Ginsenoside M1 Induces Apoptosis and Inhibits the Migration of Human Oral Cancer Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
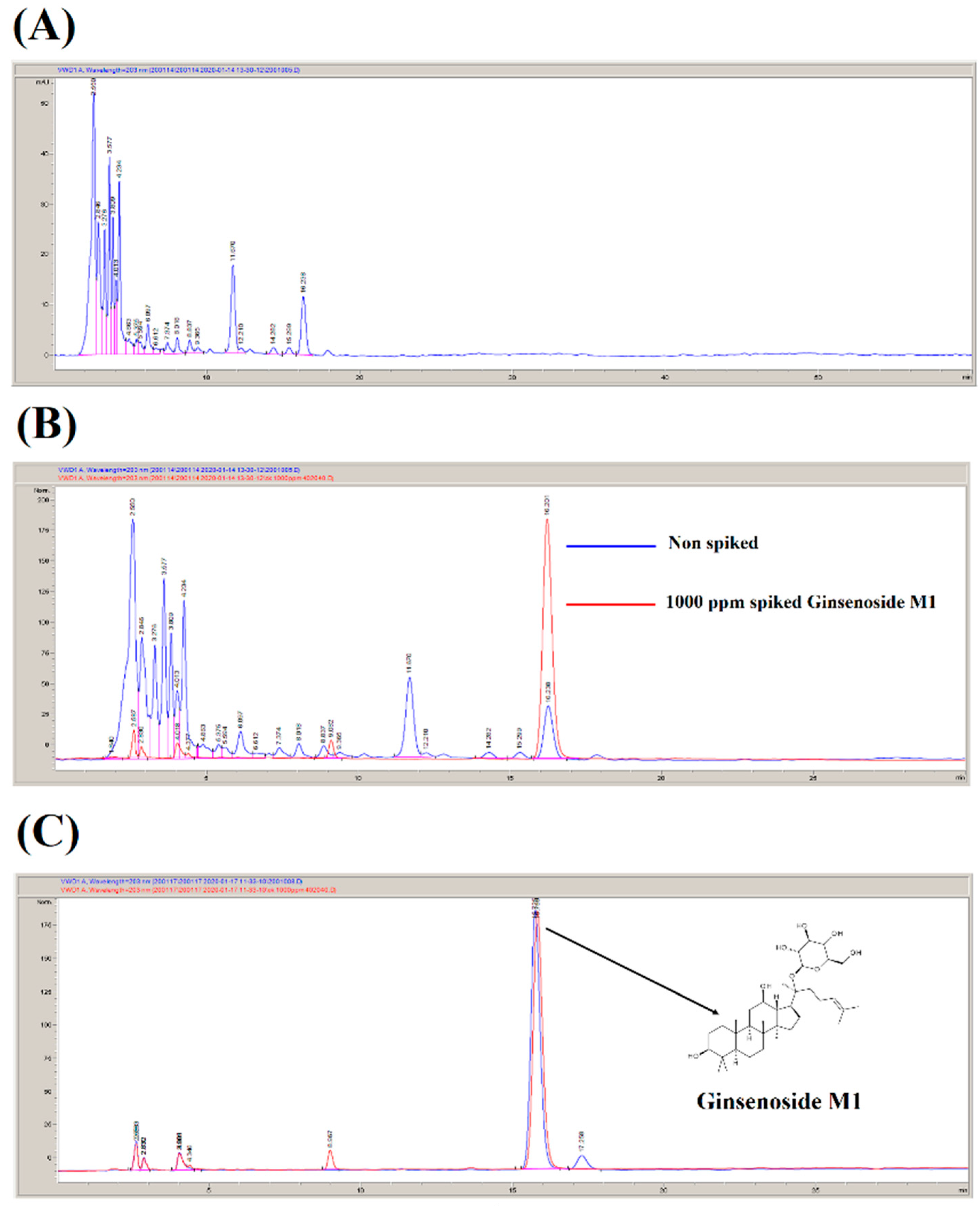
2.1. Preparation of Ginsenoside M1
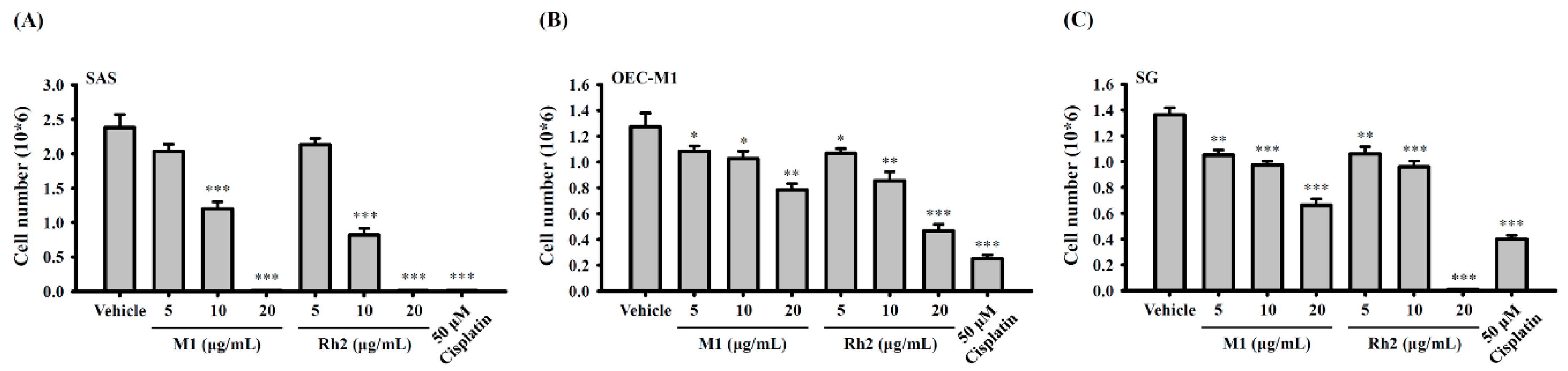
2.2. Ginsenoside M1 Induced Human Oral Cancer Cell Death
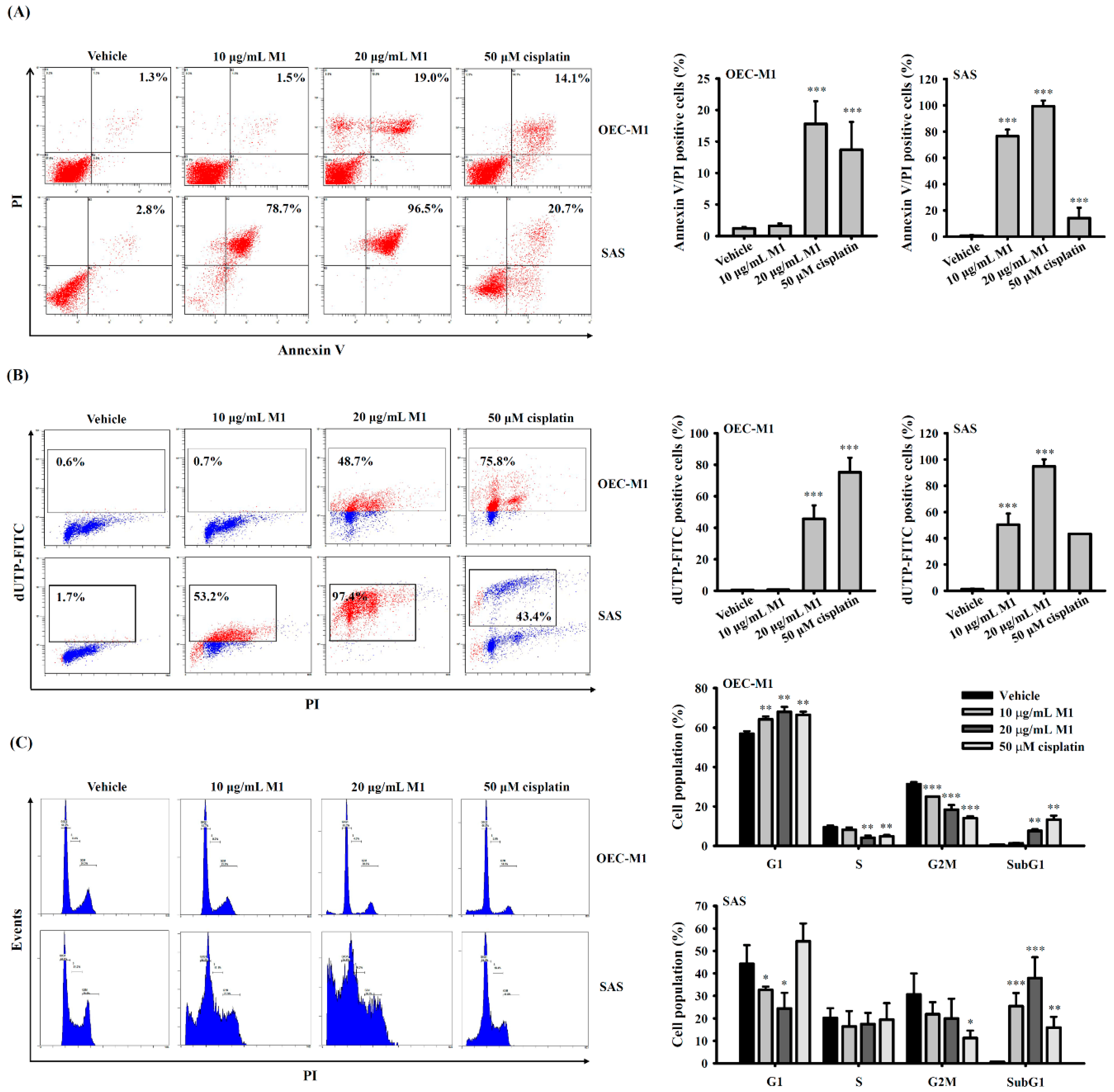
2.3. Ginsenoside M1 Induced Apoptosis in Human Oral Cancer Cells
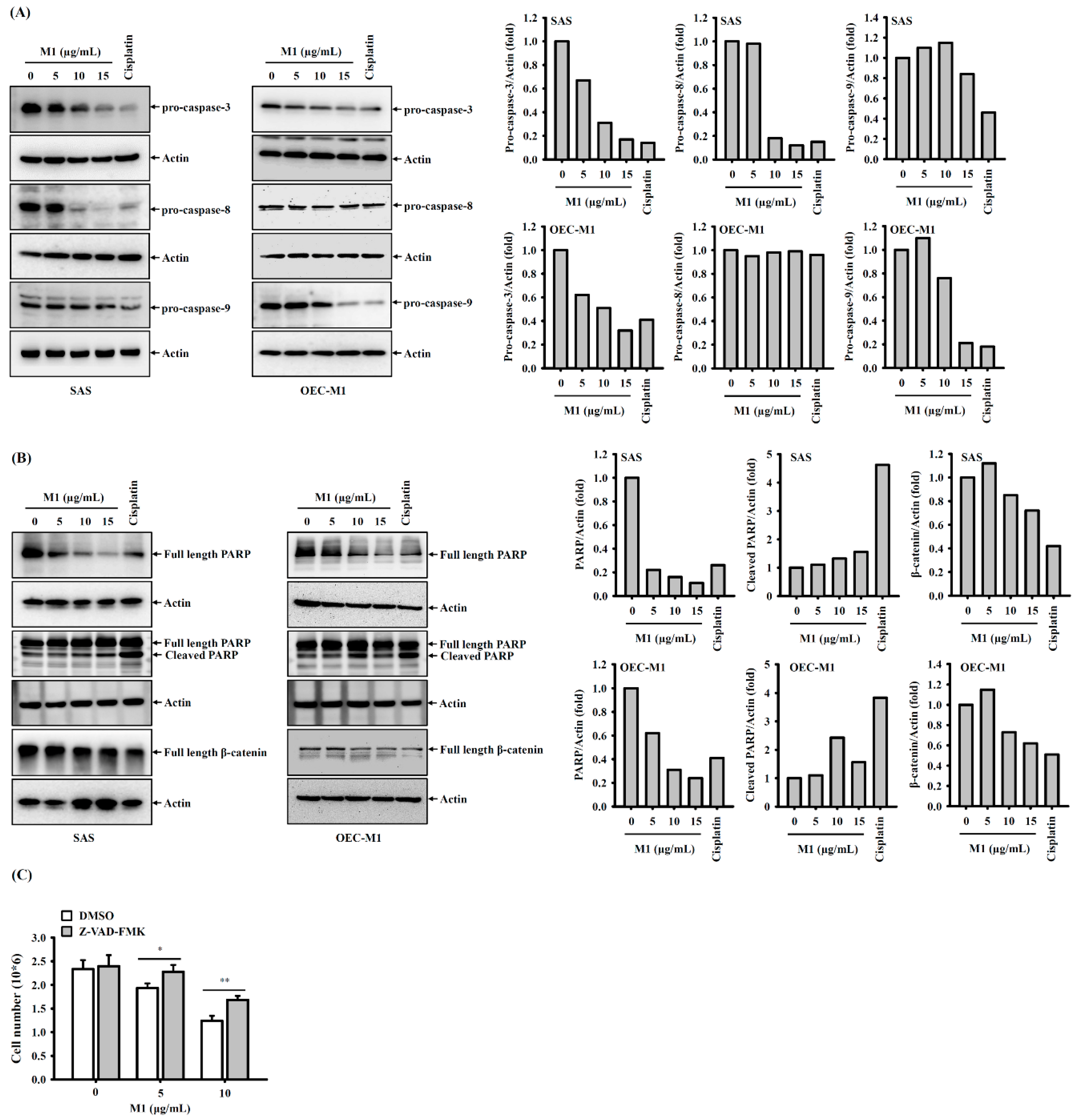
2.4. Ginsenoside M1 Induced Caspase-Dependent Cell Death
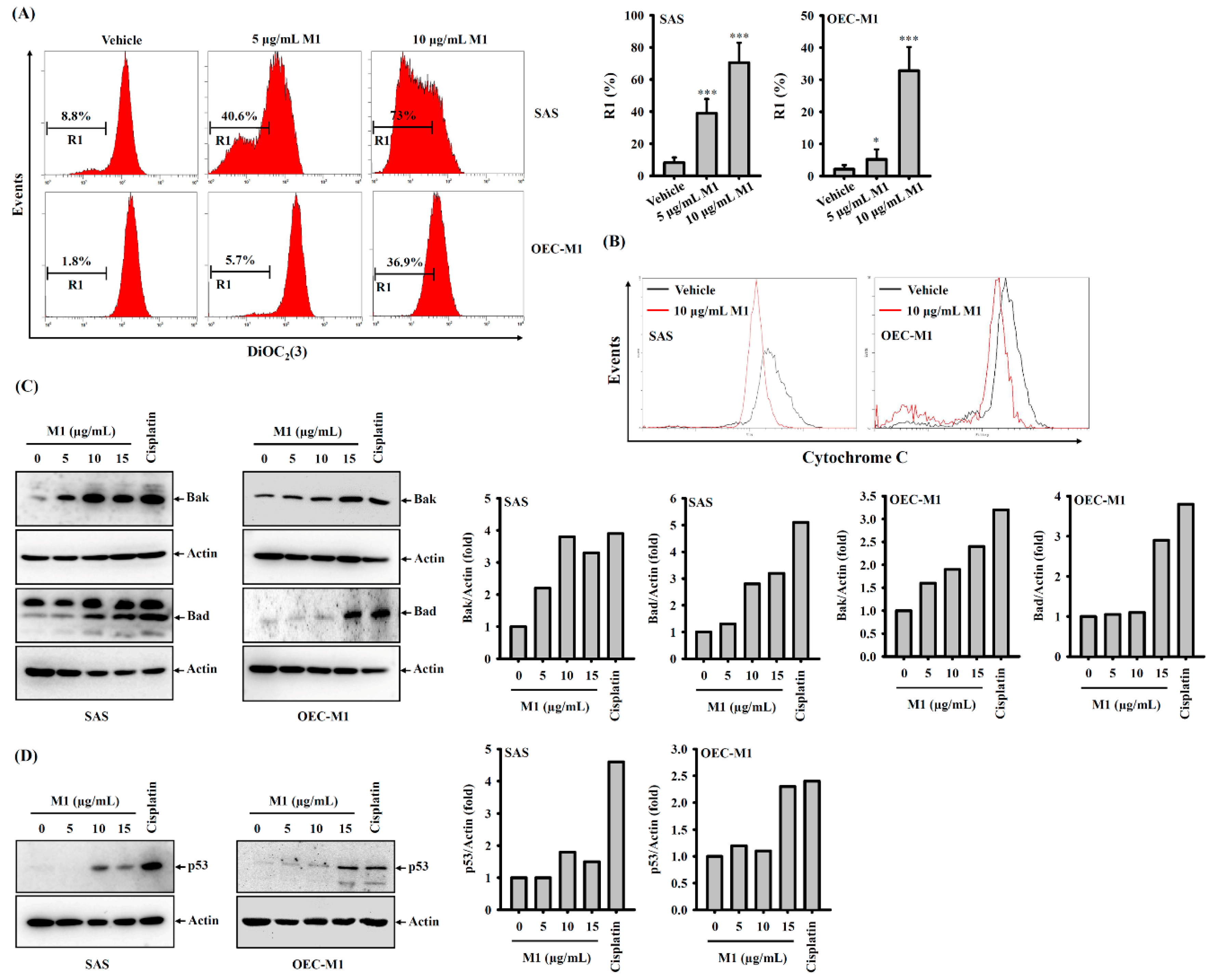
2.5. Ginsenoside M1 Induced the Mitochondrial Death Pathway
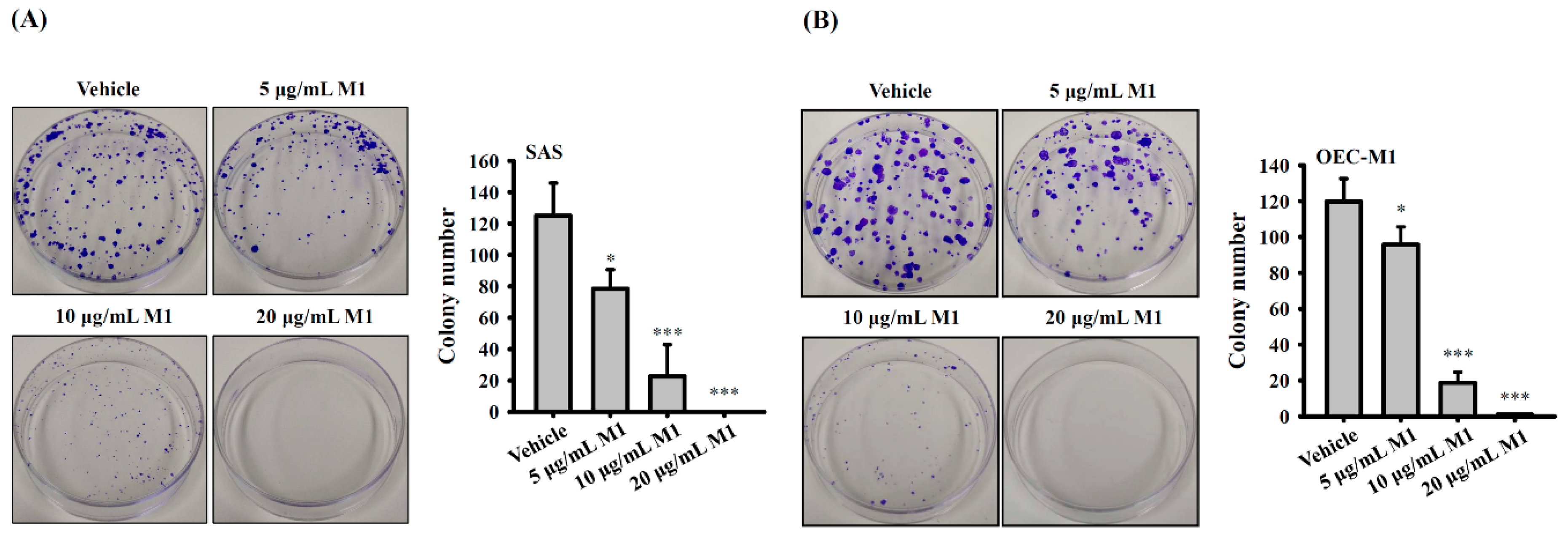
2.6. Ginsenoside M1 Reduced the Colony-Formation Ability of Human Oral Cancer Cells
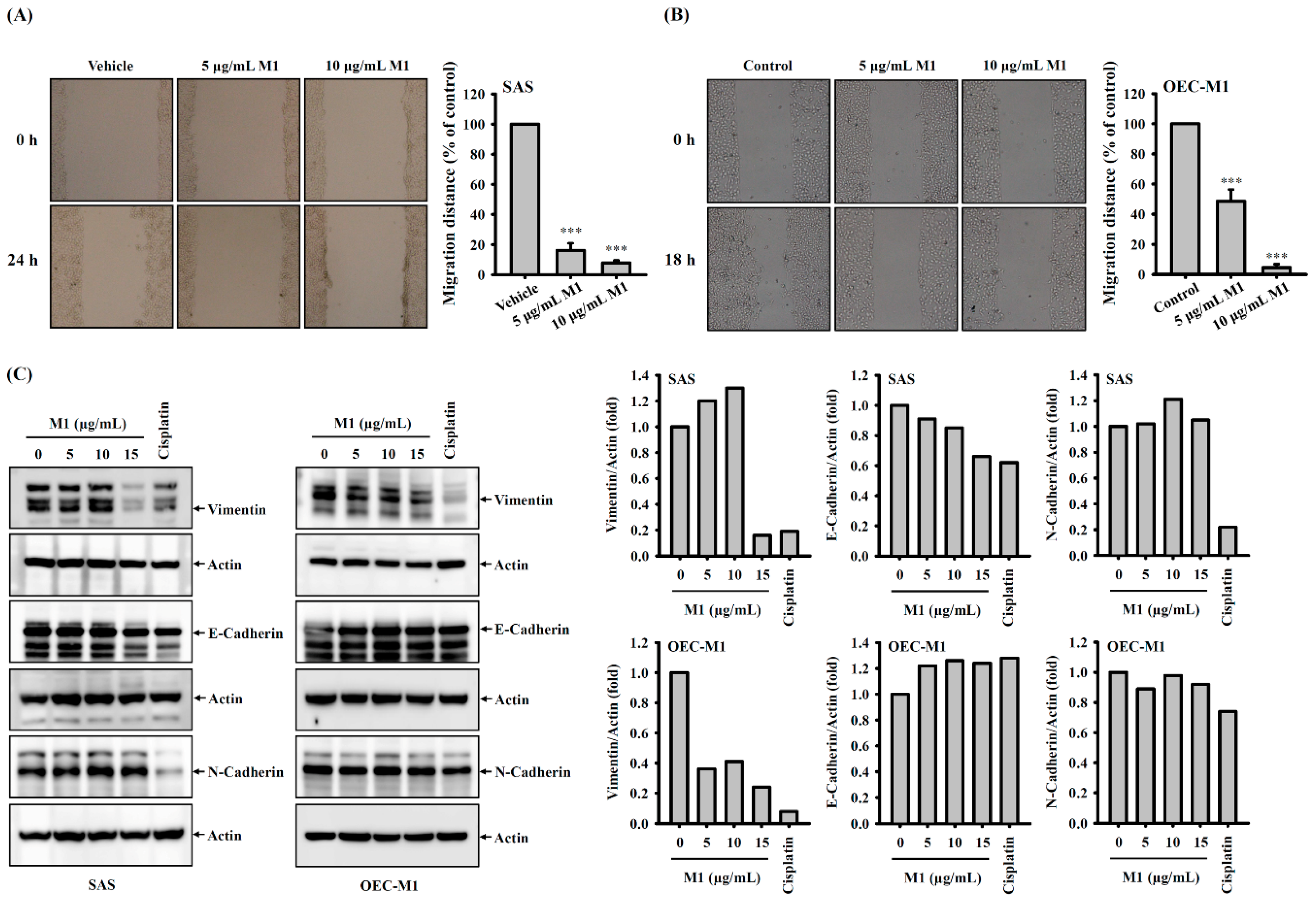
2.7. Ginsenoside M1 Reduced the Migration Ability of Human Oral Cancer Cells
2.8. Ginsenoside M1 Reduced Human Oral Cancer Growth in Mice
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Growth Assay
4.4. Cell Cycle Analysis
4.5. DNA Damage Analysis
4.6. Apoptosis Analysis
4.7. Detection of Mitochondrial Membrane Potential
4.8. Detection of Mitochondrial Cytochrome c Release
4.9. Western Blotting
4.10. Cell Colony Formation Assay
4.11. Cell Migration Assay
4.12. Antitumor Activity In Vivo
4.13. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| OSCC | Oral squamous cell carcinoma |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| Bak | The Bcl-2 homologous antagonist killer |
| Bad | The Bcl-2-associated death promoter |
| PI | Propidium iodide |
| FBS | Fetal bovine serum |
| PBS | Phosphate-buffered saline |
References
- Ghantous, Y.; Abu, I.E. Global incidence and risk factors of oral cancer. Harefuah 2017, 156, 645–649. [Google Scholar] [PubMed]
- Ahmed, N.H.; Naidoo, S. Oral cancer knowledge; attitudes; and practices among dentists in Khartoum State; Sudan. J. Cancer Educ. 2019, 34, 291–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mummudi, N.; Agarwal, J.P.; Chatterjee, S.; Mallick, I.; Ghosh-Laskar, S. Oral cavity cancer in the Indian subcontinent–challenges and opportunities. Clin. Oncol. 2019, 31, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.C.; Lin, H.W.; Yang, J.H.; Lin, P.Y.; Cheng, S.J.; Wu, Y.H.; Kuo, Y.S. Clinical outcomes of oral cancer patients who survive for more than 5 years in Taiwan. J. Formos. Med. Assoc. 2019, 118, 1616–1622. [Google Scholar] [CrossRef]
- Liang, S.Y.; Chang, T.T.; Wu, W.W.; Wang, T.J. Caring for patients with oral cancer in Taiwan: The challenges faced by family caregivers. Eur. J. Cancer Care 2019, 28, e12891. [Google Scholar] [CrossRef] [Green Version]
- Bessell, A.; Glenny, A.M.; Furness, S.; Clarkson, J.E.; Oliver, R.; Conway, D.I.; Macluskey, M.; Pavitt, S.; Sloan, P.; Worthington, H.V. Interventions for the treatment of oral and oropharyngeal cancers: Surgical treatment. Cochrane Database Syst. Rev. 2011, 7, CD006205. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.H.; Wang, T.J.; Lin, Y.P.; Lin, H.R.; Hu, W.Y.; Wung, S.H.; Liang, S.Y. The illness experience of middle-aged men with oral cancer. J. Clin. Nurs. 2013, 22, 3549–3556. [Google Scholar] [CrossRef]
- Shahid, S. Review of hematological indices of cancer patients receiving combined chemotherapy & radiotherapy or receiving radiotherapy alone. Crit. Rev. Oncol. Hematol. 2016, 105, 145–155. [Google Scholar]
- Polverini, P.J.; Nor, J.E. Apoptosis and predisposition to oral cancer. Crit. Rev. Oral Biol. Med. 1999, 10, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Kiraz, Y.; Adan, A.; Kartal Yandim, M.; Baran, Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. 2016, 37, 8471–8486. [Google Scholar] [CrossRef] [Green Version]
- Hua, K.F.; Liao, P.C.; Fang, Z.; Yang, F.L.; Yang, Y.L.; Chen, Y.L.; Chiu, Y.C.; Liu, M.L.; Lam, Y.; Wu, S.H. Generation of reactive oxygen species by polyenylpyrroles derivatives causes DNA damage leading to G2/M arrest and apoptosis in human oral squamous cell carcinoma cells. PLoS ONE 2013, 8, e67603. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Liao, P.C.; Yang, Y.L.; Yang, F.L.; Chen, Y.L.; Lam, Y.; Hua, K.F.; Wu, S.H. Synthesis and biological evaluation of polyenylpyrrole derivatives as anticancer agents acting through caspases-dependent apoptosis. J. Med. Chem. 2010, 53, 7967–7978. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Zhu, W.G. p53: Structure; function and therapeutic applications. J. Cancer Mol. 2006, 2, 141–153. [Google Scholar]
- Yang, L.; Zou, H.; Gao, Y.; Luo, J.; Xie, X.; Meng, W.; Zhou, H.; Tan, Z. Insights into gastrointestinal microbiota-generated ginsenoside metabolites and their bioactivities. Drug Metab. Rev. 2020, 52, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.D.; Yang, Y.Y.; Ouyang, D.S.; Yang, G.P. A review of biotransformation and pharmacology of ginsenoside compound K. Fitoterapia 2015, 100, 208–220. [Google Scholar] [CrossRef]
- Chen, L.; Chen, M.Y.; Shao, L.; Zhang, W.; Rao, T.; Zhou, H.H.; Huang, W.H. Panax notoginseng saponins prevent colitis-associated colorectal cancer development: The role of gut microbiota. Chin. J. Nat. Med. 2020, 18, 500–507. [Google Scholar] [CrossRef]
- Oh, J.M.; Kim, E.; Chun, S. Ginsenoside Compound K Induces Ros-Mediated Apoptosis and Autophagic Inhibition in Human Neuroblastoma Cells In Vitro and In Vivo. Int. J. Mol. Sci. 2019, 20, 4279. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Dong, Y.; Wang, L.; Xu, G.; Yang, Q.; Tang, X.; Qiao, Y.; Cong, Z. Ginsenoside metabolite compound K induces apoptosis and autophagy in non-small cell lung cancer cells via AMPK-mTOR and JNK pathways. Biochem. Cell Biol. 2019, 97, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, S.; Sun, Q.; Jiao, W.; Yan, Y.; Zhang, X. Compound K Induces Endoplasmic Reticulum Stress and Apoptosis in Human Liver Cancer Cells by Regulating STAT3. Molecules 2018, 23, 1482. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Li, Y. Effects of ginsenoside compound K combined with cisplatin on the proliferation; apoptosis and epithelial mesenchymal transition in MCF-7 cells of human breast cancer. Pharm. Biol. 2016, 54, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Song, G.; Zhang, B.; Liu, Z.; Chen, R.; Zhang, H.; Hu, T. Intestinal metabolite compound K of panaxoside inhibits the growth of gastric carcinoma by augmenting apoptosis via Bid-mediated mitochondrial pathway. J. Cell Mol. Med. 2012, 16, 96–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.Y.; Hua, K.F.; Hsu, W.H.; Suzuki, Y.; Chu, L.J.; Lee, Y.C.; Takahata, A.; Lee, S.L.; Wu, C.C.; Nikolic-Paterson, D.J.; et al. IgA Nephropathy Benefits from Compound K Treatment by Inhibiting NF-κB/NLRP3 Inflammasome and Enhancing Autophagy and SIRT1. J. Immunol. 2020, 205, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.H.; Hua, K.F.; Tuan, L.H.; Tsai, Y.L.; Chu, L.J.; Lee, Y.C.; Wong, W.T.; Lee, S.L.; Lai, J.H.; Chu, C.L.; et al. Compound K inhibits priming and mitochondria-associated activating signals of NLRP3 inflammasome in renal tubulointerstitial lesions. Nephrol. Dial. Transpl. 2020, 35, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.L.; Lee, Y.C. Method for Preparing 20-O-b-D-glucopyranosyl-20(S)-protopanaxadiol (ginsenoside M1) by Using Sanqi Leaves and Stems. U.S. Patent US7932057B2, 26 April 2011. [Google Scholar]
- Luo, H.; Vong, C.T.; Chen, H.; Gao, Y.; Lyu, P.; Qiu, L.; Zhao, M.; Liu, Q.; Cheng, Z.; Zou, J.; et al. Naturally occurring anti-cancer compounds: Shining from Chinese herbal medicine. Chin. Med. 2019, 14, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiu, Y.; Ma, L.; Zhao, H.; Sun, X.; Li, X.; Liu, S. Differentiation and identification of ginsenoside structural isomers by two-dimensional mass spectrometry combined with statistical analysis. J. Ginseng Res. 2019, 43, 368–376. [Google Scholar]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Christensen, L.P. Ginsenosides chemistry; biosynthesis; analysis; and potential health effects. Adv. Food Nutr. Res. 2009, 55, 1–99. [Google Scholar]
- Tawab, M.A.; Bahr, U.; Karas, M.; Wurglics, M.; Schubert-Zsilavecz, M. Degradation of ginsenosides in humans after oral administration. Drug Metab. Dispos. 2003, 31, 1065–1071. [Google Scholar] [CrossRef] [Green Version]
- Karikura, M.; Miyase, T.; Tanizawa, H.; Taniyama, T.; Takino, Y. Studies on absorption; distribution; excretion and metabolism of ginseng saponins. VII. Comparison of the decomposition modes of ginsenoside-Rb1 and -Rb2 in the digestive tract of rats. Chem. Pharm. Bull 1991, 39, 2357–2361. [Google Scholar]
- Chen, L.; Meng, Y.; Sun, Q.; Zhang, Z.; Guo, X.; Sheng, X.; Tai, G.; Cheng, H.; Zhou, Y. Ginsenoside compound K sensitizes human colon cancer cells to TRAIL-induced apoptosis via autophagy-dependent and -independent DR5 upregulation. Cell Death Dis. 2016, 7, e2334. [Google Scholar] [CrossRef]
- Kim, A.D.; Kang, K.A.; Kim, H.S.; Kim, D.H.; Choi, Y.H.; Lee, S.J.; Kim, H.S.; Hyun, J.W. A ginseng metabolite; compound K; induces autophagy and apoptosis via generation of reactive oxygen species and activation of JNK in human colon cancer cells. Cell Death Dis. 2013, 4, e750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Kim, J.E.; Song, N.R.; Jung, S.K.; Lee, M.H.; Park, J.S.; Yeom, M.H.; Bode, A.M.; Dong, Z.; Lee, K.W. The ginsenoside 20-O-β-D-glucopyranosyl-20(S)-protopanaxadiol induces autophagy and apoptosis in human melanoma via AMPK/JNK phosphorylation. PLoS ONE 2014, 9, e104305. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Huang, Q.; Wang, M.; Yan, X.; Song, X.; Ma, R.; Jiang, R.; Zhao, D.; Sun, L. Compound K Inhibits Autophagy-Mediated Apoptosis Through Activation of the PI3K-Akt Signaling Pathway Thus Protecting Against Ischemia/Reperfusion Injury. Cell Physiol. Biochem. 2018, 47, 2589–2601. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Lou, T.; Wang, M.; Xue, L.; Lu, J.; Zhang, H.; Zhang, Z.; Wang, H.; Jing, C.; Zhao, D.; et al. Compound K inhibits autophagy-mediated apoptosis induced by oxygen and glucose deprivation/reperfusion via regulating AMPK-mTOR pathway in neurons. Life Sci. 2020, 254, 117793. [Google Scholar] [CrossRef]
- Wang, C.Z.; Du, G.J.; Zhang, Z.; Wen, X.D.; Calway, T.; Zhen, Z.; Musch, M.W.; Bissonnette, M.; Chang, E.B.; Yuan, C.S. Ginsenoside compound K; not Rb1; possesses potential chemopreventive activities in human colorectal cancer. Int. J. Oncol. 2012, 40, 1970–1976. [Google Scholar]
- Cho, S.H.; Chung, K.S.; Choi, J.H.; Kim, D.H.; Lee, K.T. Compound K, a metabolite of ginseng saponin, induces apoptosis via caspase-8-dependent pathway in HL-60 human leukemia cells. BMC Cancer 2009, 9, 449. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.X.; Li, Y.; Sun, C.; Jiang, D.; Lin, Y.J.; Jin, F.X.; Lee, S.K.; Jin, Y.H. p53-dependent Fas expression is critical for Ginsenoside Rh2 triggered caspase-8 activation in HeLa cells. Protein Cell 2014, 5, 224–234. [Google Scholar]
- Pucci, B.; Kasten, M.; Giordano, A. Cell cycle and apoptosis. Neoplasia 2000, 2, 291. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Chen, H.G.; Yan, Q.; Deng, M.; Liu, J.; Doerge, S.; Ma, W.; Dong, Z.; Li, D.W. Protein phosphatase-2A is a target of epigallocatechin-3-gallate and modulates p53-Bak apoptotic pathway. Cancer Res. 2008, 68, 4150–4162. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Nakajima, W.; Seike, M.; Gemma, A.; Tanaka, N. Cisplatin-induced apoptosis in non-small-cell lung cancer cells is dependent on Bax- and Bak-induction pathway and synergistically activated by BH3-mimetic ABT-263 in p53 wild-type and mutant cells. Biochem. Biophys. Res. Commun. 2016, 473, 490–496. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Heese, K.; Wu, M. The Bad guy cooperates with good cop p53: Bad is transcriptionally up-regulated by p53 and forms a Bad/p53 complex at the mitochondria to induce apoptosis. Mol. Cell Biol. 2006, 26, 9071–9082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Zhao, J.; Wang, C.Z.; Searle, J.; He, T.C.; Yuan, C.S.; Du, W. Ginsenoside Rh2 induces apoptosis and paraptosis-like cell death in colorectal cancer cells through activation of p53. Cancer Lett. 2011, 301, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Wang, T.; Wang, G.; Li, G.; Sun, C.; Jiang, Z.; Yang, J.; Li, Y.; You, Y.; Wu, X.; et al. Preclinical safety of ginsenoside compound K: Acute; and 26-week oral toxicity studies in mice and rats. Food Chem. Toxicol. 2019, 131, 110578. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.H.; Min, J.W.; Jin, Y.; Wang, C.; Kim, Y.J.; Yang, D.C. Enzymatic biotransformation of ginsenoside Rb1 to compound K by recombinant β-glucosidase from Microbacterium esteraromaticum. J. Agric. Food Chem. 2012, 60, 3776–3781. [Google Scholar]
- Quan, L.H.; Piao, J.Y.; Min, J.W.; Kim, H.B.; Kim, S.R.; Yang, D.U.; Yang, D.C. Biotransformation of Ginsenoside Rb1 to Prosapogenins; Gypenoside XVII, Ginsenoside Rd; Ginsenoside F2; and Compound K by Leuconostoc mesenteroides DC102. J. Ginseng Res. 2011, 35, 344–351. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-C.; Wong, W.-T.; Li, L.-H.; Chu, L.J.; Menon, M.P.; Ho, C.-L.; Chernikov, O.V.; Lee, S.-L.; Hua, K.-F. Ginsenoside M1 Induces Apoptosis and Inhibits the Migration of Human Oral Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9704. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249704
Lee Y-C, Wong W-T, Li L-H, Chu LJ, Menon MP, Ho C-L, Chernikov OV, Lee S-L, Hua K-F. Ginsenoside M1 Induces Apoptosis and Inhibits the Migration of Human Oral Cancer Cells. International Journal of Molecular Sciences. 2020; 21(24):9704. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249704
Chicago/Turabian StyleLee, Yu-Chieh, Wei-Ting Wong, Lan-Hui Li, Lichieh Julie Chu, Mridula P. Menon, Chen-Lung Ho, Oleg V. Chernikov, Sheau-Long Lee, and Kuo-Feng Hua. 2020. "Ginsenoside M1 Induces Apoptosis and Inhibits the Migration of Human Oral Cancer Cells" International Journal of Molecular Sciences 21, no. 24: 9704. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249704