The Histamine 3 Receptor Is Expressed in the Heart and Its Activation Opposes Adverse Cardiac Remodeling in the Angiotensin II Mouse Model

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
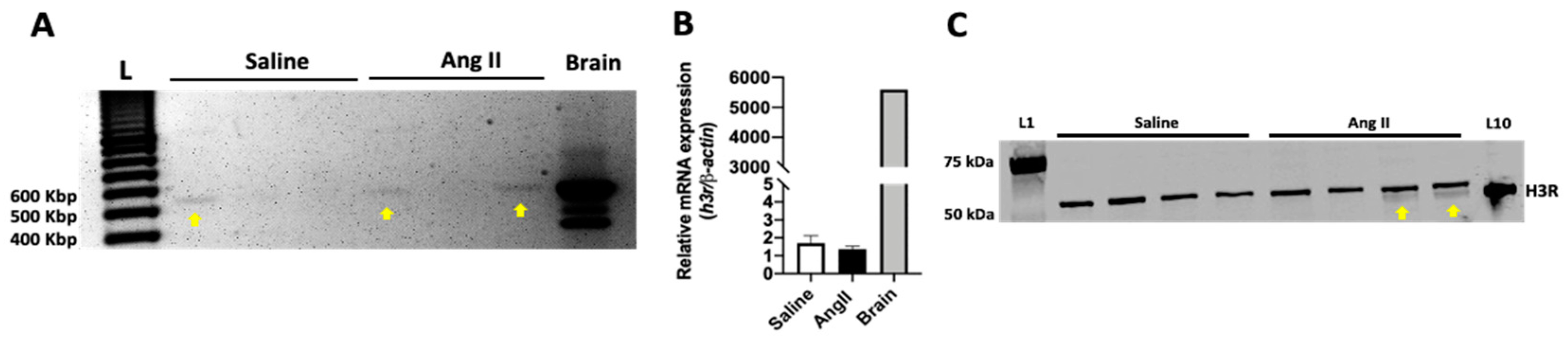
2.1. The H3R Is Expressed in the Mouse Heart
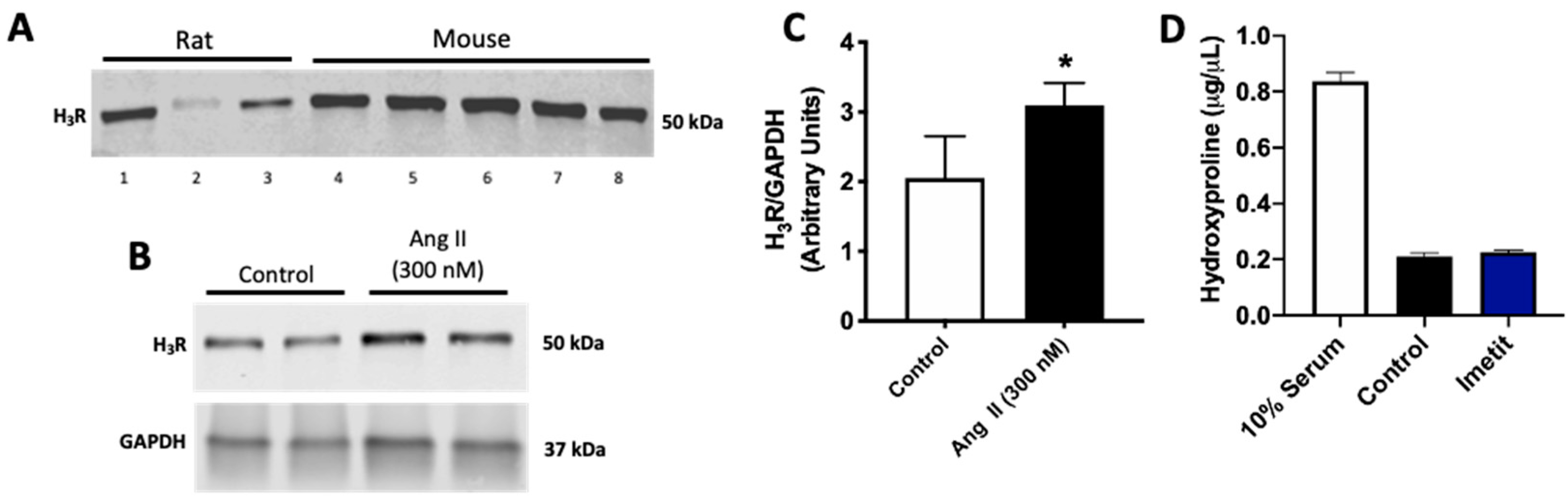
2.2. Ang II Increases H3R Levels in Isolated Cardiac Fibroblasts
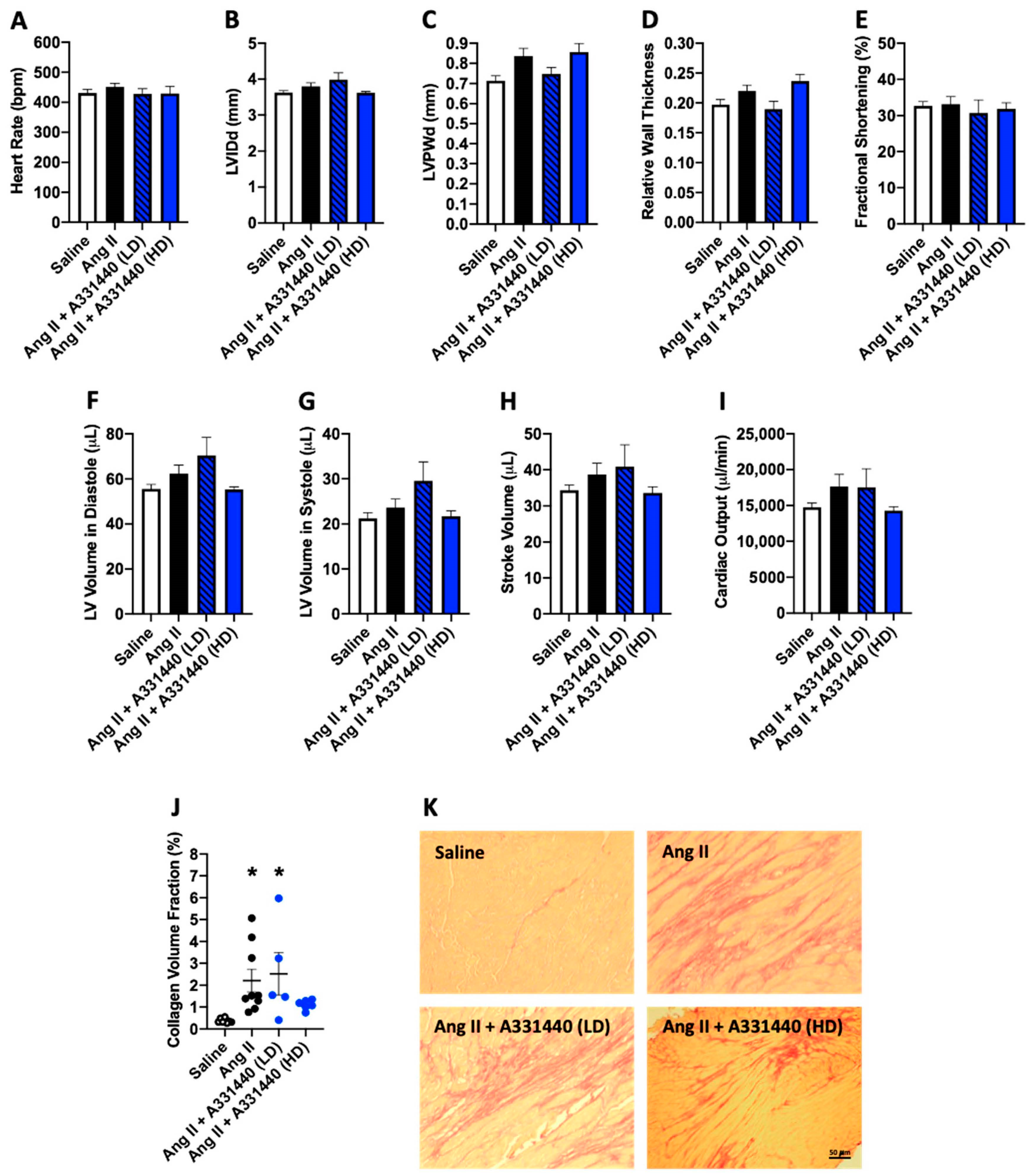
2.3. Effect of H3R Blockade on LV Structure and Function in Ang-II-Infused Mice
2.4. H3R Blockade Did Not Alter Cardiac Fibrosis
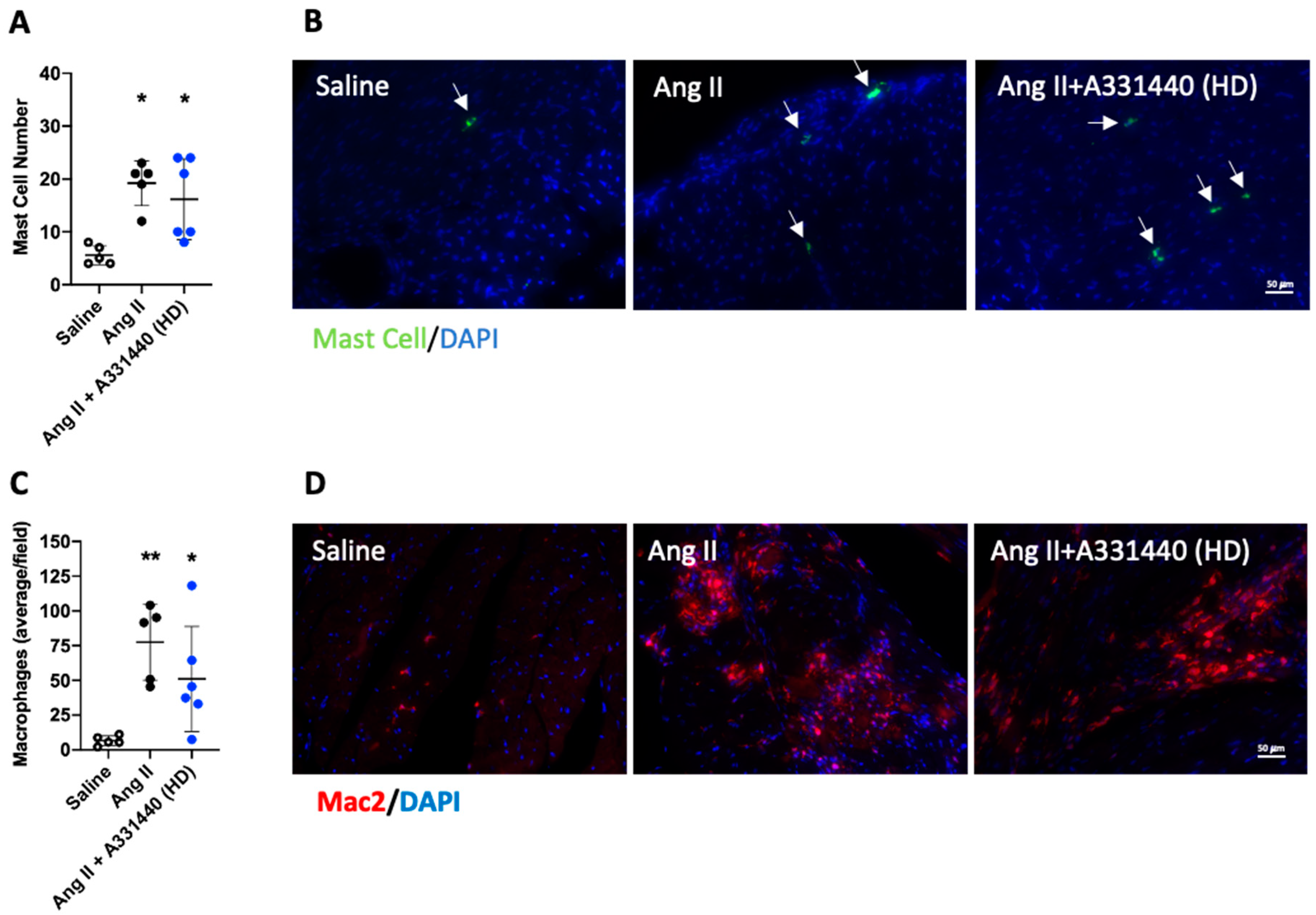
2.5. H3R Blockade Did Not Alter Inflammation
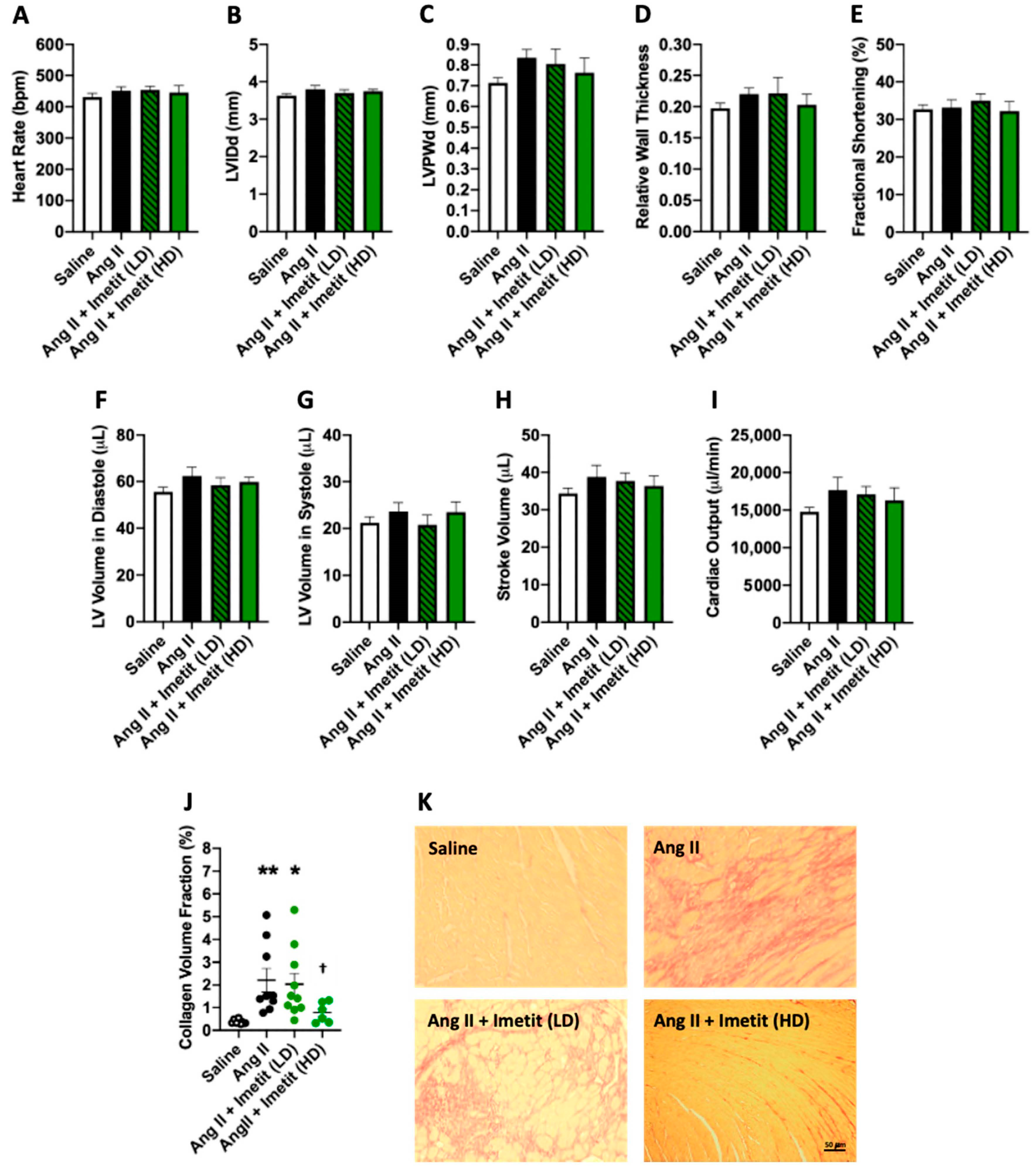
2.6. Effect of H3R Activation on LV Structure and Function in Ang-II-Infused Mice
2.7. H3R Activation Does Alter Cardiac Fibrosis
2.8. Norepinephrine Levels in the Heart
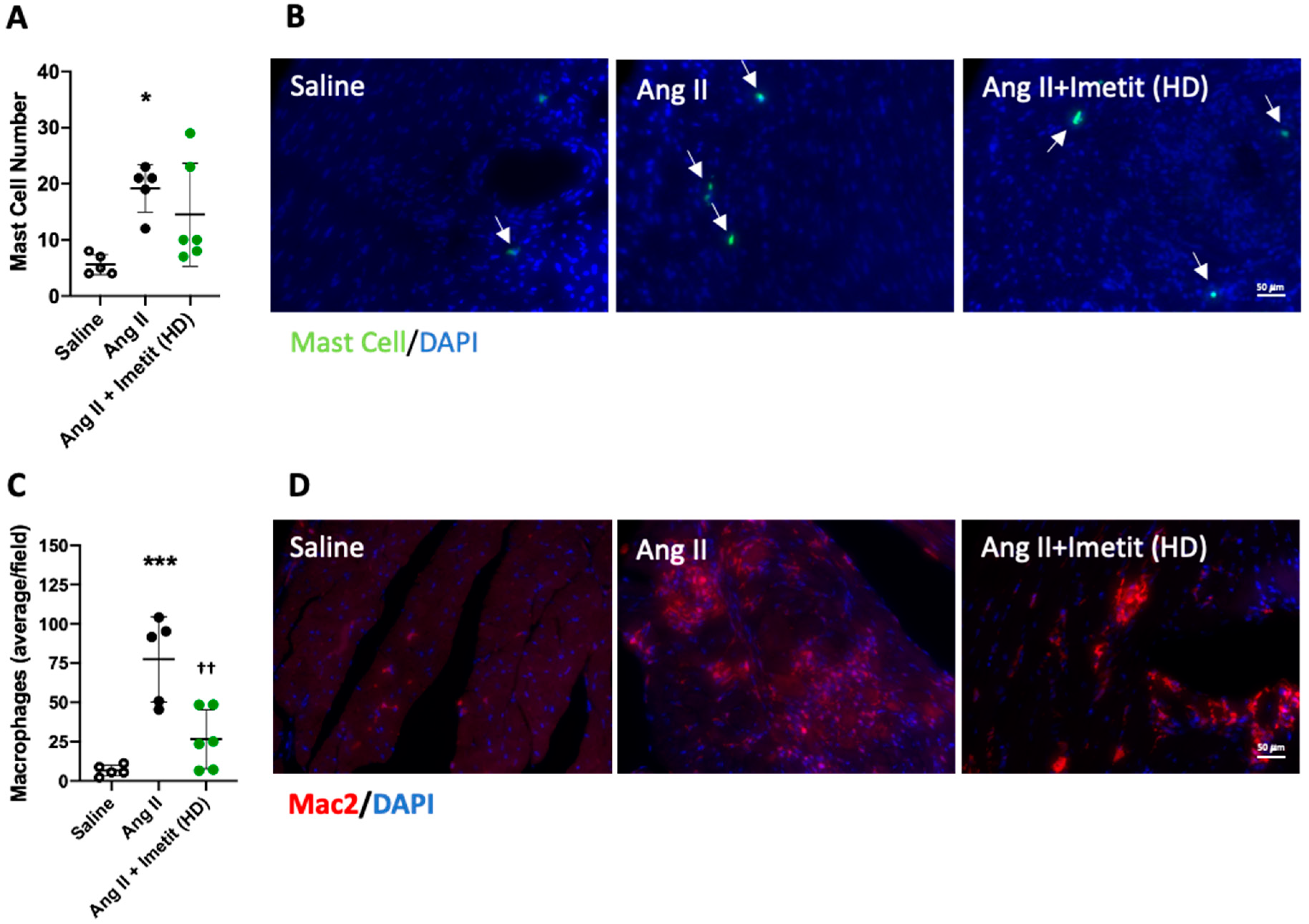
2.9. H3R Activation Reduced Inflammation
3. Discussion
4. Materials and Methods
4.1. Experimental Design
4.2. Echocardiography
4.3. Histology/Immunofluorescence
4.4. Isolation and Treatment of Cardiac Fibroblasts
4.5. PCR/Sequencing
4.6. Protein Analysis
4.7. Hydroxyproline Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| H1R | Histamine 1 receptor |
| H2R | Histamine 2 receptor |
| H3R | Histamine 3 receptor |
| H4R | Histamine 4 receptor |
| MESA | Multi-ethnic Study of Atherosclerosis |
| Ang II | Angiotensin II |
| LV | Left ventricle |
| RV | Right ventricle |
| LVIDd | Left ventricular internal diameter in diastole |
| LVPWd | Left ventricular posterior wall thickness in diastole |
References
- Levick, S.P.; McLarty, J.L.; Murray, D.B.; Freeman, R.M.; Carver, W.E.; Brower, G.L. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension 2009, 53, 1041–1047. [Google Scholar] [CrossRef]
- Levick, S.P.; Melendez, G.C.; Plante, E.; McLarty, J.L.; Brower, G.L.; Janicki, J.S. Cardiac mast cells: The centrepiece in adverse myocardial remodelling. Cardiovasc. Res. 2011, 89, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Jubair, S.; Levick, S.P.; Janicki, J.S. The autocrine role of tryptase in pressure overload-induced mast cell activation, chymase release and cardiac fibrosis. IJC Metab. Endocr. 2016, 10, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLarty, J.L.; Melendez, G.C.; Brower, G.L.; Janicki, J.S.; Levick, S.P. Tryptase/Protease-activated receptor 2 interactions induce selective mitogen-activated protein kinase signaling and collagen synthesis by cardiac fibroblasts. Hypertension 2011, 58, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Widiapradja, A.; Manteufel, E.J.; Dehlin, H.M.; Pena, J.; Goldspink, P.H.; Sharma, A.; Kolb, L.L.; Imig, J.D.; Janicki, J.S.; Lu, B.; et al. Regulation of cardiac mast cell maturation and function by the neurokinin-1 receptor in the fibrotic heart. Sci. Rep. 2019, 9, 11004. [Google Scholar] [CrossRef] [PubMed]
- Janicki, J.S.; Brower, G.L.; Levick, S.P. The emerging prominence of the cardiac mast cell as a potent mediator of adverse myocardial remodeling. Methods Mol. Biol. 2015, 1220, 121–139. [Google Scholar]
- Kanemitsu, H.; Takai, S.; Tsuneyoshi, H.; Nishina, T.; Yoshikawa, K.; Miyazaki, M.; Ikeda, T.; Komeda, M. Chymase inhibition prevents cardiac fibrosis and dysfunction after myocardial infarction in rats. Hypertens. Res. 2006, 29, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.Y.; Zhao, L.Y.; Zheng, Q.S.; Su, J.L.; Guan, H.; Shang, F.J.; Niu, X.L.; He, Y.P.; Lu, X.L. Chymase induces profibrotic response via transforming growth factor-beta 1/Smad activation in rat cardiac fibroblasts. Mol. Cell. Biochem. 2008, 310, 159–166. [Google Scholar] [CrossRef]
- Levick, S.P.; Widiapradja, A. Mast cells: Key contributors to cardiac fibrosis. Int. J. Mol. Sci. 2018, 19, 231. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Shen, L.; Li, X.; Luo, T.; Wei, X.; Zhang, J.; Cao, S.; Huang, X.; Fukushima, Y.; Bin, J.; et al. Disruption of histamine H2 receptor slows heart failure progression through reducing myocardial apoptosis and fibrosis. Clin. Sci. 2014, 127, 435–448. [Google Scholar] [CrossRef]
- Leary, P.J.; Tedford, R.J.; Bluemke, D.A.; Bristow, M.R.; Heckbert, S.R.; Kawut, S.M.; Krieger, E.V.; Lima, J.A.; Masri, C.S.; Ralph, D.D.; et al. Histamine H2 receptor antagonists, left ventricular morphology, and heart failure risk: The MESA study. J. Am. Coll. Cardiol. 2016, 67, 1544–1552. [Google Scholar] [CrossRef] [PubMed]
- Imamura, M.; Poli, E.; Omoniyi, A.T.; Levi, R. Unmasking of activated histamine H3-receptors in myocardial ischemia: Their role as regulators of exocytotic norepinephrine release. J. Pharmacol. Exp. Ther. 1994, 271, 1259–1266. [Google Scholar] [PubMed]
- Gothert, M.; Garbarg, M.; Hey, J.A.; Schlicker, E.; Schwartz, J.C.; Levi, R. New aspects of the role of histamine in cardiovascular function: Identification, characterization, and potential pathophysiological importance of H3 receptors. Can. J. Physiol. Pharmacol. 1995, 73, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Imamura, M.; Seyedi, N.; Lander, H.M.; Levi, R. Functional identification of histamine H3-receptors in the human heart. Circ. Res. 1995, 77, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Hatta, E.; Yasuda, K.; Levi, R. Activation of histamine H3 receptors inhibits carrier-mediated norepinephrine release in a human model of protracted myocardial ischemia. J. Pharmacol. Exp. Ther. 1997, 283, 494–500. [Google Scholar]
- Imamura, M.; Lander, H.M.; Levi, R. Activation of histamine H3-receptors inhibits carrier-mediated norepinephrine release during protracted myocardial ischemia. Comparison with adenosine A1-receptors and alpha2-adrenoceptors. Circ. Res. 1996, 78, 475–481. [Google Scholar] [CrossRef]
- Piera, L.; Olczak, S.; Kun, T.; Galdyszynska, M.; Ciosek, J.; Szymanski, J.; Drobnik, J. Disruption of histamine/H3 receptor signal reduces collagen deposition in cultures scar myofibroblasts. J. Physiol. Pharmacol. Off. J. Pol. Physiol. Soc. 2019, 70. [Google Scholar] [CrossRef]
- Morisset, S.; Sasse, A.; Gbahou, F.; Héron, A.; Ligneau, X.; Tardivel-Lacombe, J.; Schwartz, J.C.; Arrang, J.M. The rat H3 receptor: Gene organization and multiple isoforms. Biochem. Biophys. Res. Commun. 2001, 280, 75–80. [Google Scholar] [CrossRef]
- Rouleau, A.; Héron, A.; Cochois, V.; Pillot, C.; Schwartz, J.C.; Arrang, J.M. Cloning and expression of the mouse histamine H3 receptor: Evidence for multiple isoforms. J. Neurochem. 2004, 90, 1331–1338. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting cardiac cellular composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, B.; Bansal, S.S.; Ismahil, M.A.; Hamid, T.; Rokosh, G.; Mack, M.; Prabhu, S.D. CCR2(+) monocyte-derived infiltrating macrophages are required for adverse cardiac remodeling during pressure overload. JACC Basic Transl. Sci. 2018, 3, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chancey, A.L.; Tzeng, H.P.; Zhou, Z.; Lavine, K.J.; Gao, F.; Sivasubramanian, N.; Barger, P.M.; Mann, D.L. The development of myocardial fibrosis in transgenic mice with targeted overexpression of tumor necrosis factor requires mast cell-fibroblast interactions. Circulation 2011, 124, 2106–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlini, S.; Palladini, G.; Ferrero, I.; Tozzi, R.; Fallarini, S.; Facoetti, A.; Nano, R.; Clari, F.; Busca, G.; Fogari, R.; et al. Sympathectomy or doxazosin, but not propranolol, blunt myocardial interstitial fibrosis in pressure-overload hypertrophy. Hypertension 2005, 46, 1213–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lameris, T.W.; de Zeeuw, S.; Duncker, D.J.; Alberts, G.; Boomsma, F.; Verdouw, P.D.; van den Meiracker, A.H. Exogenous angiotensin II does not facilitate norepinephrine release in the heart. Hypertension 2002, 40, 491–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henegar, J.R.; Schwartz, D.D.; Janicki, J.S. Ang II-related myocardial damage: Role of cardiac sympathetic catecholamines and beta-receptor regulation. Am. J. Physiol. 1998, 275, H534–H541. [Google Scholar] [CrossRef] [Green Version]
- Kabour, A.; Henegar, J.R.; Devineni, V.R.; Janicki, J.S. Prevention of angiotensin II induced myocyte necrosis and coronary vascular damage by lisinopril and losartan in the rat. Cardiovasc. Res 1995, 29, 543–548. [Google Scholar] [CrossRef]
- Dehlin, H.M.; Manteufel, E.J.; Monroe, A.L.; Reimer, M.H., Jr.; Levick, S.P. Substance P acting via the neurokinin-1 receptor regulates adverse myocardial remodeling in a rat model of hypertension. Int. J. Cardiol. 2013, 168, 4643–4651. [Google Scholar] [CrossRef] [Green Version]
- Melendez, G.C.; McLarty, J.L.; Levick, S.P.; Du, Y.; Janicki, J.S.; Brower, G.L. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 2010, 56, 225–231. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Saline | Ang II | Ang II + A331440 (LD) | Ang II + A331440 (HD) | Ang II + Imetit (LD) | Ang II + Imetit (HD) | |
|---|---|---|---|---|---|---|
| Body Weight (g) | 23.5 ± 1.9 | 25.3 ± 1.7 | 23.3 ± 1.9 | 20.3 ± 1.3 **,††††,‡‡‡ | 24.2 ± 1.4 | 20.9 ± 0.9 *,††††,‡‡ |
| LV mass (mg) | 77.4 ± 10.0 | 98.4 ± 8.5 *** | 91.2 ± 12.1 | 76.8 ± 7.3 ††,‡‡‡ | 100.2 ± 12.4 *** | 76.8 ± 4.8 ††,‡‡‡ |
| LV/Tibia (mg/mm) | 4.0 ± 0.6 | 4.9 ± 0.4 ** | 4.7 ± 0.7 | 3.3 ± 0.4 ††,‡‡‡ | 5.1 ± 0.6 *** | 3.9 ± 0.2 ††,‡‡‡ |
| RV mass (mg) | 21.3 ± 1.5 | 22.2 ± 3.5 | 26.4 ± 10.0 | 24.7 ± 3.4 | 22.8 ± 3.7 | 23.3 ± 2.5 |
| RV/Tibia (mg/mm) | 1.1 ± 0.1 | 1.1 ± 0.2 | 1.4 ± 0.5 | 1.3 ± 0.2 | 1.2 ± 0.2 | 1.2 ± 0.1 |
| Lung mass (mg) | 115.1 ± 8.5 | 125.7 ± 10.9 | 130.2 ± 17.9 | 130.5 ± 7.2 | 130.2 ± 10.9 | 115.8 ± 10.5 |
| Lung/Tibia (mg/mm) | 6.5 ± 1.5 | 6.3 ± 0.6 | 6.7 ± 0.7 | 6.6 ± 0.4 | 6.7 ± 0.5 | 5.9 ± 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCaffrey, S.L.; Lim, G.; Bullock, M.; Kasparian, A.O.; Clifton-Bligh, R.; Campbell, W.B.; Widiapradja, A.; Levick, S.P. The Histamine 3 Receptor Is Expressed in the Heart and Its Activation Opposes Adverse Cardiac Remodeling in the Angiotensin II Mouse Model. Int. J. Mol. Sci. 2020, 21, 9757. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249757
McCaffrey SL, Lim G, Bullock M, Kasparian AO, Clifton-Bligh R, Campbell WB, Widiapradja A, Levick SP. The Histamine 3 Receptor Is Expressed in the Heart and Its Activation Opposes Adverse Cardiac Remodeling in the Angiotensin II Mouse Model. International Journal of Molecular Sciences. 2020; 21(24):9757. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249757
Chicago/Turabian StyleMcCaffrey, Samuel L., Grace Lim, Martyn Bullock, Ainsley O. Kasparian, Roderick Clifton-Bligh, William B. Campbell, Alexander Widiapradja, and Scott P. Levick. 2020. "The Histamine 3 Receptor Is Expressed in the Heart and Its Activation Opposes Adverse Cardiac Remodeling in the Angiotensin II Mouse Model" International Journal of Molecular Sciences 21, no. 24: 9757. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21249757