Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction



Division of Nephrology, Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(24), 9456; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249456
Submission received: 18 November 2020
/
Revised: 6 December 2020
/
Accepted: 8 December 2020
/
Published: 11 December 2020
(This article belongs to the Special Issue Molecular Mechanisms Involved in Diabetic Nephropathy)
{kind=link}
{kind=link}
Abstract
:Diabetic kidney disease (DKD) is the leading cause of end-stage renal disease (ESRD), with prevalence increasing at an alarming rate worldwide and today, there are no known cures. The pathogenesis of DKD is complex, influenced by genetics and the environment. However, the underlying molecular mechanisms that contribute to DKD risk in about one-third of diabetics are still poorly understood. The early stage of DKD is characterized by glomerular hyperfiltration, hypertrophy, podocyte injury and depletion. Recent evidence of glomerular endothelial cell injury at the early stage of DKD has been suggested to be critical in the pathological process and has highlighted the importance of glomerular intercellular crosstalk. A potential mechanism may include reactive oxygen species (ROS), which play a direct role in diabetes and its complications. In this review, we discuss different cellular sources of ROS in diabetes and a new emerging paradigm of endothelial cell dysfunction as a key event in the pathogenesis of DKD.
1. Introduction
Diabetes is the leading primary cause of end-stage renal disease (ESRD) [1] and is increasing in prevalence at an alarming rate worldwide [2]. The economic burden is substantial, as the costs of diabetes management in 2019 were estimated to be approximately US $760 billion and expected to increase to US $845 billion by 2045, the majority of which will be spent on the treatment of diabetic complications [3]. Therefore, new therapeutic approaches to prevent the progression of DKD are an urgent unmet medical need and subject to intense efforts by the medical research community and pharmaceutical industry.
Approximately 20–40% of diabetic patients develop DKD even with comparable blood glucose levels. Clinical diagnosis of DKD relies on the presence of persistent albuminuria, reduced estimated glomerular filtration rate (eGFR) and the presence of other diabetes-related complications such as retinopathy [4,5]. Metabolic dysregulation, including hyperglycemia and dyslipidemia, drives the early pathological changes in DKD. The glomerulus is the primary site of diabetic injury in the kidney, and hallmarks of progressive DKD include glomerular hyperfiltration and alterations in the production and composition of the extracellular matrix, leading to mesangial matrix expansion and increased thickness of the glomerular basement membrane (GBM), hence decreasing glomerular filtration surface area [6]. These changes are all early predictors of DKD progression [6,7,8]. Furthermore, the degree of podocyte damage and depletion also correlates closely with the severity of the disease, and this process is proceeded by albuminuria, glomerular sclerosis and eventual declining renal function [9,10,11].
Although hyperglycemia and hypertension are known to drive the onset and progression of DKD, intensive glycemic control has only had modest effects and fails to stop DKD progression to ESRD and death [1,12]. Although new therapies are emerging today, the absolute risk of renal and cardiovascular morbidity and mortality, as well as the need for renal replacement therapy, remains high. Therefore, elucidation of the mechanisms underlying DKD and the development of new and more effective approaches to the prevention of renal dysfunction and treatment requires a better understanding of disease mechanisms. The next sections will present up to date literature on the emerging evidence of endothelial cell dysfunction and the potential mechanisms involved in early DKD pathogenesis.
2. The Diabetic Milieu Affects Structure and Function of the Interconnected Glomerular Filtration Barrier
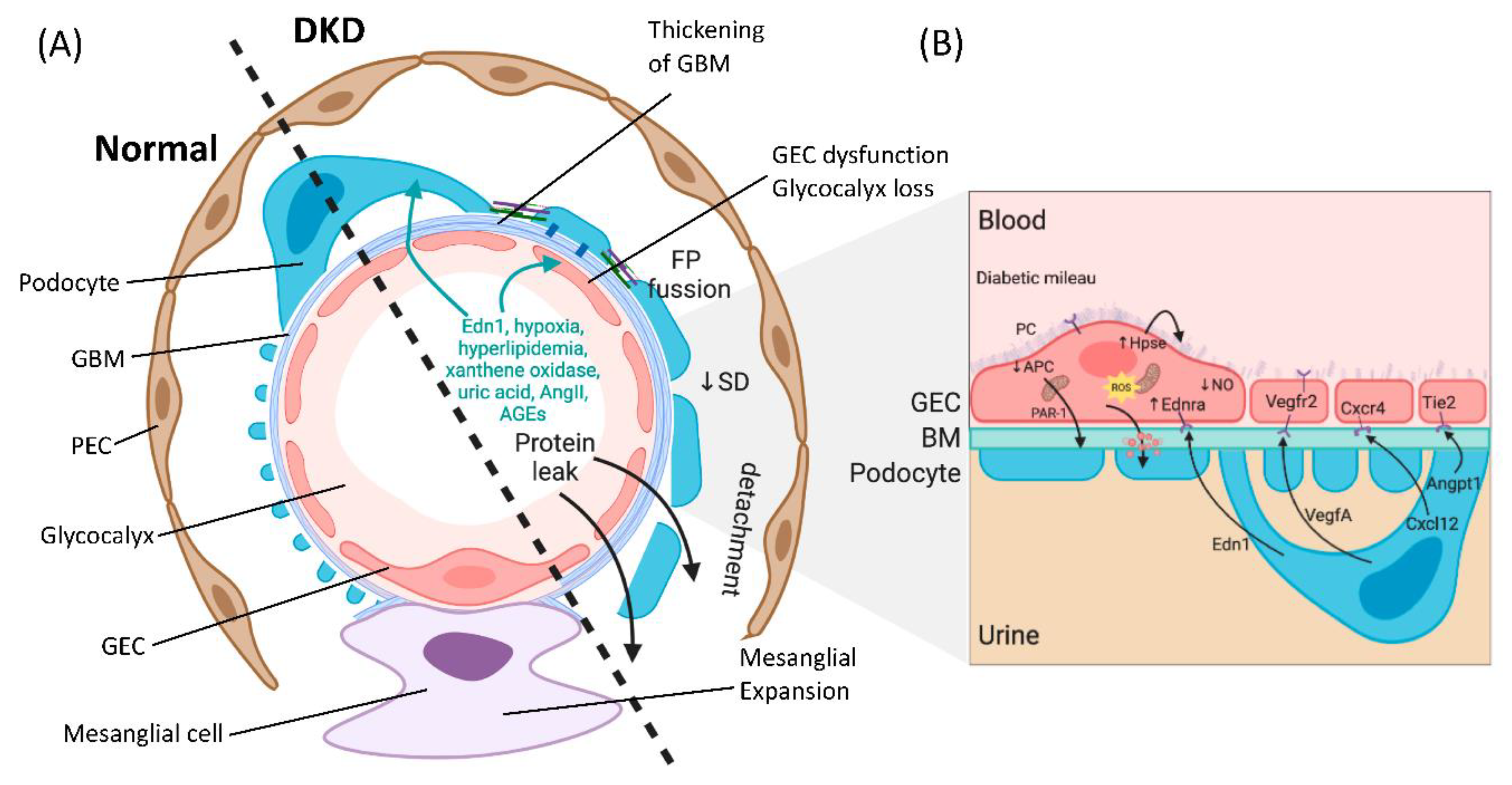
The glomerulus consists of four different cell types: parietal epithelial cells, podocytes (visceral epithelial cells), glomerular endothelial cells (GECs) and mesangial cells (Figure 1A). Parietal epithelial cells line the Bowman’s capsule, where the pre-urine is collected and forwarded to the proximal tubule. Mesangial cells are contractile cells that make up the mesangium and structurally support the glomerular tuft. Podocytes tightly wrap around and support the glomerular capillary vessels by an elaborate net of interdigitating foot processes. Between the foot processes are slit diaphragm proteins (e.g., nephrin and podocin), allowing contact between the podocytes and forming a size selectivity barrier for the passage of molecules and maintenance of glomerular filtration [13]. GECs cover the luminal surface of glomerular capillaries and are the cells of the glomerulus in direct contact with the blood. GECs and podocytes share a common extracellular matrix, the glomerular basement membrane (GBM), and together they form an interconnected glomerular filtration barrier.
Podocyte depletion associated with the progression of DKD has been extensively studied for mechanistic delineation in the breakdown of the glomerular filtration barrier [14]. Disease progression affects the intricate structure of the podocytes and leads to foot process effacement (FPE) [13]. There are several alterations in podocyte structure and function associated with FPE in DKD, including dedifferentiation (epithelial-to-mesenchymal transition), cytoskeletal rearrangement, impaired autophagy and apoptosis, which have been reviewed elsewhere [15,16,17,18]. Although podocytes have been studied extensively as primary targets in DKD, more recently, GEC dysfunction has been attributed to the pathogenesis of glomerular sclerotic diseases, including DKD [19,20,21,22].
The glomerular endothelium is among the unique vascular structures in the body. GECs are highly fenestrated, with fenestrae that are about 17 times larger than the diameter of albumin [23]. This structure allows for high water permeability, or hydraulic conductivity, needed for the large filtration volumes handled by the glomerulus [24]. The apical surface of the endothelial cells is covered by the negatively charged endothelial glycocalyx and endothelial surface layer (ESL), which cover and floats into the lumen of the capillary vessels, and is a key player in the integrity of the glomerular filtration barrier. The endothelial glycocalyx and ESL consist of glycoproteins and proteoglycans covalently linked to the glucosaminoglycans heparan sulfate, chondroitin sulfate and hyaluronic acid [25]. The importance of the glycocalyx in homeostasis has progressively been recognized, due in part to its high charge selectivity restricting the passage of negatively charged molecules such as albumin [23]. The presence of the glycocalyx also creates a space between blood and the endothelium and thereby controls vessel permeability, which leads to the regulation of water efflux [26]. The glycocalyx additionally restricts leukocyte and platelet adhesion to the endothelium, thus moderating inflammation and thrombosis, and allows an appropriate GEC response to flow variation through mechanosensing [26,27,28].
Endothelial cells are exposed to circulating high blood glucose levels and are particularly vulnerable to hyperglycemia-induced injury in diabetes; they then undergo a phenotypic switch that modifies their intracellular signaling leading to dysfunction [29]. Endothelial cell dysfunction is characterized by one or more of the following features: decreased nitric oxide (NO) bioavailability, reduced endothelium-mediated vasorelaxation, hemodynamic deregulation, impaired fibrinolytic ability, enhanced turnover and overproduction of growth factors. There can also be increased expression of adhesion molecules and inflammatory genes, excessive generation of reactive oxygen species (ROS), and enhanced permeability of the cell layer [30]. In diabetes, there is also evidence of a deficiency in endothelial progenitor cells involved in vascular regeneration [31]. Although different endothelial cell surfaces differ in the way they regulate glucose uptake, in GECs, hyperglycemia leads to saturation of glucose metabolism and results in activation of deleterious pathways such as the polyol pathway, the hexosamine pathway, the AGE/RAGE axis and the PKC pathway, leading to endogenous ROS overproduction [8,32]. In early DKD, activation of endothelial nitric oxide synthase (eNOS) that produces the vasodilator NO has been reported, while the progressive disease is associated with a deficiency of NO [33]. Reduced NO triggers uncoupling of eNOS in GECs and the formation of superoxide and peroxynitrite (ONOO−) [34,35]. This reaction can then oxidize the eNOS cofactor tetrahydrobiopterin (BH4), leading to more ROS and eNOS uncoupling [36]. Experimental models of T1DM and T2DM with eNOS knockout (eNOS−/−) have been shown to recapitulate glomerular lesions seen in human DKD, such as mesangial expansion, podocyte injury and depletion, albuminuria and focal segmental glomerular sclerosis (FSGS) [37,38]. These studies also suggest that podocytes, as well as mesangial cells, receive pathogenic signals from GECs in diabetes, indicating crosstalk among the cells in the glomerulus and interdependence.
3. Crosstalk between GECs and Podocytes Is Essential for Filtration Barrier Function and Is Disturbed in DKD
Balanced intercellular crosstalk between podocytes and endothelial cells is required for maintenance of the filtration barrier (Figure 1B). The best-studied mechanism of this crosstalk involves podocyte production of vascular endothelial growth factor-A (VEGFA), acting through paracrine signaling on VEGF receptors (VEGFR1 and VEGFR2) on GECs under physiological conditions. Eremina et al. elegantly demonstrated how conditional deletion of the VEGF gene in podocytes led to a loss of endothelial fenestrae, microangiopathy and proteinuria in otherwise healthy mice [39]. Others have shown that in VEGF-stimulated murine GECs, there is an increase in phosphorylation of eNOS, relating VEGF signaling to NO synthesis [40]. Today with the help of genetic models, we have a greater appreciation of the importance of VEGF-A imbalance in the diabetic setting. Studies using podocyte-specific VEGF-A ablation in STZ-diabetic mice resulted in the development of heavy proteinuria, marked glomerulosclerosis and glomerular cell apoptosis [41]. In contrast, earlier studies have suggested pharmacological inhibitors of VEGF activity to be beneficial in preventing kidney disease [42,43]. On the other hand, increased podocyte-derived VEGF-A was shown to be deleterious in non-diabetic mice, and the injury was further exacerbated with diabetes induction, resulting in advanced glomerulopathy with massive proteinuria [44]. Another example of podocyte-to-endothelial crosstalk is angiopoietins produced by podocytes, which are important for endothelial cell function [45]. They are critical for modulating the vascular response after the onset of diabetes and function as endothelial cell-protective factors in diabetes [46]. In addition, human biopsies, as well as experimental models of FSGS, have uncovered a causative role for the endothelin-1/endothelin receptor type A (Ednra) axis in promoting endothelial cell dysfunction and loss of the glomerular endothelial glycocalyx by increased degradation of glucosaminoglycans [47,48,49]. In diabetes, an increase in circulating as well as local production of endothelin-1 was demonstrated to activate Ednra in glomerular endothelial cells, resulting in mitochondrial stress and endothelial dysfunction [22] (Figure 1B). Podocytes or infiltrating macrophages may also promote the production of the heparan sulfate degrading enzyme heparanase, resulting in loss of glycocalyx [49,50].
When considering endothelial cells-derived intercellular signals, Isermann et al. have shown that the thrombomodulin-dependent formation of activated protein C by glomerular endothelial cells can support podocyte viability via protease-activated receptor1 (PAR-1) and the endothelial protein C receptor (EPCR) [51]. In diabetes, however, this pathway is disturbed and causes mitochondrial apoptosis pathway activation in podocytes and increased glomerular damage, leading to DKD [51]. GEC to podocyte crosstalk is regulated by shear stress, and studies have shown a critical pathway involving an ERK5 mediated increase in expression of KLF2 and downstream molecules, promoting an anti-coagulant, anti-inflammatory phenotype, which has been shown to directly affect podocyte function in co-cultures [52]. A study showed that endothelial cell knockout of KLF2 resulted in podocyte injury in diabetes and reduction of the endothelial glycocalyx [53]. More recently, the GEC and podocyte-specific contribution of TGF-β signaling in the progression of DKD has been examined; the study further highlights the importance of intricate crosstalk between injured glomerular cells [54]. Histological assessment of human biopsies from patients with T1DM and T2DM have unequivocally demonstrated podocyte as well as GEC injury [55,56]. Systematic assessment of glomerular capillaries using experimental models in early diabetes have provided insights suggesting that GEC injury may precede podocyte FP fusion [22] and may predispose to albuminuria either directly or indirectly by communication with neighboring podocytes via secreted mediators of exosomes [57,58]. Importantly, GEC injury and dysfunction were demonstrated to be absolutely necessary for subsequent podocyte depletion [22,47,48]. These studies support the importance of podocyte and GEC crosstalk in the maintenance, as well as the breakdown of the filtration barrier observed in DKD.
4. Oxidative Stress in DKD
A potential mechanism for GEC injury in DKD pathogenesis are reactive oxygen species (ROS), which play a direct role in diabetes and its complications, including nephropathy [59]. ROS are oxygen-derived, highly reactive molecules. Among them are free radicals such as superoxide (O2−) or hydroxyl (•OH) and non-radical ROS such as hydrogen peroxide (H2O2) [60]. ROS have a physiological role in cell signaling concerning cell proliferation and survival, which is under tight regulation and balanced by the cell’s antioxidant response [61]. Unless high levels of ROS are locally deployed for fighting against pathogens, excess ROS leads to oxidative stress, resulting in several cellular changes that can lead to organ dysfunction. Oxidative stress derived from the major sources of ROS has emerged as a causative mechanism in vascular dysregulation in disease states, including diabetes [59,62].
Metabolic changes (e.g., hyperglycemia, dyslipidemia) accompanying the diabetic pathology leads to increased circulation of noxious substances (e.g., glycated proteins and free fatty acids), a saturation of glucose metabolism pathways, and disturbed cellular redox balance. In the glomerulus, in particular, the diabetic milieu triggers oxidative stress responses in all cells via several endogenous pathways, including oxidative phosphorylation in mitochondria, NADPH oxidases (NOX), cytochrome P450, xanthine oxidase and uncoupled eNOS. Studies using increased extracellular glucose (30 mmol/L) found that it can rapidly stimulate intracellular ROS generation of conditionally immortalized podocytes via NADPH oxidase [63]. Another enzymatic source of extracellular and intracellular ROS in diabetes comes from increased xanthine oxidoreductase (XOR) activity [64]. An increase in mitochondrial ROS and mitochondrial dysfunction also plays a critical role in the pathogenesis of DKD [65]. There is now accumulating evidence that supports that the diverse sources of ROS, the timing, the location and the type of oxidative damage generated are important in the initiation and progression of kidney diseases. The next sections will focus on the major ROS generating pathways in DKD, such as NADPH oxidase, XOR and mitochondrial-derived ROS, as well as their potential interplay as it pertains to GECs injury in early DKD.
4.1. Active Enzymatic ROS Generation in DKD
4.1.1. NADPH Oxidase (NOX)
NADPH Oxidases (NOX) have been suggested to contribute to the initiation and progression of DKD and other diabetic complications, and their activity is elevated significantly following the onset of hyperglycemia and increased circulating angiotensin II (Ang II), advanced glycation end products (AGEs) and TGFβ1 [66]. The biological function of NOX enzymes is to generate ROS by transferring electrons across biological membranes [67]. There are seven isoforms of NADPH oxidases, NOX 1-5 and dual oxidases (DUOX) 1 and 2. All isoforms catalyze the reduction of molecular oxygen (O2) to superoxide (O2−) using NADPH as an electron donor [60]. NOX4, however, has been shown to produce hydrogen peroxide (H2O2) rather than superoxide in vitro [68,69]. The regulatory machinery of the NOX isoforms differs. For example, ROS production by NOX 1–4, but not 5 requires binding to the subunit p22phox [70]. NOX1 and NOX3 additionally bind the regulatory proteins NOX organizer 1, NOX activator 1 and Rac, although Rac appears more important for activation of NOX1 than of NOX3 [70]. NOX4, on the other hand, has high constitutive activity and may require only p22phox for activation [60,70]. In contrast, regulation of NOX5, the most recently identified member of the NADPH oxidases, is the only NOX dependent on intracellular calcium [71,72,73,74]. The kidney has a distinct profile for NOX expression within the renal tubular cells, glomerular cells and in the vasculature [67,75].
There has been a significant research effort in the past decade focused on NOX function in the diabetic kidney. NOX4 has been of particular interest due to its enrichment in kidney tissue. In the glomerulus, NOX4 upregulation in glomerular mesangial cells in response to Ang II was shown to be associated with hypertrophy and fibronectin accumulation [76], and NOX4 together with NOX1 and CYP4A were shown by the same research group to mediate increased ROS production in podocytes in response to high glucose [77]. In contrast, other studies suggest that NOX4 expression levels were down rather than upregulated in tubular epithelial cells in chronic kidney disease (CKD) [78], as well as other reports showing a protective effect of NOX4 in the vasculature after ischemia-induced or inflammatory injury [79]. A study by Zhao et al. showed that hyperglycemia induced the upregulation of hedgehog interacting protein in GECs, which stimulated fibrosis through TGFβ-signaling, inducing apoptosis of GECs by NOX4 generation of H2O2 [80]. These studies in aggregate suggest that the timing and localization of ROS production by NOX4 are important in determining the effect of NOX4 enzymatic activity. Interestingly, overexpression of human NOX2 in endothelial cells of Akita T1DM mice led to increased superoxide production, decreased thickness of the endothelial glycocalyx, mesangial matrix expansion and increased podocyte damage with proteinuria [81], despite the mice having a C57BL/6 background; known to be a relatively resistant strain to development of DKD [82]. Importantly, a recent study by the Jandeleit-Dahm research group presented evidence for deleterious effects of endothelial NOX5 expression resulting in increased ROS production in non-diabetic conditions, which was exacerbated by diabetes [83]. These studies suggest that GEC specific NOX-derived ROS can result in glomerular injury in DKD.
Considering the evidence for increased NOX activity and associated ROS production in DKD, NOX’s have been explored as potential therapeutic targets. The dual specific NOX1/NOX4 inhibitor GKT137831 was successful in ameliorating glomerular structural changes, albuminuria and fibrotic signaling in diabetic mice models [84,85]. However, in humans, a phase II clinical trial did not show a reduction of proteinuria compared to placebo [86]. GKT137831 has since been tested as a regulator of fibrosis in the autoimmune liver disease primary biliary cholangitis with promising results [87]. The pan NOX inhibitor APX115 has also demonstrated renal protective effects in preclinical studies involving a T2DM murine model [88], and a clinical trial is currently underway with T2DM patients [89].
4.1.2. Xanthine Oxidoreductase (XOR)
Xanthine oxidoreductases (XOR) are xanthine dehydrogenase (XDH) and xanthine oxidase (XO), interchangeable forms of the same enzyme encoded by the XDH gene. They catalyze the oxidation of purine substrates, xanthine and hypoxanthine and use NAD+ as an electron acceptor. In humans, the enzymatic oxidation of hypoxanthine to xanthine and further to uric acid by XO uses O2 as an electron acceptor and generates H2O2 and O2− [90,91]. Uric acid (UA) is the endpoint of purine metabolism in humans, and both hyperuricemia and hypouricemia can have negative consequences for renal health [91,92].
High XOR activity was shown to be correlated with high serum UA levels, as well as with insulin resistance, adiposity, and subclinical inflammation [93], and was an independent predictor of diabetic complications among T2DM patients [94]. Importantly, increased XOR in circulation is strongly associated with ESRD [95] and have been shown to be risk factors for cardiovascular diseases and DKD [96,97]. Although the role of UA as a risk factor for CKD has been largely debated, there are many studies supporting its role in the development and progression of kidney fibrosis, vascular dysfunction, as well as the benefits of XOR inhibitors in these conditions [96,97,98,99,100,101,102,103,104].
Prospective studies involving patients with type 1 diabetes have shown that higher serum urate levels are associated with an increased risk of rapid GFR decline [105,106]. Hence, efforts to decrease serum UA have been assessed for efficacy in CKD. Outcomes from two recent high profile trials; the Preventing Early Renal Loss in Diabetes (PERL) trial and the Controlled Trial of Slowing of Kidney Disease Progression from the Inhibition of Xanthine Oxidase (CKD-FIX) over 3 and 2 years, respectively, did not show benefit in T1DM patients with mild to moderate kidney disease [107,108]. Although the hypothesis tested was to lower UA, effects on XOR-derived ROS were not examined, despite the many reports supporting that the tested substance allopurinol causes ROS through self-oxidation to form oxypurinol, resulting in the reduction of O2 [109,110]. This initial reaction drives substrates such as xanthine to donate electrons with enzyme turnover reactions that result in excess ROS formation before inhibition of UA is attained [111]. This undesirable action of allo/oxypurinol ROS generation has led to significant misinterpretation of ROS-driven pathology where XOR is a contributor. Febuxostat, a non-purine XOR inhibitor [112], was shown in experimental DKD to decrease ROS damage. The anti-albuminuric and the renoprotective effects observed were shown to be attributed to attenuation of the inflammatory and oxidative stress [113]. Febuxostat was able to slow the decline in eGFR in CKD stages three and four compared to placebo in a smaller trial [114], with no adverse events observed [115]. These studies and trials highlight the need to understand the chemistries of XOR inhibitors and for future studies to focus on ROS production in patients.
4.1.3. Mitochondrial ROS
Mitochondria are the energy-producing organelles in cells via the generation of ATP through oxidative phosphorylation. The kidney consumes a large amount of energy for the reabsorption of large quantities of fluid and solutes across the renal tubular epithelium. Hence it is not so surprising that renal disease can be observed in inherited mitochondrial disorders, including Kearns–Sayre syndrome, Pearson syndrome, DIDMOAD (Wolfram’s syndrome), and Leigh syndrome [116]. Mitochondrial mutations have been associated with childhood-onset FSGS and steroid-resistant nephrotic syndrome [117,118,119,120], and mitochondrial function seems to be crucial for the maintenance of the glomerular filtration barrier [121]. This is even though podocytes under physiological conditions rely on anaerobic glycolysis as the predominant energy source [122]. Endothelial cells similarly generate >75% of their ATP via glycolysis, despite abundant access to oxygen [123,124]. Mutations in genes involved in coenzyme Q10 biosynthesis in podocytes, important in supporting electron transport of oxidative phosphorylation, or in complex IV assembly cofactor heme A: farnesyltransferase in cells of the developing nephrons, were sufficient to cause FSGS [125,126,127], underscoring the importance of mitochondria for and beyond energy production.
Mitochondria conduct other key cellular functions, such as homeostasis of calcium and iron, regulation of tissue oxygen gradients, H2O2 signaling and fatty acid uptake [128,129], as well as biosynthesis of heme, pyrimidines, steroids and modulation of programmed cell death [130,131]. ROS are byproducts from the oxidative phosphorylation reaction, and a significant portion of electrons (0.2% of the oxygen consumed) normally escape the electron transport chain as superoxide anions (O2−). This figure can increase to up to 2% under conditions of oxidative stress [132,133,134], resulting in damage to mitochondria and activating a vicious cycle of more ROS generation, eventually resulting in loss of cell function and tissue abnormality. The deleterious impact of excess mitochondrial ROS, together with a decrease in antioxidative defense systems, are involved in the pathophysiology of DKD, affecting both the glomerulus and the tubular system [135,136,137].
Although there are relatively few studies exploring the effects of mitochondrial dysfunction and ROS production specifically in glomerular endothelial cells [138], research from our lab has shown that genes involved in oxidative phosphorylation and mitochondrial dysfunction were the most enriched in a transcriptomic comparison of mice susceptible and resistant to DKD [22]. Importantly, mitochondrial oxidative stress and DNA damage in DKD susceptible mice was specific to endothelial cells, resulting in loss of endothelial fenestrae and subsequent podocyte depletion [22]. Other studies have shown that the mRNA profiles of isolated GECs and podocytes from diabetic mice kidneys demonstrated distinct upregulated pathways involving mitochondrial function and oxidative stress in the endothelium compartment. Meanwhile, changes in the regulation of actin cytoskeleton-related genes were the major pathways affected in podocytes isolated from diabetic mice [139]. Furthermore, deleterious effects on the endothelial glycocalyx associated with increased mitochondrial ROS exposure contribute to the breakdown of the glomerular filtration barrier [48,140]. More recently, we have demonstrated that diabetic milieu-mediated GEC mitochondrial oxidative stress and impaired autophagy resulted in oxidative damage accumulation in vitro, while exposure of podocytes to the same diabetic milieu resulted in minimal oxidative stress [141]. Interestingly, factors secreted by the stressed GECs caused podocyte apoptosis, while the effect was blocked by the addition of the mitochondrial ROS scavenger MitoTEMPO [141]. These findings provide evidence of endothelial cell mitochondrial dysfunction and overproduction of ROS as early insults can trigger podocyte injury and, therefore, breakdown of glomerular filtration barrier through intercellular crosstalk in DKD. Importantly, cell-specific ROS overproduction could have cells specific distinct and important roles in the glomerulus.
Much remains to be elucidated in defining the functional role of mitochondria in DKD. However, restoration of mitochondrial function could be beneficial. Some mitochondrial interventions currently being explored include the Szeto–Schiller peptide elamipretide (MTP-131), a tetrapeptide that targets mitochondrial cardiolipin, which demonstrated benefits in rodent models of DKD by improving mitochondrial bioenergetics [142,143] and protected mitochondrial cristae structure in both GECs and podocytes [135,144]. The peptide is currently being evaluated for cardiac and renal effects in hospitalized heart failure patients. The efficacy of coenzyme Q10 supplementation was reported to be promising for the treatment of DKD [145] and shown to improve mitochondrial function and decrease oxidative stress in patients receiving hemodialysis [146]. Due to coenzyme Q10 being lipophilic in nature, transport to the mitochondrial inner-membrane is limited. Thus a more hydrophilic intermediate such as 2,4-dihydroxybenzoic acid, which is found naturally in certain foods, can reactivate coenzyme Q10 levels [147,148] and could be of benefit to DKD patients. The mitochondrial-targeted ROS scavenger MitoQ, a form of coenzyme Q with a lipophilic cation for enrichment in mitochondria, have been shown to convey renoprotective effects, with improved albuminuria and hyperfiltration, but not hypertrophy and mesangial expansion, in T2DM mice [149]. MitoQ is currently being evaluated clinically to examine microvascular function in patients with moderate to severe CKD. Finally, boosting antioxidant and mitochondrial biogenesis pathways by activation of the transcription factor, nuclear factor erythroid-2 related factor 2 (NRF-2), has been shown to improve kidney function in a number of glomerular diseases, although there was no reduction in proteinuria [150]. Clinical trials are underway to evaluate the cardiac and renal benefits of mitochondria stabilizing agents in patients. The question of whether or not mitochondrial stabilization and mitochondrial ROS inhibition can improve patient endpoints in large, randomized DKD clinical trials still remains.
4.2. ROS Interplay
In diabetes, oxidative stress may result from an interplay between different ROS sources resulting in a vicious cycle in glomerular cells, and as discussed above, this can result in impaired GEC function (Figure 2). “Redox switches” have been identified in different sources of superoxide, hydrogen peroxide, and peroxynitrite, for example, for the conversion of XDH to the XO form or for the uncoupling process of eNOS [151]. Both ROS and UA are products of XOR reaction and have been shown to induce mitochondrial dysfunction and reduced mitochondrial mass and ATP production in diabetes [100,152]. XOR products can also downregulate mitochondrial metabolism by increasing mitochondrial calcium and stimulating superoxide production [104]. Mitochondrial permeability transition pore is affected by ROS produced by non-mitochondrial sources and result in an increase in peroxynitrite (ONOO−) with eNOS uncoupling, as well as mitochondrial protein, RNA and DNA damage [153,154]. In addition, mitochondrial ROS scavengers can influence XO activity, as demonstrated by the improvement of cardiac complications and XO activation with MitoQ [155]. Moreover, increased angiotensin-II-dependent NADPH oxidase activation in diabetes can mediate mitochondrial dysfunction with subsequent mitochondrial-derived ROS formation [156]. Importantly, the Nox4 isoform was reported to be localized in mitochondria in diabetes and could contribute to processes that are associated with mitochondrial oxidative stress [157]. However, to this date, there is only limited evidence for redox-based activation pathways of NOX, XO and for the role of mitochondrial ROS in DKD. Understanding these interactions is important, as not all ROS are the same. ROS are produced both under physiological and pathological conditions. Hence general antioxidant therapy approaches have failed in large clinical trials with DKD patients [158,159]. More research is needed to further the general understanding of the contribution of redox processes in DKD.
4.3. Current Clinical Approaches for DKD and Their Effects on ROS
In addition to the pharmacological agents targeting the different sources of ROS described above, there are ongoing investigations of other primary pharmaceutical targets that impact ROS in DKD. For instance, the selective endothelin 1 receptor A (Ednra) antagonist atrasentan has been demonstrated to have beneficial effects on renal function and proteinuria in T2DM patients in phase 3 randomized clinical trial SONAR [160]. Despite adverse effects, including fluid retention and heart failure accompanying Ednra inhibition in DKD [161], encouraging outcomes from the phase 2 DUET trial in people with primary FSGS with sparsentan combined with angiotensin receptor blocker [162] are being further explored in the Phase 3 DUPLEX trial. The proven antiproteinuric effects of Ednra blockade could be attributed to the prevention of pathologic crosstalk between podocytes and GECs in DKD, where increased Ednra signaling in GECs leads to mitochondrial oxidative stress and damage of GECs [22]. As previously discussed, GEC stress and dysfunction mediated podocyte injury and depletion [47], and inhibition of Ednra was found to be beneficial in maintaining the endothelial glycocalyx [48,49,50]. Another mechanism that indirectly ameliorates oxidative stress and is being explored in patients with DKD is the selective induction of ATP-binding cassette A1 (ABCA1) [163,164]. ABCA1 inducers promote the removal of excess cholesterol from podocytes and therefore stabilizes mitochondrial cardiolipin in podocytes in DKD [163].
Exciting outcomes from recent trials testing the effectiveness of sodium–glucose cotransporter 2 (SGLT2) inhibitors have produced great expectations with positive effects on hyperglycemia control, as well as on cardiovascular and renal outcomes of T2DM. SGLT2 inhibitors have systematic pleiotropic effects, including the normalization of altered tubuloglomerular feedback, and hence can control diabetes-induced hyperfiltration and intraglomerular pressure. Interestingly, other effects of SGLT2 inhibitors that are independent of blood glucose control or bodyweight reduction are the amelioration of mitochondrial damage in tubular cells [165] as well as reduction of UA [166]. In experimental DKD, these agents reduced mesangial expansion, accumulation of extracellular matrix proteins, and podocyte injury [167]. A study showed that canagliflozin inhibited high-glucose-induced activation of the protein kinase C-NOX pathway and ROS production in cultured mesangial cells [168]. Although no studies have reported the effect of SGLT2 inhibitors on ROS in GECs, empagliflozin and dapagliflozin were shown to restore barrier function, adhesion molecules expression, NO bioavailability and inhibit ROS production in endothelial cells under inflammatory conditions [169], and these effects were independent of SGLT2 expression in these cells. A recent study also suggested that the glucagon-like peptide 1 agonist liraglutide can ameliorate uncoupling of the VEGF-NO axis by activation of the AMPK–eNOS pathway in glomeruli of mice with obesity-related kidney disease [170]. Studies assessing their effects on GECs are expected to provide valuable tools in DKD therapy in the upcoming years.
5. Conclusions
The diabetic state plays a pivotal role in the stimulation of excess ROS generation in the kidney. Increased ROS production and accumulation of ROS damage products promote injury in all glomerular cells, especially in GECs, resulting in dysfunction. Impaired endothelial function in DKD is now recognized to cause functional and structural changes within the glomerulus through crosstalk [20,138,171], and this may be central to early pathogenesis and help drive disease progression. Though being generated from many sources in diabetes, ROS from NADPH oxidase, XORs and mitochondria are thought to cause the onset of albuminuria followed by a progression of renal damage. Still, the validation of findings in experimental models and translation to humans remains a point of controversy. Better understanding of the context which results in ROS overproduction, timing, and cell–cell interactions may lead to greater insights regarding the potentially reversible events that lead to DKD progression and inspire novel therapeutic approaches.
Author Contributions
Conceptualization, E.L. and I.S.D.; writing—original draft preparation, writing—review and editing, E.L. and I.S.D.; funding acquisition, I.S.D. All authors have read and agreed to the published version of the manuscript.
Funding
I.S.D. is supported by the National Institutes of Health grant R01DK097253 and the Department of Defense CDMRP grant E01 W81XWH2010836.
Acknowledgments
We thank John Cijiang He for his helpful comments. Illustrations were created using BioRender.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ABCA1 | ATP-binding cassette A1 |
| AGE | Advanced glycation end product |
| AMPK | AMP-activated protein kinase |
| Ang II | Angiotensin II |
| Angpt1 | Angiopoietin 1 |
| APC | Activated protein C |
| ATP | Adenosine triphosphate |
| CKD | Chronic kidney disease |
| CYP | Cytochrome P450 |
| DKD | Diabetic kidney disease |
| Ednra | Endothelin receptor A |
| Edn1 | Endothelin 1 |
| eNOS | Endothelial nitric oxide synthase |
| EPCR | Endothelial protein C receptor |
| ESL | Endothelial surface layer |
| ESRD | End-stage renal disease |
| FP | Foot process |
| FSGS | Focal segmental glomerulosclerosis |
| GBM | Glomerular basement membrane |
| GEC | Glomerular endothelial cell |
| GFR | Glomerular filtration rate |
| KLF2 | Krüppel-like factor 2 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NO | Nitric oxide |
| NOX | NADPH oxidase |
| PAR-1 | Protease-activated receptor 1 |
| PEC | Parietal epithelial cell |
| PKC | Protein kinase C |
| RAAS | Renin angiotensin aldosterone system |
| ROS | Reactive oxygen species |
| SD | Slit diaphragm |
| SGLT2 | Sodium glucose cotransporter 2 |
| STZ | Streptozotocin |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| TGFβ | Transforming growth factor β |
| UA | Uric acid |
| VEGFA | Vascular endothelial growth factor A |
| VEGFR | Vascular endothelial growth factor receptor |
| XDH | Xanthine dehydrogenase |
| XO | Xanthine oxidase |
| XOR | Xanthine oxidoreductase |
References
- Chapter 1: Incidence, Prevalence, Patient Characteristics, and Treatment Modalities. Am. J. Kidney Dis. 2019, 73, S291–S332. [CrossRef]
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.; Cavan, D.; Shaw, J.; Makaroff, L.E. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.; Karuranga, S.; Malanda, B.; Saeedi, P.; Basit, A.; Besançon, S.; Bommer, C.; Esteghamati, A.; Ogurtsova, K.; Zhang, P.; et al. Global and regional estimates and projections of diabetes-related health expenditure: Results from the International Diabetes Federation Diabetes Atlas. Diabetes Res. Clin. Pract. 2020, 162, 108072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Diabetes Association 11. Microvascular Complications and Foot Care: Standards of Medical Care in Diabetes-2020. Diabetes Care 2019, 43, S135–S151. [Google Scholar] [CrossRef] [Green Version]
- Anders, H.-J.; Huber, T.B.; Isermann, B.; Schiffer, M. CKD in diabetes: Diabetic kidney disease versus nondiabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 361–377. [Google Scholar] [CrossRef]
- Caramori, M.L.; Parks, A.; Mauer, M. Renal Lesions Predict Progression of Diabetic Nephropathy in Type 1 Diabetes. J. Am. Soc. Nephrol. 2013, 24, 1175–1181. [Google Scholar] [CrossRef] [Green Version]
- Drummond, K.N.; Kramer, M.S.; Suissa, S.; Lévy-Marchal, C.; Dell’Aniello, S.; Sinaiko, A.; Mauer, M. Effects of duration and age at onset of type 1 diabetes on preclinical manifestations of nephropathy. Diabetes 2003, 52, 1818–1824. [Google Scholar] [CrossRef]
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Investig. 2014, 124, 2333–2340. [Google Scholar] [CrossRef]
- Steffes, M.W.; Schmidt, D.; McCrery, R.; Basgen, J.M. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int. 2001, 59, 2104–2113. [Google Scholar] [CrossRef] [Green Version]
- Meyer, T.W.; Bennett, P.H.; Nelson, R.G. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with Type II diabetes and microalbuminuria. Diabetologia 1999, 42, 1341–1344. [Google Scholar] [CrossRef] [Green Version]
- Pagtalunan, M.; Miller, P.L.; Jumping-Eagle, S.; Nelson, R.G.; Myers, B.D.; Rennke, H.G.; Coplon, N.S.; Sun, L.; Meyer, T.W. Podocyte loss and progressive glomerular injury in type II diabetes. J. Clin. Investig. 1997, 99, 342–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dounousi, E.; Duni, A.; Leivaditis, K.; Vaios, V.; Eleftheriadis, T.; Liakopoulos, V. Improvements in the Management of Diabetic Nephropathy. Rev. Diabet. Stud. 2015, 12, 119–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katalin, S.; Susztak, K. Podocytes: The Weakest Link in Diabetic Kidney Disease? Curr. Diabetes Rep. 2016, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Stieger, N.; Worthmann, K.; Teng, B.; Engeli, S.; Das, A.M.; Haller, H.; Schiffer, M. Impact of high glucose and transforming growth factor–β on bioenergetic profiles in podocytes. Metab. Clin. Exp. 2012, 61, 1073–1086. [Google Scholar] [CrossRef]
- Kravets, I.; Mallipattu, S.K. The Role of Podocytes and Podocyte-Associated Biomarkers in Diagnosis and Treatment of Diabetic Kidney Disease. J. Endocr. Soc. 2020, 4, bvaa029. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.; Liu, Q.; Liu, B. Research Progress on Mechanism of Podocyte Depletion in Diabetic Nephropathy. J. Diabetes Res. 2017, 2017, 2615286. [Google Scholar] [CrossRef]
- Brosius, F.C.; Coward, R.J. Podocytes, Signaling Pathways, and Vascular Factors in Diabetic Kidney Disease. Adv. Chronic Kidney Dis. 2014, 21, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Mathieson, P.W. The podocyte as a target for therapies—New and old. Nat. Rev. Nephrol. 2011, 8, 52–56. [Google Scholar] [CrossRef]
- Zheng, X.; Soroush, F.; Long, J.; Hall, E.T.; Adishesha, P.K.; Bhattacharya, S.; Kiani, M.F.; Bhalla, V. Murine glomerular transcriptome links endothelial cell-specific molecule-1 deficiency with susceptibility to diabetic nephropathy. PLoS ONE 2017, 12, e0185250. [Google Scholar] [CrossRef]
- Sol, M.; Kamps, J.A.A.M.; Born, J.V.D.; Heuvel, M.C.V.D.; Van Der Vlag, J.; Krenning, G.; Hillebrands, J.-L. Glomerular Endothelial Cells as Instigators of Glomerular Sclerotic Diseases. Front. Pharmacol. 2020, 11, 573557. [Google Scholar] [CrossRef]
- Kuwabara, A.; Satoh, M.; Tomita, N.; Sasaki, T.; Kashihara, N. Deterioration of glomerular endothelial surface layer induced by oxidative stress is implicated in altered permeability of macromolecules in Zucker fatty rats. Diabetologia 2010, 53, 2056–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, H.; Casalena, G.; Shi, S.; Yu, L.; Ebefors, K.; Sun, Y.; Zhang, W.; D’Agati, V.; Schlondorff, D.; Haraldsson, B.; et al. Glomerular Endothelial Mitochondrial Dysfunction Is Essential and Characteristic of Diabetic Kidney Disease Susceptibility. Diabetes 2016, 66, 763–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraldsson, B.; Nyström, J.; Deen, W.M. Properties of the Glomerular Barrier and Mechanisms of Proteinuria. Physiol. Rev. 2008, 88, 451–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jen, K.Y.; Laszik, Z.G. Endotheliopathies: Hemolytic Uremic Syndrome, Thrombotic Thrombocytopenic Purpura, and Preeclampsia. In Pathobiology of Human Disease; McManus, L.M., Mitchell, R.N., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 2767–2787. [Google Scholar] [CrossRef]
- Weinbaum, S.; Cancel, L.M.; Fu, B.M.; Tarbell, J.M. The Glycocalyx and Its Role in Vascular Physiology and Vascular Related Diseases. Cardiovasc. Eng. Technol. 2020, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, S.; Vink, H.; Hiramatsu, O.; Kajita, T.; Shigeto, F.; Spaan, J.A.E.; Kajiya, F. Role of hyaluronic acid glycosaminoglycans in shear-induced endothelium-derived nitric oxide release. Am. J. Physiol. Circ. Physiol. 2003, 285, H722–H726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florian, J.A.; Kosky, J.R.; Ainslie, K.; Pang, Z.; Dull, R.O.; Tarbell, J.M. Heparan Sulfate Proteoglycan Is a Mechanosensor on Endothelial Cells. Circ. Res. 2003, 93, e136–e142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarbell, J.M.; Ebong, E.E. The Endothelial Glycocalyx: A Mechano-Sensor and -Transducer. Sci. Signal. 2008, 1, pt8. [Google Scholar] [CrossRef]
- Tabit, C.E.; Chung, W.B.; Hamburg, N.M.; Vita, J.A. Endothelial dysfunction in diabetes mellitus: Molecular mechanisms and clinical implications. Rev. Endocr. Metab. Disord. 2010, 11, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Popov, D. Endothelial cell dysfunction in hyperglycemia: Phenotypic change, intracellular signaling modification, ultrastructural alteration, and potential clinical outcomes. Int. J. Diabetes Mellit. 2010, 2, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Pearsall, E.A.; Cheng, R.; Matsuzaki, S.; Zhou, K.; Ding, L.; Ahn, B.; Kinter, M.; Humphries, K.M.; Quiambao, A.B.; Farjo, R.A.; et al. Neuroprotective effects of PPARα in retinopathy of type 1 diabetes. PLoS ONE 2019, 14, e0208399. [Google Scholar] [CrossRef] [Green Version]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.-A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, S.S. Role of nitric oxide in diabetic nephropathy. Semin. Nephrol. 2004, 24, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Sato, W.; Sautin, Y.Y.; Glushakova, O.; Croker, B.; Atkinson, M.A.; Tisher, C.C.; Johnson, R.J. Uncoupling of Vascular Endothelial Growth Factor with Nitric Oxide as a Mechanism for Diabetic Vasculopathy. J. Am. Soc. Nephrol. 2006, 17, 736–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaimes, E.A.; Hua, P.; Tian, R.-X.; Raij, L. Human glomerular endothelium: Interplay among glucose, free fatty acids, angiotensin II, and oxidative stress. Am. J. Physiol. Physiol. 2010, 298, F125–F132. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Zhao, H.J.; Wang, S.; Cheng, H.; Zhang, M.-Z.; Takahashi, T.; Fogo, A.B.; Breyer, M.D.; Harris, R.C. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J. Am. Soc. Nephrol. 2006, 17, 2664–2669. [Google Scholar] [CrossRef]
- Nakagawa, T.; Sato, W.; Glushakova, O.; Heinig, M.; Clarke, T.; Campbell-Thompson, M.; Yuzawa, Y.; Atkinson, M.A.; Johnson, R.J.; Croker, B. Diabetic Endothelial Nitric Oxide Synthase Knockout Mice Develop Advanced Diabetic Nephropathy. J. Am. Soc. Nephrol. 2007, 18, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas, M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx, P.H.; et al. VEGF Inhibition and Renal Thrombotic Microangiopathy. N. Engl. J. Med. 2008, 358, 1129–1136. [Google Scholar] [CrossRef]
- Feliers, D.; Chen, X.; Akis, N.; Ghosh-Choudhury, G.; Madaio, M.; Kasinath, B.S. VEGF regulation of endothelial nitric oxide synthase in glomerular endothelial cells. Kidney Int. 2005, 68, 1648–1659. [Google Scholar] [CrossRef] [Green Version]
- Sivaskandarajah, G.A.; Jeansson, M.; Maezawa, Y.; Eremina, V.; Baelde, H.J.; Quaggin, S.E. Vegfa Protects the Glomerular Microvasculature in Diabetes. Diabetes 2012, 61, 2958–2966. [Google Scholar] [CrossRef] [Green Version]
- De Vriese, A.S.; Tilton, R.G.; Elger, M.; Stephan, C.C.; Kriz, W.; Lameire, N.H. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J. Am. Soc. Nephrol. 2001, 12, 993–1000. [Google Scholar] [PubMed]
- Sung, S.H.; Ziyadeh, F.N.; Wang, A.; Pyagay, P.E.; Kanwar, Y.S.; Chen, S. Blockade of Vascular Endothelial Growth Factor Signaling Ameliorates Diabetic Albuminuria in Mice. J. Am. Soc. Nephrol. 2006, 17, 3093–3104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veron, D.; Reidy, K.J.; Bertuccio, C.; Teichman, J.; Villegas, G.; Jimenez, J.; Shen, W.; Kopp, J.B.; Thomas, D.B.; Tufro, A. Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int. 2010, 77, 989–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satchell, S.C.; Harper, S.J.; Tooke, J.; Kerjaschki, N.; Saleem, M.; Mathieson, P.W. Human podocytes express angiopoietin 1, a potential regulator of glomerular vascular endothelial growth factor. J. Am. Soc. Nephrol. 2002, 13, 544–550. [Google Scholar] [PubMed]
- Jeansson, M.; Gawlik, A.; Anderson, G.; Li, C.; Kerjaschki, D.; Henkelman, M.; Quaggin, S.E. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J. Clin. Investig. 2011, 121, 2278–2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daehn, I.; Casalena, G.; Zhang, T.; Shi, S.; Fenninger, F.; Barasch, N.; Yu, L.; D’Agati, V.; Schlondorff, D.; Kriz, W.; et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J. Clin. Investig. 2014, 124, 1608–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebefors, K.; Wiener, R.J.; Yu, L.; Azeloglu, E.U.; Yi, Z.; Jia, F.; Zhang, W.; Baron, M.H.; He, J.C.; Haraldsson, B.; et al. Endothelin receptor-A mediates degradation of the glomerular endothelial surface layer via pathologic crosstalk between activated podocytes and glomerular endothelial cells. Kidney Int. 2019, 96, 957–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boels, M.G.S.; Avramut, M.C.; Koudijs, A.; Dane, M.J.; Lee, D.H.; Van Der Vlag, J.; Koster, A.J.; Van Zonneveld, A.J.; Van Faassen, E.; Gröne, H.-J.; et al. Atrasentan Reduces Albuminuria by Restoring the Glomerular Endothelial Glycocalyx Barrier in Diabetic Nephropathy. Diabetes 2016, 65, 2429–2439. [Google Scholar] [CrossRef] [Green Version]
- Garsen, M.; Lenoir, O.; Rops, A.L.; Dijkman, H.B.; Willemsen, B.; Van Kuppevelt, T.H.; Rabelink, T.J.; Berden, J.H.; Tharaux, P.-L.; Van Der Vlag, J. Endothelin-1 Induces Proteinuria by Heparanase-Mediated Disruption of the Glomerular Glycocalyx. J. Am. Soc. Nephrol. 2016, 27, 3545–3551. [Google Scholar] [CrossRef] [Green Version]
- Isermann, B.; Vinnikov, I.A.; Madhusudhan, T.; Herzog, S.; Kashif, M.; Blautzik, J.; Corat, M.A.; Zeier, M.; Blessing, E.; Oh, J.; et al. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat. Med. 2007, 13, 1349–1358. [Google Scholar] [CrossRef]
- Slater, S.C.; Ramnath, R.D.; Uttridge, K.; Saleem, M.A.; Cahill, P.; Mathieson, P.W.; Welsh, G.I.; Satchell, S.C. Chronic exposure to laminar shear stress induces Kruppel-like factor 2 in glomerular endothelial cells and modulates interactions with co-cultured podocytes. Int. J. Biochem. Cell Biol. 2012, 44, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Chen, H.; Wei, C.; Zhang, W.; Li, Z.; Jain, M.K.; Chuang, P.Y.; Chen, H.; Wang, Y.; Mallipattu, S.K.; et al. Reduced Krüppel-like factor 2 expression may aggravate the endothelial injury of diabetic nephropathy. Kidney Int. 2015, 87, 382–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, H.; Chen, A.; Cai, H.; Fu, J.; Salem, F.; Li, Y.; He, J.C.; Schlondorff, D.; Lee, K. Podocyte and endothelial-specific elimination of BAMBI identifies differential transforming growth factor-β pathways contributing to diabetic glomerulopathy. Kidney Int. 2020, 98, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Weil, E.J.; Lemley, K.V.; Mason, C.C.; Yee, B.; Jones, L.I.; Blouch, K.; Lovato, T.; Richardson, M.; Myers, B.D.; Nelson, R.G. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 2012, 82, 1010–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyoda, M.; Najafian, B.; Kim, Y.; Caramori, M.L.; Mauer, M. Podocyte Detachment and Reduced Glomerular Capillary Endothelial Fenestration in Human Type 1 Diabetic Nephropathy. Diabetes 2007, 56, 2155–2160. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.-M.; Gao, Y.-B.; Cui, F.-Q.; Zhang, N. Exosomes from high glucose-treated glomerular endothelial cells activate mesangial cells to promote renal fibrosis. Biol. Open 2016, 5, 484–491. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Lee, K.; Chuang, P.Y.; Liu, Z.; He, J.C. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am. J. Physiol. Physiol. 2015, 308, F287–F297. [Google Scholar] [CrossRef] [Green Version]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Jha, J.C.; Banal, C.; Chow, B.S.; Cooper, M.E.; Jandeleit-Dahm, K.A. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Widlansky, M.E.; Gutterman, D.D. Regulation of Endothelial Function by Mitochondrial Reactive Oxygen Species. Antioxid. Redox Signal. 2011, 15, 1517–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susztak, K.; Raff, A.C.; Schiffer, M.; Bottinger, E.P. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy 2. Diabetes 2006, 55, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Alicigüzel, Y.; Ozen, I.; Aslan, M.; Karayalcin, U. Activities of xanthine oxidoreductase and antioxidant enzymes in different tissues of diabetic rats. J. Lab. Clin. Med. 2003, 142, 172–177. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nat. Cell Biol. 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Kakehi, T.; Yabe-Nishimura, C. NOX enzymes and diabetic complications. Semin. Immunopathol. 2008, 30, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Fórró, L.; Schlegel, W.; Krause, K.-H. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S.I.; Dikalova, A.E.; Bikineyeva, A.T.; Schmidt, H.H.; Harrison, D.G.; Griendling, K.K. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free. Radic. Biol. Med. 2008, 45, 1340–1351. [Google Scholar] [CrossRef] [Green Version]
- Lambeth, J.D.; Kawahara, T.; Diebold, B. Regulation of Nox and Duox enzymatic activity and expression. Free. Radic. Biol. Med. 2007, 43, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Cao, Z.; Xu, X.; Meir, E.G.; Lambeth, J. Homologs of gp91 phox: Cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 2001, 269, 131–140. [Google Scholar] [CrossRef]
- Bánfi, B.; Molnár, G.; Maturana, A.D.; Steger, K.; Hegedûs, B.; Demaurex, N.; Krause, K.-H. A Ca2+-activated NADPH Oxidase in Testis, Spleen, and Lymph Nodes. J. Biol. Chem. 2001, 276, 37594–37601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holterman, C.E.; Thibodeau, J.-F.; Towaij, C.; Gutsol, A.; Montezano, A.C.; Parks, R.J.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R. Nephropathy and Elevated BP in Mice with Podocyte-Specific NADPH Oxidase 5 Expression. J. Am. Soc. Nephrol. 2013, 25, 784–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, J.C.; Banal, C.; Okabe, J.; Gray, S.P.; Hettige, T.; Chow, B.S.; Thallas-Bonke, V.; De Vos, L.; Holterman, C.E.; Coughlan, M.T.; et al. NADPH Oxidase Nox5 Accelerates Renal Injury in Diabetic Nephropathy. Diabetes 2017, 66, 2691–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Østergaard, J.A.; Cooper, M.E.; Jandeleit-Dahm, K.A.M. Targeting oxidative stress and anti-oxidant defence in diabetic kidney disease. J. Nephrol. 2020, 33, 1–13. [Google Scholar] [CrossRef]
- Block, K.; Eid, A.; Griendling, K.K.; Lee, D.-Y.; Wittrant, Y.; Gorin, Y. Nox4 NAD(P)H oxidase mediates Src-dependent tyrosine phosphorylation of PDK-1 in response to angiotensin II: Role in mesangial cell hypertrophy and fibronectin expression. J. Biol. Chem. 2008, 283, 24061–24076. [Google Scholar] [CrossRef] [Green Version]
- Eid, A.A.; Gorin, Y.; Fagg, B.M.; Maalouf, R.; Barnes, J.L.; Block, K.; Abboud, H.E. Mechanisms of Podocyte Injury in Diabetes: Role of Cytochrome P450 and NADPH Oxidases. Diabetes 2009, 58, 1201–1211. [Google Scholar] [CrossRef] [Green Version]
- Rajaram, R.D.; Dissard, R.; Faivre, A.; Ino, F.; Delitsikou, V.; Jaquet, V.; Cagarelli, T.; Lindenmeyer, M.; Jansen-Duerr, P.; Cohen, C.; et al. Tubular NOX4 expression decreases in chronic kidney disease but does not modify fibrosis evolution. Redox Biol. 2019, 26, 101234. [Google Scholar] [CrossRef]
- Schröder, R.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 Is a Protective Reactive Oxygen Species Generating Vascular NADPH Oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.-P.; Chang, S.-Y.; Liao, M.-C.; Lo, C.-S.; Chenier, I.; Luo, H.; Chiasson, J.-L.; Ingelfinger, J.R.; Chan, J.S.D.; Zhang, S.-L. Hedgehog Interacting Protein Promotes Fibrosis and Apoptosis in Glomerular Endothelial Cells in Murine Diabetes. Sci. Rep. 2018, 8, 5958. [Google Scholar] [CrossRef]
- Nagasu, H.; Satoh, M.; Kiyokage, E.; Kidokoro, K.; Toida, K.; Channon, K.M.; Kanwar, Y.S.; Sasaki, T.; Kashihara, N. Activation of endothelial NAD(P)H oxidase accelerates early glomerular injury in diabetic mice. Lab. Investig. 2016, 96, 25–36. [Google Scholar] [CrossRef]
- Breyer, M.D.; Bottinger, E.; Brosius, F.C.; Coffman, T.M.; Harris, R.C.; Heilig, C.W.; Sharma, K. Mouse Models of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2004, 16, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Dai, A.; Holterman, C.E.; Cooper, M.E.; Touyz, R.M.; Kennedy, C.R.; Jandeleit-Dahm, K.A.M. Endothelial or vascular smooth muscle cell-specific expression of human NOX5 exacerbates renal inflammation, fibrosis and albuminuria in the Akita mouse. Diabetologia 2019, 62, 1712–1726. [Google Scholar] [CrossRef] [PubMed]
- Gorin, Y.; Cavaglieri, R.C.; Khazim, K.; Lee, D.-Y.; Bruno, F.; Thakur, S.; Fanti, P.; Szyndralewiez, C.; Barnes, J.L.; Block, K.; et al. Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type 1 diabetes. Am. J. Physiol. Physiol. 2015, 308, F1276–F1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, J.C.; Gray, S.P.; Barit, D.; Okabe, J.; El-Osta, A.; Namikoshi, T.; Thallas-Bonke, V.; Wingler, K.; Szyndralewiez, C.; Heitz, F.; et al. Genetic Targeting or Pharmacologic Inhibition of NADPH Oxidase Nox4 Provides Renoprotection in Long-Term Diabetic Nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1237–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genkyotex Announces Top-Line Results of Phase 2 Clinical Program. Available online: https://www.businesswire.com/news/home/20150909005080/en/Genkyotex-Announces-Top-Line-Results-Phase-2-Clinical (accessed on 12 November 2020).
- Genkyotex Provides New Clinical Data from the PBC Phase 2 Trial Providing Further Evidence of the Anti-Fibrotic Activity of Setanaxib. Available online: https://www.businesswire.com/news/home/20200617005839/en/Genkyotex-provides-new-clinical-data-from-the-PBC-Phase-2-trial-providing-further-evidence-of-the-anti-fibrotic-activity-of-setanaxib (accessed on 12 November 2020).
- Cha, J.J.; Min, H.S.; Kim, K.T.; Kim, J.E.; Ghee, J.Y.; Kim, H.W.; Lee, J.E.; Han, J.Y.; Lee, G.; Ha, H.J.; et al. APX-115, a first-in-class pan-NADPH oxidase (Nox) inhibitor, protects db/db mice from renal injury. Lab. Investig. 2017, 97, 419–431. [Google Scholar] [CrossRef]
- Safety, Tolerability and Renal Effects of APX-115 in Subjects with Type 2 Diabetes and Nephropathy. Available online: https://clinicaltrials.gov/ct2/show/NCT04534439 (accessed on 12 November 2020).
- Nishino, T.; Okamoto, K. Mechanistic insights into xanthine oxidoreductase from development studies of candidate drugs to treat hyperuricemia and gout. JBIC J. Biol. Inorg. Chem. 2015, 20, 195–207. [Google Scholar] [CrossRef] [Green Version]
- Furuhashi, M. New insights into purine metabolism in metabolic diseases: Role of xanthine oxidoreductase activity. Am. J. Physiol. Metab. 2020, 319, E827–E834. [Google Scholar] [CrossRef]
- Dissanayake, L.V.; Spires, D.R.; Palygin, O.; Staruschenko, A. Effects of uric acid dysregulation on the kidney. Am. J. Physiol. Physiol. 2020, 318, F1252–F1257. [Google Scholar] [CrossRef]
- Washio, K.; Kusunoki, Y.; Murase, T.; Nakamura, T.; Osugi, K.; Ohigashi, M.; Sukenaga, T.; Ochi, F.; Matsuo, T.; Katsuno, T.; et al. Xanthine oxidoreductase activity is correlated with insulin resistance and subclinical inflammation in young humans. Metab. Clin. Exp. 2017, 70, 51–56. [Google Scholar] [CrossRef]
- Miric, D.J.; Kisic, B.; Filipovic-Danic, S.; Grbic, R.; Dragojevic, I.; Miric, M.B.; Puhalo-Sladoje, D. Xanthine Oxidase Activity in Type 2 Diabetes Mellitus Patients with and without Diabetic Peripheral Neuropathy. J. Diabetes Res. 2016, 2016, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Boban, M.; Kocic, G.; Radenkovic, S.; Pavlovic, R.; Cvetkovic, T.; Deljanin-Ilic, M.; Ilic, S.; Bobana, M.D.; Djindjic, B.; Stojanovic, D.; et al. Circulating purine compounds, uric acid, and xanthine oxidase/dehydrogenase relationship in essential hypertension and end stage renal disease. Ren. Fail. 2014, 36, 613–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosla, U.M.; Zharikov, S.; Finch, J.L.; Nakagawa, T.; Roncal, C.; Mu, W.; Krotova, K.; Block, E.R.; Prabhakar, S.; Johnson, R.J. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005, 67, 1739–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalal, D.; Maahs, D.M.; Hovind, P.; Nakagawa, T. Uric Acid as a Mediator of Diabetic Nephropathy. Semin. Nephrol. 2011, 31, 459–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanbay, M.; Yilmaz, M.I.; Sonmez, A.; Turgut, F.; Sağlam, M.; Cakir, E.; Yenicesu, M.; Covic, A.; Jalal, D.; Johnson, R.J. Serum uric acid level and endothelial dysfunction in patients with nondiabetic chronic kidney disease. Am. J. Nephrol. 2011, 33, 298–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patschan, D.; Patschan, S.; Gobe, G.G.; Chintala, S.; Goligorsky, M.S. Uric Acid Heralds Ischemic Tissue Injury to Mobilize Endothelial Progenitor Cells. J. Am. Soc. Nephrol. 2007, 18, 1516–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Lozada, L.G.; Lanaspa, M.A.; Cristóbal-García, M.; García-Arroyo, F.; Soto, V.; Cruz-Robles, D.; Nakagawa, T.; Yu, M.; Kang, D.-H.; Johnson, R.J. Uric Acid-Induced Endothelial Dysfunction Is Associated with Mitochondrial Alterations and Decreased Intracellular ATP Concentrations. Nephron 2013, 121, e71–e78. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.-A.; Sánchez-Lozada, L.G.; Johnson, R.J.; Kang, D.-H. Oxidative stress with an activation of the renin–angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J. Hypertens. 2010, 28, 1234–1242. [Google Scholar] [CrossRef]
- Zharikov, S.; Krotova, K.; Hu, H.; Baylis, C.; Johnson, R.J.; Block, E.R.; Patel, J. Uric acid decreases NO production and increases arginase activity in cultured pulmonary artery endothelial cells. Am. J. Physiol. Physiol. 2008, 295, C1183–C1190. [Google Scholar] [CrossRef] [Green Version]
- Rajesh, M.; Mukhopadhyay, P.; Bátkai, S.; Mukhopadhyay, B.; Patel, V.; Haskó, G.; Szabó, C.; Mabley, J.G.; Liaudet, L.; Pacher, P. Xanthine oxidase inhibitor allopurinol attenuates the development of diabetic cardiomyopathy. J. Cell. Mol. Med. 2009, 13, 2330–2341. [Google Scholar] [CrossRef]
- Hong, Q.; Qi, K.; Feng, Z.; Huang, Z.; Cui, S.; Wang, L.; Fu, B.; Ding, R.; Yang, J.; Chen, X.; et al. Hyperuricemia induces endothelial dysfunction via mitochondrial Na+/Ca2+ exchanger-mediated mitochondrial calcium overload. Cell Calcium 2012, 51, 402–410. [Google Scholar] [CrossRef]
- Ficociello, L.H.; Rosolowsky, E.T.; Niewczas, M.A.; Maselli, N.J.; Weinberg, J.; Aschengrau, A.; Eckfeldt, J.H.; Stanton, R.C.; Galecki, A.T.; Doria, A.; et al. High-Normal Serum Uric Acid Increases Risk of Early Progressive Renal Function Loss in Type 1 Diabetes. Diabetes Care 2010, 33, 1337–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovind, P.; Rossing, P.; Tarnow, L.; Johnson, R.J.; Parving, H.-H. Serum Uric Acid as a Predictor for Development of Diabetic Nephropathy in Type 1 Diabetes: An Inception Cohort Study. Diabetes 2009, 58, 1668–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doria, A.; Galecki, A.T.; Spino, C.; Pop-Busui, R.; Cherney, D.Z.; Lingvay, I.; Parsa, A.; Rossing, P.; Sigal, R.J.; Afkarian, M.; et al. Serum Urate Lowering with Allopurinol and Kidney Function in Type 1 Diabetes. N. Engl. J. Med. 2020, 382, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Badve, S.V.; Pascoe, E.M.; Tiku, A.; Boudville, N.; Brown, F.G.; Cass, A.; Clarke, P.; Dalbeth, N.; Day, R.O.; De Zoysa, J.R.; et al. Effects of Allopurinol on the Progression of Chronic Kidney Disease. N. Engl. J. Med. 2020, 382, 2504–2513. [Google Scholar] [CrossRef] [PubMed]
- Galbusera, C.; Orth, P.; Fedida, D.; Spector, T. Superoxide radical production by allopurinol and xanthine oxidase. Biochem. Pharmacol. 2006, 71, 1747–1752. [Google Scholar] [CrossRef]
- Haberland, A.; Luther, H.; Schimke, I. Does allopurinol prevent superoxide radical production by xanthine oxidase (XOD)? Inflamm. Res. 1991, 32, 96–97. [Google Scholar] [CrossRef]
- Massey, V.; Komai, H.; Palmer, G.; Elion, G.B. On the mechanism of inactivation of xanthine oxidase by allopurinol and other pyrazolo[3,4-d]pyrimidines. J. Biol. Chem. 1970, 245, 2837–2844. [Google Scholar]
- Takano, Y.; Hase-Aoki, K.; Horiuchi, H.; Zhao, L.; Kasahara, Y.; Kondo, S.; Becker, M.A. Selectivity of febuxostat, a novel non-purine inhibitor of xanthine oxidase/xanthine dehydrogenase. Life Sci. 2005, 76, 1835–1847. [Google Scholar] [CrossRef]
- Lee, H.-J.; Jeong, K.H.; Kim, Y.G.; Moon, J.Y.; Lee, S.H.; Ihm, C.G.; Sung, J.Y.; Lee, T.W. Febuxostat Ameliorates Diabetic Renal Injury in a Streptozotocin-Induced Diabetic Rat Model. Am. J. Nephrol. 2014, 40, 56–63. [Google Scholar] [CrossRef]
- Sircar, D.; Chatterjee, S.; Waikhom, R.; Golay, V.; Raychaudhury, A.; Chatterjee, S.; Pandey, R. Efficacy of Febuxostat for Slowing the GFR Decline in Patients With CKD and Asymptomatic Hyperuricemia: A 6-Month, Double-Blind, Randomized, Placebo-Controlled Trial. Am. J. Kidney Dis. 2015, 66, 945–950. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.; Hosoya, T.; Uchida, S.; Inaba, M.; Makino, H.; Maruyama, S.; Ito, S.; Yamamoto, T.; Tomino, Y.; Ohno, I.; et al. Febuxostat Therapy for Patients With Stage 3 CKD and Asymptomatic Hyperuricemia: A Randomized Trial. Am. J. Kidney Dis. 2018, 72, 798–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niaudet, P. Mitochondrial disorders and the kidney. Arch. Dis. Child. 1998, 78, 387–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinzii, C.; Naini, A.; Salviati, L.; Trevisson, E.; Navas, P.; DiMauro, S.; Hirano, M. A Mutation in Para-Hydroxybenzoate-Polyprenyl Transferase (COQ2) Causes Primary Coenzyme Q10 Deficiency. Am. J. Hum. Genet. 2006, 78, 345–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heeringa, S.F.; Chernin, G.; Chaki, M.; Zhou, W.; Sloan, A.J.; Ji, Z.; Xie, L.X.; Salviati, L.; Hurd, T.W.; Vega-Warner, V.; et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J. Clin. Investig. 2011, 121, 2013–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, L.C.; Schuelke, M.; Quinzii, C.M.; Kanki, T.; Rodenburg, R.J.T.; Naini, A.; DiMauro, S.; Hirano, M. Leigh Syndrome with Nephropathy and CoQ10 Deficiency Due to decaprenyl diphosphate synthase subunit 2 (PDSS2) Mutations. Am. J. Hum. Genet. 2006, 79, 1125–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashraf, S.; Gee, H.Y.; Woerner, S.; Xie, L.X.; Vega-Warner, V.; Lovric, S.; Fang, H.; Song, X.; Cattran, D.C.; Avila-Casado, C.; et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J. Clin. Investig. 2013, 123, 5179–5189. [Google Scholar] [CrossRef] [Green Version]
- Mã¼Ller-Deile, J.; Schiffer, M.; Müller-Deile, J. The Podocyte Power-Plant Disaster and Its Contribution to Glomerulopathy. Front. Endocrinol. 2014, 5, 209. [Google Scholar] [CrossRef] [Green Version]
- Brinkkoetter, P.T.; Bork, T.; Salou, S.; Liang, W.; Mizi, A.; Özel, C.; Koehler, S.; Hagmann, H.H.; Ising, C.; Kuczkowski, A.; et al. Anaerobic Glycolysis Maintains the Glomerular Filtration Barrier Independent of Mitochondrial Metabolism and Dynamics. Cell Rep. 2019, 27, 1551–1566.e5. [Google Scholar] [CrossRef] [Green Version]
- Culic, O.; Gruwel, M.L.; Schrader, J. Energy turnover of vascular endothelial cells. Am. J. Physiol. Physiol. 1997, 273, C205–C213. [Google Scholar] [CrossRef]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Widmeier, E.; Airik, M.; Hugo, H.; Schapiro, D.; Wedel, J.; Ghosh, C.C.; Nakayama, M.; Schneider, R.; Awad, A.M.; Nag, A.; et al. Treatment with 2,4-Dihydroxybenzoic Acid Prevents FSGS Progression and Renal Fibrosis in Podocyte-Specific Coq6 Knockout Mice. J. Am. Soc. Nephrol. 2019, 30, 393–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, J.-H.; Gomez, I.G.; Wada, Y.; Roach, A.; Mahad, D.; Duffield, J.S. Deletion of the Mitochondrial Complex-IV Cofactor Heme A:Farnesyltransferase Causes Focal Segmental Glomerulosclerosis and Interferon Response. Am. J. Pathol. 2018, 188, 2745–2762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widmeier, E.; Yu, S.; Nag, A.; Chung, Y.W.; Nakayama, M.; Fernández-Del-Río, L.; Hugo, H.; Schapiro, D.; Buerger, F.; Choi, W.-I.; et al. ADCK4 Deficiency Destabilizes the Coenzyme Q Complex, Which Is Rescued by 2,4-Dihydroxybenzoic Acid Treatment. J. Am. Soc. Nephrol. 2020, 31, 1191–1211. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; Yucel, N.; Kim, B.; Arany, Z. Local Mitochondrial ATP Production Regulates Endothelial Fatty Acid Uptake and Transport. Cell Metab. 2020, 32, 309–319.e7. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Garrido, C.; Kroemer, G. Life’s smile, death’s grin: Vital functions of apoptosis-executing proteins. Curr. Opin. Cell Biol. 2004, 16, 639–646. [Google Scholar] [CrossRef]
- Kroemer, G.; Reed, J.C. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar] [CrossRef]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of Superoxide Production from Different Sites in the Mitochondrial Electron Transport Chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [Green Version]
- Tahara, E.B.; Navarete, F.D.T.; Kowaltowski, A.J. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free. Radic. Biol. Med. 2009, 46, 1283–1297. [Google Scholar] [CrossRef]
- Ricci, J.-E.; Gottlieb, R.A.; Green, D.R. Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J. Cell Biol. 2003, 160, 65–75. [Google Scholar] [CrossRef]
- Wei, P.Z.; Szeto, C.C. Mitochondrial dysfunction in diabetic kidney disease. Clin. Chim. Acta 2019, 496, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial Fission Triggered by Hyperglycemia Is Mediated by ROCK1 Activation in Podocytes and Endothelial Cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daehn, I.S. Glomerular Endothelial Cell Stress and Cross-Talk with Podocytes in Early Diabetic Kidney Disease. Front. Med. 2018, 5, 76. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wei, C.; Lee, K.; Zhang, W.; He, W.; Chuang, P.; Liu, J.; He, J.C. Comparison of Glomerular and Podocyte mRNA Profiles in Streptozotocin-Induced Diabetes. J. Am. Soc. Nephrol. 2015, 27, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Ramnath, R.D.; Foster, R.R.; Wylie, E.C.; Fridén, V.; Dasgupta, I.; Haraldsson, B.; Welsh, G.I.; Mathieson, P.W.; Satchell, S.C. Reactive Oxygen Species Modulate the Barrier Function of the Human Glomerular Endothelial Glycocalyx. PLoS ONE 2013, 8, e55852. [Google Scholar] [CrossRef] [Green Version]
- Casalena, G.; Yu, L.; Gil, R.; Rodriguez, S.; Sosa, S.; Janssen, W.G.M.; Azeloglu, E.U.; Leventhal, J.S.; Daehn, I.S. The diabetic microenvironment causes mitochondrial oxidative stress in glomerular endothelial cells and pathological crosstalk with podocytes. Cell Commun. Signal. 2020, 18, 1–15. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The Mitochondrial-Targeted Compound SS-31 Re-Energizes Ischemic Mitochondria by Interacting with Cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- Hou, Y.; Li, S.; Wu, M.; Wei, J.; Ren, Y.; Du, C.; Wu, H.; Han, C.; Duan, H.; Shi, Y. Mitochondria-targeted peptide SS-31 attenuates renal injury via an antioxidant effect in diabetic nephropathy. Am. J. Physiol. Physiol. 2016, 310, F547–F559. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Luk, A.O.Y.; Tam, C.H.T.; Xie, F.; Carstensen, B.; Lau, E.S.H.; Lim, C.K.P.; Lee, H.M.; Ng, A.C.W.; Ng, M.C.Y.; et al. Progression of diabetic kidney disease and trajectory of kidney function decline in Chinese patients with Type 2 diabetes. Kidney Int. 2019, 95, 178–187. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, Z.; Liu, Q.; Quan, H.; Cheng, X. Effects of coenzyme Q10 intervention on diabetic kidney disease. Medicine 2019, 98, e15850. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.K.; Billings, I.F.T.; Claessens, A.J.; Roshanravan, B.; Linke, L.; Sundell, M.B.; Ahmad, S.; Shao, B.; Shen, D.; Ikizler, T.A.; et al. Coenzyme Q10 dose-escalation study in hemodialysis patients: Safety, tolerability, and effect on oxidative stress. BMC Nephrol. 2015, 16, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierrel, F. Impact of Chemical Analogs of 4-Hydroxybenzoic Acid on Coenzyme Q Biosynthesis: From Inhibition to Bypass of Coenzyme Q Deficiency. Front. Physiol. 2017, 8, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doimo, M.; Trevisson, E.; Airik, R.; Bergdoll, M.; Santos-Ocaña, C.; Hildebrandt, F.; Navas, P.; Pierrel, F.; Salviati, L. Effect of vanillic acid on COQ6 mutants identified in patients with coenzyme Q10 deficiency. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.S.; Flemming, N.B.; Gallo, L.A.; Fotheringham, A.K.; McCarthy, D.A.; Zhuang, A.; Tang, P.H.; Borg, D.J.; Shaw, H.; Harvie, B.; et al. Targeted mitochondrial therapy using MitoQ shows equivalent renoprotection to angiotensin converting enzyme inhibition but no combined synergy in diabetes. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Baigent, C.; Lennon, R. Should We Increase GFR with Bardoxolone in Alport Syndrome? J. Am. Soc. Nephrol. 2018, 29, 357–359. [Google Scholar] [CrossRef]
- Schulz, E.; Wenzel, P.; Münzel, T.; Daiber, A. Mitochondrial Redox Signaling: Interaction of Mitochondrial Reactive Oxygen Species with Other Sources of Oxidative Stress. Antioxid. Redox Signal. 2014, 20, 308–324. [Google Scholar] [CrossRef]
- Peraçoli, M.T.S.; Bannwart, C.F.; Cristofalo, R.; Borges, V.T.M.; Costa, R.A.A.; Witkin, S.S.; Peraçoli, J.C. Increased Reactive Oxygen Species and Tumor Necrosis Factor-Alpha Production by Monocytes are Associated with Elevated Levels of Uric Acid in Pre-Eclamptic Women. Am. J. Reprod. Immunol. 2011, 66, 460–467. [Google Scholar] [CrossRef]
- D’Ischia, M.; Napolitano, A.; Manini, P.; Panzella, L. Secondary Targets of Nitrite-Derived Reactive Nitrogen Species: Nitrosation/Nitration Pathways, Antioxidant Defense Mechanisms and Toxicological Implications. Chem. Res. Toxicol. 2011, 24, 2071–2092. [Google Scholar] [CrossRef]
- Daiber, A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 897–906. [Google Scholar] [CrossRef] [Green Version]
- Gladden, J.D.; Zelickson, B.R.; Wei, C.-C.; Ulasova, E.; Zheng, J.; Ahmed, M.I.; Chen, Y.; Bamman, M.; Ballinger, S.; Darley-Usmar, V.; et al. Novel insights into interactions between mitochondria and xanthine oxidase in acute cardiac volume overload. Free. Radic. Biol. Med. 2011, 51, 1975–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular Mechanisms of Angiotensin II–Mediated Mitochondrial Dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonn, E.; Yusuf, S.; Hoogwerf, B.; Pogue, J.; Yi, Q.; Zinman, B.; Bosch, J.; Dagenais, G.; Mann, J.F.; Gerstein, H.C. Effects of Vitamin E on Cardiovascular and Microvascular Outcomes in High-Risk Patients With Diabetes: Results of the HOPE Study and MICRO-HOPE Substudy. Diabetes Care 2002, 25, 1919–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Heerspink, H.J.L.; et al. Bardoxolone Methyl in Type 2 Diabetes and Stage 4 Chronic Kidney Disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heerspink, H.J.L.; Parving, H.-H.; Andress, D.L.; Bakris, G.; Correa-Rotter, R.; Hou, F.-F.; Kitzman, D.W.; Kohan, D.; Makino, H.; McMurray, J.J.V.; et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 2019, 393, 1937–1947. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, B.; Fernandez-Prado, R.; Górriz, J.L.; Martinez-Castelao, A.; Navarro-González, J.F.; Porrini, E.; Soler, M.J.; Ortiz, A. Canagliflozin and Renal Events in Diabetes with Established Nephropathy Clinical Evaluation and Study of Diabetic Nephropathy with Atrasentan: What was learned about the treatment of diabetic kidney disease with canagliflozin and atrasentan? Clin. Kidney J. 2019, 12, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Trachtman, H.; Nelson, P.; Adler, S.G.; Campbell, K.N.; Chaudhuri, A.; Derebail, V.K.; Gambaro, G.; Gesualdo, L.; Gipson, D.S.; Hogan, J.; et al. DUET: A Phase 2 Study Evaluating the Efficacy and Safety of Sparsentan in Patients with FSGS. J. Am. Soc. Nephrol. 2018, 29, 2745–2754. [Google Scholar] [CrossRef] [Green Version]
- Ducasa, G.M.; Mitrofanova, A.; Mallela, S.K.; Liu, X.; Molina, J.; Sloan, A.; Pedigo, C.E.; Ge, M.; Santos, J.V.; Hernandez, Y.; et al. ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J. Clin. Investig. 2019, 129, 3387–3400. [Google Scholar] [CrossRef] [Green Version]
- Pedigo, C.E.; Ducasa, G.M.; Leclercq, F.; Sloan, A.; Mitrofanova, A.; Hashmi, T.; Molina-David, J.; Ge, M.; Lassenius, M.I.; Forsblom, C.; et al. Local TNF causes NFATc1-dependent cholesterol-mediated podocyte injury. J. Clin. Investig. 2016, 126, 3336–3350. [Google Scholar] [CrossRef] [Green Version]
- Takagi, S.; Li, J.; Takagaki, Y.; Kitada, M.; Nitta, K.; Takasu, T.; Kanasaki, K.; Koya, D. Ipragliflozin improves mitochondrial abnormalities in renal tubules induced by a high-fat diet. J. Diabetes Investig. 2018, 9, 1025–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Xu, L.; Tian, D.; Xia, P.; Zheng, H.; Wang, L.; Chen, L. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: A meta-analysis of randomized controlled trials. Diabetes, Obes. Metab. 2018, 20, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Levi, J.; Luo, Y.; Myakala, K.; Herman-Edelstein, M.; Qiu, L.; Wang, D.; Peng, Y.; Grenz, A.; Lucia, S.; et al. SGLT2 Protein Expression Is Increased in Human Diabetic Nephropathy. J. Biol. Chem. 2017, 292, 5335–5348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, T.; Maeno, S.; Maeda, Y.; Yamato, M.; Sonoda, N.; Ogawa, Y.; Wakisaka, M.; Inoguchi, T. Amelioration of diabetic nephropathy by SGLT2 inhibitors independent of its glucose-lowering effect: A possible role of SGLT2 in mesangial cells. Sci. Rep. 2019, 9, 4703. [Google Scholar] [CrossRef]
- Uthman, L.; Homayr, A.; Juni, R.P.; Spin, E.L.; Kerindongo, R.; Boomsma, M.; Hollmann, M.W.; Preckel, B.; Koolwijk, P.; Van Hinsbergh, V.W.M.; et al. Empagliflozin and Dapagliflozin Reduce ROS Generation and Restore NO Bioavailability in Tumor Necrosis Factor α-Stimulated Human Coronary Arterial Endothelial Cells. Cell. Physiol. Biochem. 2019, 53, 865–886. [Google Scholar] [CrossRef]
- Li, K.; Sun, J.; Huang, N.; Ma, Y.; Han, F.; Liu, Y.; Hou, N.; Sun, X. Liraglutide improves obesity-induced renal injury by alleviating uncoupling of the glomerular VEGF–NO axis in obese mice. Clin. Exp. Pharmacol. Physiol. 2020, 47, 1978–1984. [Google Scholar] [CrossRef]
- Siddiqi, F.S.; Advani, A. Endothelial-Podocyte Crosstalk: The Missing Link between Endothelial Dysfunction and Albuminuria in Diabetes. Diabetes 2013, 62, 3647–3655. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
The glomerular filtration barrier. (A) Schematic showing a glomerular capillary loop that forms the primary filtering unit of the kidney, illustrating normal (left) and diabetic kidney disease (DKD) (right) morphology features. The glomerular capillary loop is comprised of specialized structures: parietal epithelial cells (PECs), the mesangial cells, visceral epithelial cells (podocytes) with interdigitating foot processes (FP) attached to the glomerular basement membrane (GBM), fenestrated glomerular endothelial cells (GEC), and the glycocalyx. Between podocyte foot processes is the slit diaphragm (SD), forming a bridge and tethering foot processes together. In DKD, there are profound morphological changes to the glomerulus, including thickening of the GBM, mesangial matrix expansion and increases in extracellular matrix deposition occluding its capillaries. Additional changes are GEC dysfunction, degradation of the glycocalyx, fusion of foot processes, disrupted architecture of the slit diaphragm, podocytes depletion and consequential protein leak. (B) There is disrupted bidirectional signaling among the cells in the filtration barrier in DKD, including increased podocyte-derived vascular endothelial growth factor a (VEGFA) in early DKD and decrease of VEGF in progressive disease. There is a loss of angiopoietin-1 (Angpt1) and tyrosine-protein kinase receptor (Tie2) interaction in diabetes, and production of activated protein C (APC) in the glomerulus is reduced because of suppression of thrombomodulin expression. Decreased functional activity of APC affects the permeability of the glomerular capillary wall and enhances apoptosis of GECs and podocytes. Endothelin-1 (Edn1) and endothelin receptor A (Ednra) pathway activation in DKD lead to mitochondrial oxidative stress, endothelial nitric oxide (NO) depletion, and degradation of the endothelial glycocalyx.
Figure 1.
The glomerular filtration barrier. (A) Schematic showing a glomerular capillary loop that forms the primary filtering unit of the kidney, illustrating normal (left) and diabetic kidney disease (DKD) (right) morphology features. The glomerular capillary loop is comprised of specialized structures: parietal epithelial cells (PECs), the mesangial cells, visceral epithelial cells (podocytes) with interdigitating foot processes (FP) attached to the glomerular basement membrane (GBM), fenestrated glomerular endothelial cells (GEC), and the glycocalyx. Between podocyte foot processes is the slit diaphragm (SD), forming a bridge and tethering foot processes together. In DKD, there are profound morphological changes to the glomerulus, including thickening of the GBM, mesangial matrix expansion and increases in extracellular matrix deposition occluding its capillaries. Additional changes are GEC dysfunction, degradation of the glycocalyx, fusion of foot processes, disrupted architecture of the slit diaphragm, podocytes depletion and consequential protein leak. (B) There is disrupted bidirectional signaling among the cells in the filtration barrier in DKD, including increased podocyte-derived vascular endothelial growth factor a (VEGFA) in early DKD and decrease of VEGF in progressive disease. There is a loss of angiopoietin-1 (Angpt1) and tyrosine-protein kinase receptor (Tie2) interaction in diabetes, and production of activated protein C (APC) in the glomerulus is reduced because of suppression of thrombomodulin expression. Decreased functional activity of APC affects the permeability of the glomerular capillary wall and enhances apoptosis of GECs and podocytes. Endothelin-1 (Edn1) and endothelin receptor A (Ednra) pathway activation in DKD lead to mitochondrial oxidative stress, endothelial nitric oxide (NO) depletion, and degradation of the endothelial glycocalyx.

Figure 2.
Generation of excess reactive oxygen species (ROS) in the GECs. Pathological metabolic conditions associated with diabetes mellitus (including hyperglycemia, increased circulation of advanced glycation end products (AGEs), endothelin-1 (Edn1) and free fatty acids (FFAs), as well as activation of RAAS leading to release of angiotensin II (Ang II)) can exacerbate ROS production over the cell’s antioxidant capacity, and this imbalance contributes to endothelial dysfunction in DKD. ROS are generated by enzymatic and nonenzymatic redox reactions during cellular metabolism under normal and pathological conditions. The superoxide anion (O2•−), generated in mitochondria, plasma membrane, peroxisomes, and cytosol, becomes the precursor free radical for the generation of other ROS molecules. Next, cytosolic CuZN superoxide dismutase (SOD) and mitochondrial MnSOD convert O2•− to H2O2, which yields highly reactive hydroxyl radicals (OH•) by interaction with reduced transition metal ions (such as Fe and Cu) in a Fenton reaction. In addition to ROS, cells also generate reactive nitrogen species (RNS). The major RNS include nitric oxide (•NO), peroxynitrite (ONOO−), and nitrogen dioxide (•NO2). Nitric oxide (•NO) is produced by three isoforms of nitric oxide synthase (NOS). Finally, the excess ROS produced causes oxidative damage to mitochondrial and nuclear DNA, lipid and protein oxidation, protein nitration, and mitochondrial dysfunction.
Figure 2.
Generation of excess reactive oxygen species (ROS) in the GECs. Pathological metabolic conditions associated with diabetes mellitus (including hyperglycemia, increased circulation of advanced glycation end products (AGEs), endothelin-1 (Edn1) and free fatty acids (FFAs), as well as activation of RAAS leading to release of angiotensin II (Ang II)) can exacerbate ROS production over the cell’s antioxidant capacity, and this imbalance contributes to endothelial dysfunction in DKD. ROS are generated by enzymatic and nonenzymatic redox reactions during cellular metabolism under normal and pathological conditions. The superoxide anion (O2•−), generated in mitochondria, plasma membrane, peroxisomes, and cytosol, becomes the precursor free radical for the generation of other ROS molecules. Next, cytosolic CuZN superoxide dismutase (SOD) and mitochondrial MnSOD convert O2•− to H2O2, which yields highly reactive hydroxyl radicals (OH•) by interaction with reduced transition metal ions (such as Fe and Cu) in a Fenton reaction. In addition to ROS, cells also generate reactive nitrogen species (RNS). The major RNS include nitric oxide (•NO), peroxynitrite (ONOO−), and nitrogen dioxide (•NO2). Nitric oxide (•NO) is produced by three isoforms of nitric oxide synthase (NOS). Finally, the excess ROS produced causes oxidative damage to mitochondrial and nuclear DNA, lipid and protein oxidation, protein nitration, and mitochondrial dysfunction.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lassén, E.; Daehn, I.S. Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction. Int. J. Mol. Sci. 2020, 21, 9456. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249456
AMA Style
Lassén E, Daehn IS. Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction. International Journal of Molecular Sciences. 2020; 21(24):9456. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249456
Chicago/Turabian StyleLassén, Emelie, and Ilse S. Daehn. 2020. "Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction" International Journal of Molecular Sciences 21, no. 24: 9456. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249456
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.