray, L. Structural Basis of Ribosomal S6 Kinase 1 (RSK1) Inhibition by S100B Protein: Modulation of the Extracellular Signal-Regulated Kinase (ERK) Signaling Cascade in a Calcium-Dependent Way. J. Biol. Chem. 2016, 291, 11–27. [Google Scholar] [CrossRef] [Green Version]
- Soond, S.M.; Everson, B.; Riches, D.W.; Murphy, G. ERK-mediated phosphorylation of Thr735 in TNFalpha-converting enzyme and its potential role in TACE protein trafficking. J. Cell Sci. 2005, 118, 2371–2380. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.H.; Yu, M.C.; Tung, W.H.; Chen, T.T.; Yu, C.C.; Weng, C.M.; Tsai, Y.J.; Bai, K.J.; Hong, C.Y.; Chien, M.H.; et al. Connective tissue growth factor induces collagen I expression in human lung fibroblasts through the Rac1/MLK3/JNK/AP-1 pathway. Biochim. Biophys. Acta 2013, 1833, 2823–2833. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ou, S.-C.; Bai, K.-J.; Cheng, W.-H.; Chen, J.-Y.; Lin, C.-H.; Wen, H.-C.; Chen, B.-C. TGF-β Induced CTGF Expression in Human Lung Epithelial Cells through ERK, ADAM17, RSK1, and C/EBPβ Pathways. Int. J. Mol. Sci. 2020, 21, 9084. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239084
Ou S-C, Bai K-J, Cheng W-H, Chen J-Y, Lin C-H, Wen H-C, Chen B-C. TGF-β Induced CTGF Expression in Human Lung Epithelial Cells through ERK, ADAM17, RSK1, and C/EBPβ Pathways. International Journal of Molecular Sciences. 2020; 21(23):9084. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239084
Chicago/Turabian StyleOu, Shu-Ching, Kuan-Jen Bai, Wun-Hao Cheng, Jing-Yun Chen, Chien-Huang Lin, Heng-Ching Wen, and Bing-Chang Chen. 2020. "TGF-β Induced CTGF Expression in Human Lung Epithelial Cells through ERK, ADAM17, RSK1, and C/EBPβ Pathways" International Journal of Molecular Sciences 21, no. 23: 9084. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21239084