Interplay between Thyroid Hormones and Stearoyl-CoA Desaturase 1 in the Regulation of Lipid Metabolism in the Heart

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Hypothyroidism Decreases Heart Weight and Increases Adiposity in Wildtype and SCD1−/− Mice
2.2. SCD1 Deficiency Affects Genomic and Non-Genomic TH Pathways in the Heart
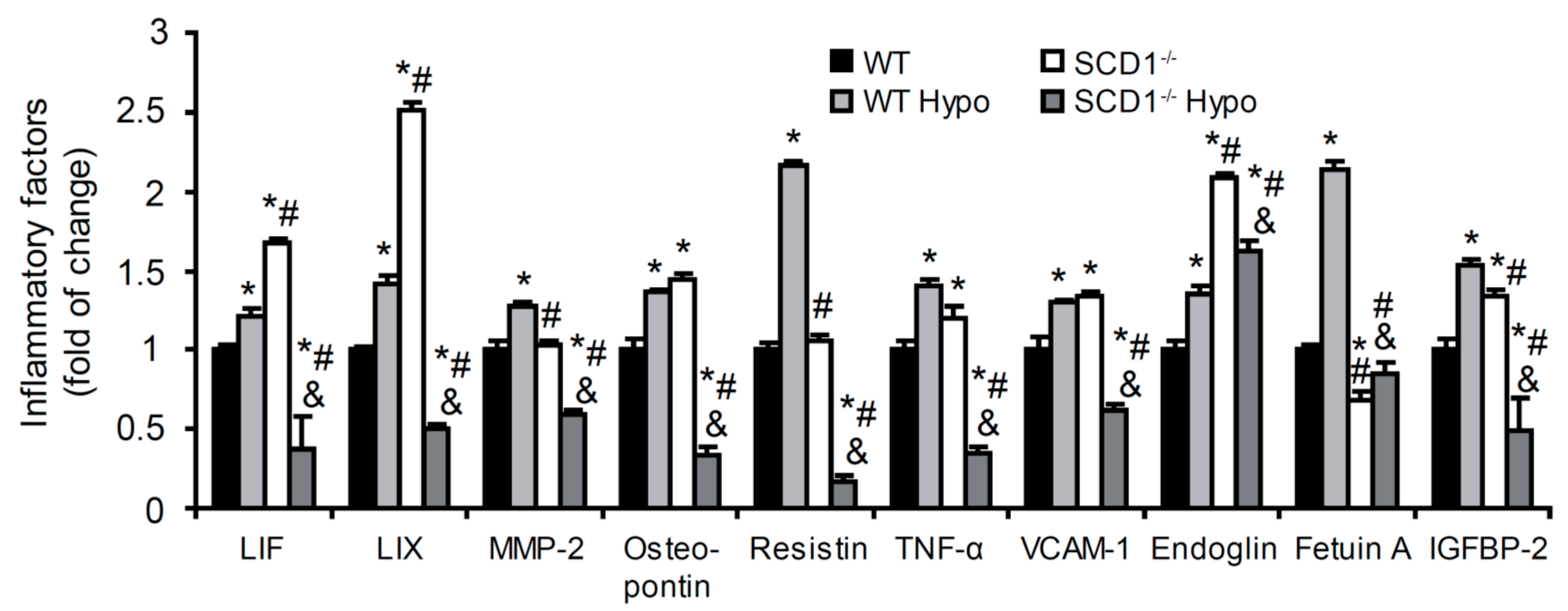
2.3. Effect of SCD1 Deficiency and Hypothyroidism on Inflammatory Factor Content in the Heart
2.4. Loss of SCD1 Exacerbates Hypothyroidism Induced Cardiac Steatosis
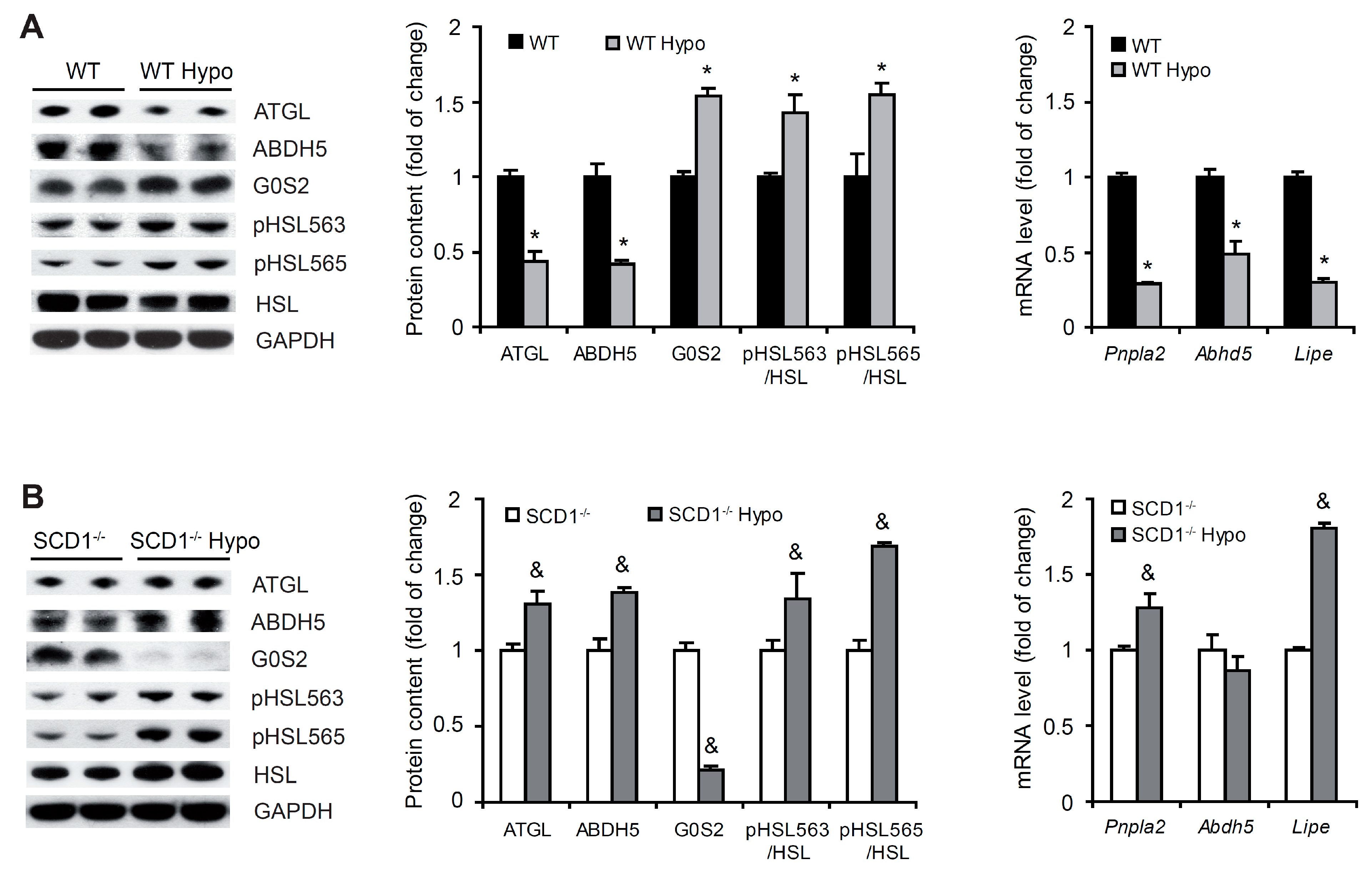
2.5. SCD1 Deficiency Upregulates Lipolysis in Cardiomyocytes in Hypothyroidism
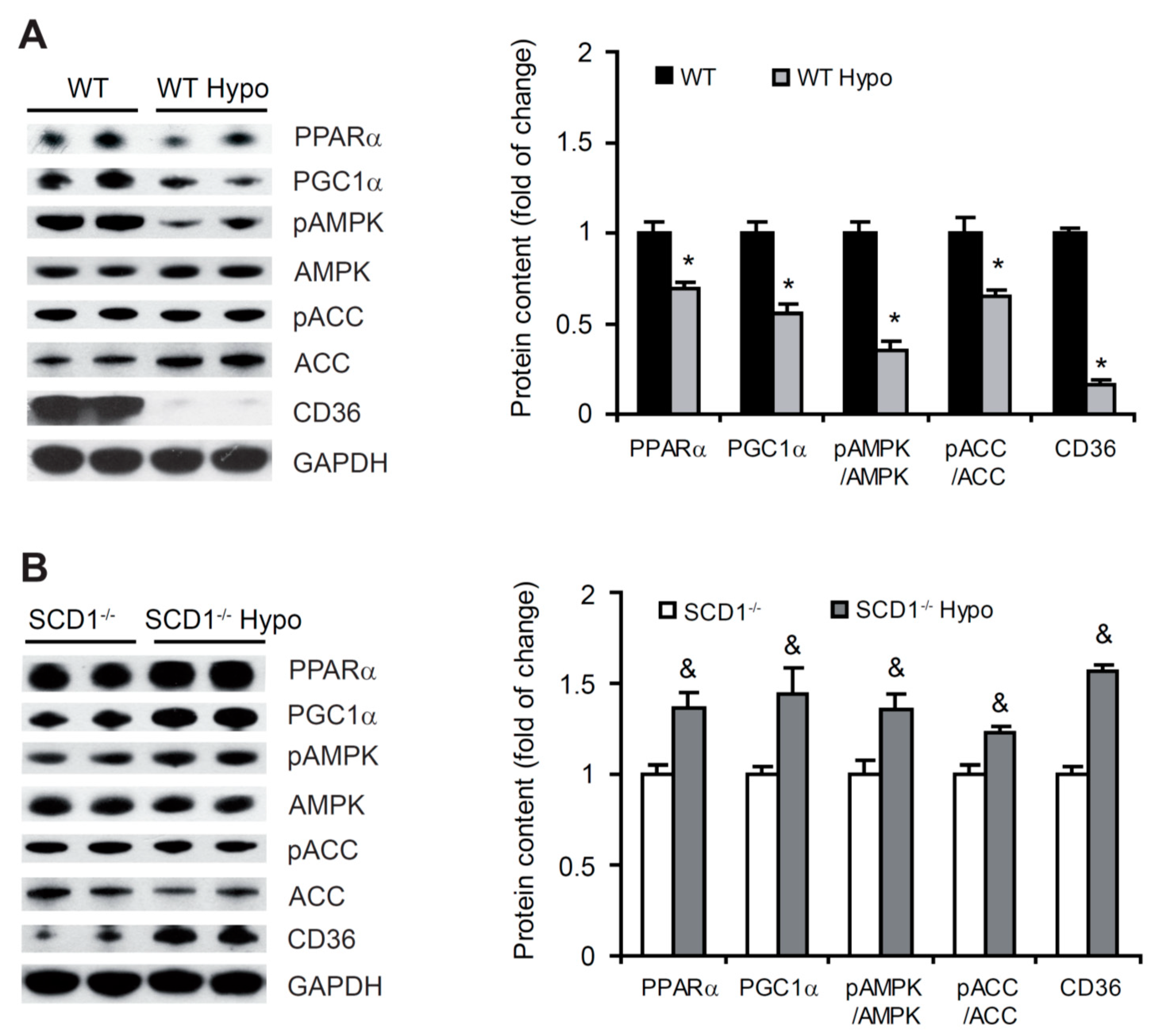
2.6. Higher FA β-Oxidation in the Heart in Hypothyroid SCD1−/− Mice
2.7. Inhibition of Deiodinases Increases TG Accumulation in HL-1 Cardiomyocytes
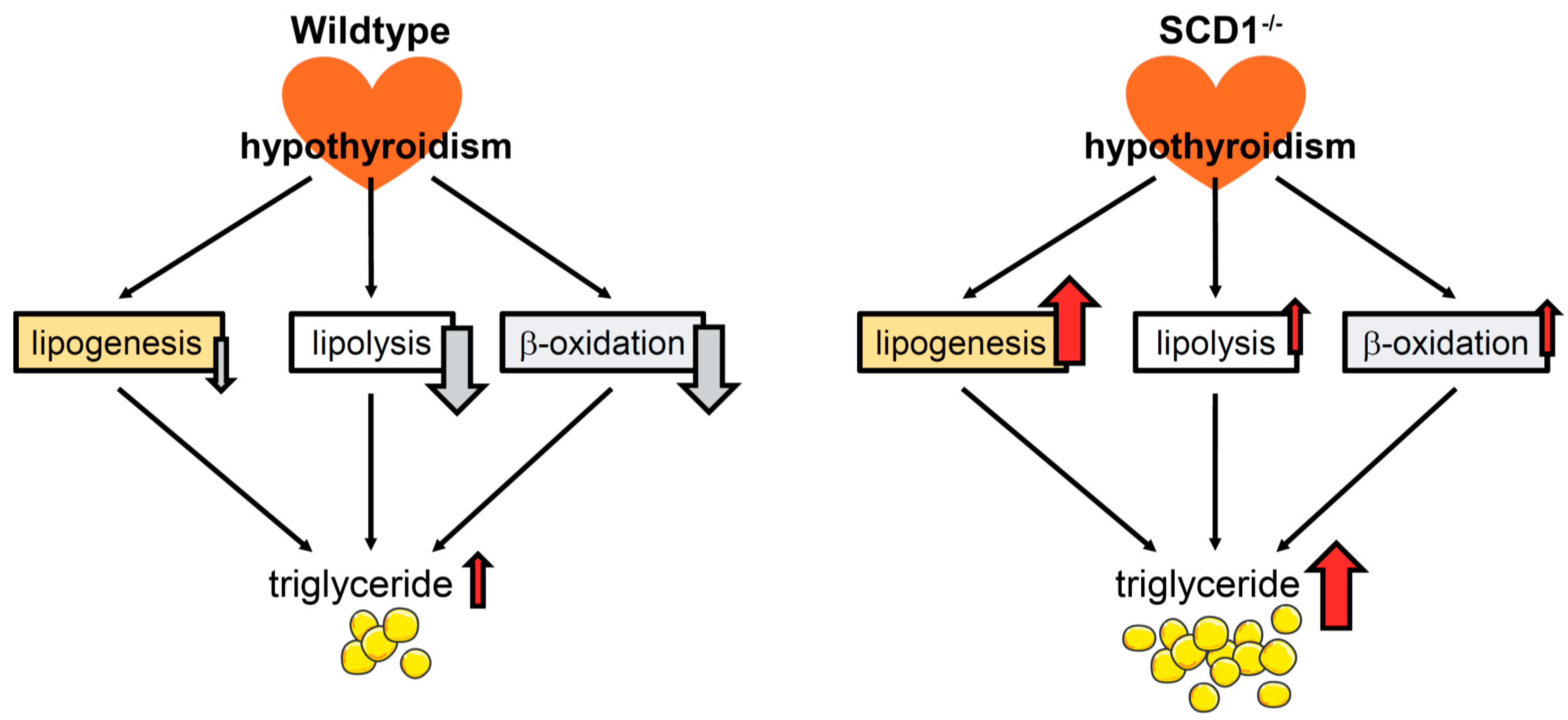
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Blood and Tissue Sampling
4.4. Plasma Lipid, TH, and Glucose Concentrations
4.5. Inflammatory Factors
4.6. Gene Expression Analysis
4.7. Culture of HL-1 Cardiomyocytes
4.8. Western Blot Analysis
4.9. Measurement of Lipids
4.10. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABDH5 | α/β-hydrolase domain containing 5 |
| AKT | protein kinase B |
| AMPK | 5′-adenosine monophosphate-activated protein kinase |
| ATGL | adipose triglyceride lipase |
| DAG | diacylglycerol |
| DGAT | diacylglycerol acyltransferase |
| DIO | deiodinase |
| ERK1/2 | extracellular signal-regulated kinases 1/2 |
| FA | fatty acids |
| FFA | free fatty acids |
| fT3 | free triiodothyronine |
| G0S2 | G0/G1 switch protein 2 |
| GPAT | glycerol-3-phosphate acyltransferase |
| GSK3 | glycogen synthase kinase 3 |
| HSL | hormone-sensitive lipase |
| IGFBP-2 | insulin-like growth factor-binding protein-2 |
| inhDIO | deiodinases inhibitor (iopanoic acid) |
| inhSCD1 | stetaroyl-CoA desaturase 1 inhibitor (A939573) |
| LIF | leukemia inhibitory factor |
| LIX | C-X-C motif chemokine 5 |
| MCT8 | monocarboxylate transporter 8 |
| MMP-2 | matrix metalloproteinase 2 |
| mTOR | mammalian target of rapamycin |
| PGC1α | peroxisome proliferator-activated receptor gamma coactivator 1α |
| PPARα | peroxisome proliferator-activated receptor α |
| S6K | S6 kinase |
| SCD1 | stearoyl-CoA desaturase 1 |
| SREBP1 | sterol regulatory element-binding protein 1 |
| T3 | triiodothyronine |
| T4 | thyroxine |
| TG | triglyceride |
| TH | thyroid hormones |
| TNFα | tumor necrosis factor α |
| TR | thyroid receptor |
| TSH | thyroid stimulate hormone |
| VCAM-1 | vascular cell adhesion protein 1 |
| WT | wildtype mice |
References
- Grais, I.R.; Sowers, J.R. Thyroid and the heart. Am. J. Med. 2014, 127, 691–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Accorroni, A.; Saponaro, F.; Zucchi, R. Tissue thyroid hormones and thyronamines. Heart Fail. Rev. 2016, 21, 373–390. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Matsumoto, S.; Yamada, M.; Satoh, T.; Mori, M. Liver X receptor-α gene expression is positively regulated by thyroid hormone. Endocrinology 2007, 148, 4667–4675. [Google Scholar] [CrossRef]
- Hashimoto, K.; Ishida, E.; Matsumoto, S.; Okada, S.; Yamada, M.; Satoh, T.; Monden, T.; Mori, M. Carbohydrate response element binding protein gene expression is positively regulated by thyroid hormone. Endocrinology 2009, 150, 417–3424. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Viscarra, J.; Kim, S.J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell. Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat. Rev. Endocrinol. 2018, 14, 259–269. [Google Scholar] [CrossRef]
- Radenne, A.; Akpa, M.; Martel, C.; Sawadogo, S.; Mauvoisin, D.; Mounier, C. Hepatic regulation of fatty acid synthase by insulin and T3: Evidence for T3 genomic and nongenomic actions. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E884–E894. [Google Scholar] [CrossRef] [Green Version]
- Dang, A.Q.; Faas, F.H.; Carter, W.J. Influence of hypo- and hyperthyroidism on rat liver glycerophospholipid metabolism. Lipids 1985, 20, 897–902. [Google Scholar] [CrossRef]
- Yao, X.; Hou, S.; Zhang, D.; Xia, H.; Wang, Y.C.; Jiang, J.; Yin, H.; Ying, H. Regulation of fatty acid composition and lipid storage by thyroid hormone in mouse liver. Cell. Biosci. 2014, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Klieverik, L.P.; Coomans, C.P.; Endert, E.; Sauerwein, H.P.; Havekes, L.M.; Voshol, P.J.; Rensen, P.C.; Romijn, J.A.; Kalsbeek, A.; Fliers, E. Thyroid hormone effects on whole-body energy homeostasis and tissue-specific fatty acid uptake in vivo. Endocrinology 2009, 150, 5639–5648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppenheimer, J.H.; Schwartz, H.L.; Lane, J.T.; Thompson, M.P. Functional relationship of thyroid hormone-induced lipogenesis, lipolysis, and thermogenesis in the rat. J. Clin. Investig. 1991, 87, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugden, M.C.; Priestman, D.A.; Orfali, K.A.; Holness, M.J. Hyperthyroidism facilitates cardiac fatty acid oxidation through altered regulation of cardiac carnitine palmitoyltransferase: Studies in vivo and with cardiac myocytes. Horm. Metab. Res. 1999, 31, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Gjedde, S.; Gormsen, L.C.; Rungby, J.; Nielsen, S.; Jorgensen, J.O.; Pedersen, S.B.; Riis, A.L.; Weeke, J.; Moller, N. Decreased lipid intermediate levels and lipid oxidation rates despite normal lipolysis in patients with hypothyroidism. Thyroid 2010, 20, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Scherer, T.; Wolf, P.; Winhofer, Y.; Duan, H.; Einwallner, E.; Gessl, A.; Luger, A.; Trattnig, S.; Hoffmann, M.; Niessner, A.; et al. Levothyroxine replacement in hypothyroid humans reduces myocardial lipid load and improves cardiac function. J. Clin. Endocrinol. Metab. 2014, 99, E2341–E2346. [Google Scholar] [CrossRef] [Green Version]
- Dobrzyn, P.; Bednarski, T.; Dobrzyn, A. Metabolic reprogramming of the heart through stearoyl-CoA desaturase. Prog. Lipid Res. 2015, 57, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dobrzyn, P.; Jazurek, M.; Dobrzyn, A. Stearoyl-CoA desaturase and insulin signaling—What is the molecular switch? Biochim. Biophys. Acta 2010, 1797, 1189–1194. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Strable, M.S.; Ntambi, J.M. Stearoyl-CoA desaturase 1: Role in cellular inflammation and stress. Adv. Nutr. 2011, 2, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Dobrzyn, P.; Sampath, H.; Dobrzyn, A.; Miyazaki, M.; Ntambi, J.M. Loss of stearoyl-CoA desaturase 1 inhibits fatty acid oxidation and increases glucose utilization in the heart. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E357–E364. [Google Scholar] [CrossRef] [Green Version]
- Dobrzyn, P.; Dobrzyn, A.; Miyazaki, M.; Ntambi, J.M. Loss of stearoyl-CoA desaturase 1 rescues cardiac function in obese leptin-deficient mice. J. Lipid. Res. 2010, 51, 2202–2210. [Google Scholar] [CrossRef] [Green Version]
- Bednarski, T.; Olichwier, A.; Opasinska, A.; Pyrkowska, A.; Gan, A.M.; Ntambi, J.M.; Dobrzyn, P. Stearoyl-CoA desaturase 1 deficiency reduces lipid accumulation in the heart by activating lipolysis independently of peroxisome proliferator-activated receptor α. Biochim. Biophys. Acta 2016, 1861, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Waters, K.M.; Miller, C.W.; Ntambi, J.M. Localization of a negative thyroid hormone-response region in hepatic stearoyl-CoA desaturase gene 1. Biochem. Biophys. Res. Commun. 1997, 233, 838–843. [Google Scholar] [CrossRef]
- Hashimoto, K.; Ishida, E.; Miura, A.; Ozawa, A.; Shibusawa, N.; Satoh., T.; Okada, S.; Yamada, M.; Mori, M. Human stearoyl-CoA desaturase 1 (SCD-1) gene expression is negatively regulated by thyroid hormone without direct binding of thyroid hormone receptor to the gene promoter. Endocrinology 2013, 154, 537–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, A.; Yamamoto, N.; Morisawa, Y.; Uchimoto, T.; Yukioka, M.; Morisawa, S. Unesterified long-chain fatty acids inhibit thyroid hormone binding to the nuclear receptor Solubilized receptor and the receptor in cultured cells. Eur. J. Biochem. 1989, 183, 565–572. [Google Scholar] [CrossRef]
- Li, Q.L.; Yamamoto, N.; Inoue, A.; Morisawa, S. Fatty acyl-CoAs are potent inhibitors of the nuclear thyroid hormone receptor in vitro. J. Biochem. 1990, 107, 699–702. [Google Scholar] [CrossRef] [PubMed]
- Planck, T.; Shahida, B.; Sjogren, M.; Groop, L.; Hallengren, B.; Lantz, M. Association of BTG2, CYR61, ZFP36, and SCD gene polymorphisms with Graves’ disease and ophthalmopathy. Thyroid 2014, 24, 1156–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonen, A.; Heynen, M.; Hatta, H. Distribution of monocarboxylate transporters MCT1-MCT8 in rat tissues and human skeletal muscle. Appl. Physiol. Nutr. Metab. 2006, 31, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Heier, C.; Radner, F.P.; Moustafa, T.; Schreiber, R.; Grond, S.; Eichmann, T.O.; Schweiger, M.; Schmidt, A.; Cerk, I.K.; Oberer, M.; et al. G0/G1 switch Gene 2 regulates cardiac lipolysis. J. Biol. Chem. 2015, 290, 26141–26150. [Google Scholar] [CrossRef] [Green Version]
- Anthonsen, M.W.; Rönnstrand, L.; Wernstedt, C.; Degerman, E.; Holm, C. Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J. Biol. Chem. 1998, 273, 215–221. [Google Scholar] [CrossRef] [Green Version]
- Madrazo, J.A.; Kelly, D.P. The PPAR trio: Regulators of myocardial energy metabolism in health and disease. J. Mol. Cell. Cardiol. 2008, 44, 968–975. [Google Scholar] [CrossRef]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing glucose as well as cellular energy status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St Germain, D.L.; Galton, V.A.; Hernandez, A. Minireview: Defining the roles of the iodothyronine deiodinases: Current concepts and challenges. Endocrinology 2009, 150, 1097–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koulouri, O.; Gurnell, M. How to interpret thyroid function. Clin. Med. 2013, 13, 282–286. [Google Scholar]
- Freake, H.C.; Schwartz, H.L.; Oppenheimer, J.H. The regulation of lipogenesis by thyroid hormone and its contribution to thermogenesis. Endocrinology 1989, 125, 2868–2874. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Baro, M.R.; Lewin, T.M.; Coleman, R.A. Regulation of triglyceride metabolism. II. Function of mitochondrial GPAT1 in the regulation of triacylglycerol biosynthesis and insulin action. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1195–G1199. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Wang, Q.; Lu, M.; Chen, W.; Song, Y.; Jing, F.; Guan, Y.; Wang, L.; Lin, Y.; Bo, T.; et al. Thyrotropin increases hepatic triglyceride content through upregulation of SREBP-1c activity. J. Hepatol. 2014, 61, 1358–1364. [Google Scholar] [CrossRef]
- Araki, O.; Ying, H.; Zhu, X.G.; Willingham, M.C.; Cheng, S.Y. Distinct dysregulation of lipid metabolism by unliganded thyroid hormone receptor isoforms. Mol. Endocrinol. 2009, 23, 308–315. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Tang, T.; Abbott, M.; Viscarra, J.A.; Wang, Y.; Sul, H.S. AMPK phosphorylates desnutrin/ATGL and hormone-sensitive lipase to regulate lipolysis and fatty acid oxidation within adipose tissue. Mol. Cell. Biol. 2016, 36, 1961–1976. [Google Scholar] [CrossRef] [Green Version]
- Watt, M.; Holmes, A.G.; Pinnamaneni, S.K.; Garnham, A.P.; Steinberg, G.R.; Kemp, B.E.; Febbraio, M.A. Regulation of HSL serine phosphorylation in skeletal muscle and adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E500–E508. [Google Scholar] [CrossRef]
- Brenta, G.; Berg, G.; Miksztowicz, V.; Lopez, G.; Lucero, D.; Faingold, C.; Murakami, M.; Machima, T.; Nakajima, K.; Schreier, L. Atherogenic lipoproteins in subclinical hypothyroidism and their relationship with hepatic lipase activity: Response to replacement treatment with levothyroxine. Thyroid 2016, 26, 365–372. [Google Scholar] [CrossRef]
- Kraemer, F.B.; Shen, W.J. Hormone-sensitive lipase: Control of intracellular tri-(di-)acylglycerol and cholesteryl ester hydrolysis. J. Lipid Res. 2002, 43, 1585–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.Y.; Heymann, R.S.; Moatamed, F.; Schultz, J.J.; Sobel, D.; Brent, G.A. A mutant thyroid hormone receptor α antagonizes peroxisome proliferator-activated receptor α signaling in vivo and impairs fatty acid oxidation. Endocrinology 2007, 148, 1206–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, M.; Kambe, F.; Cao, X.; Lu, X.; Kozaki, Y.; Oiso, Y.; Seo, Y. Thyroid hormone activates adenosine 5′-monophosphate-activated protein kinase via intracellular calcium mobilization and activation of calcium/calmodulin-dependent protein kinase kinase-beta. Mol. Endocrinol. 2008, 22, 893–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irrcher, I.; Walkinshaw, D.R.; Sheehan, T.E.; Hood, D.A. Thyroid hormone (T3) rapidly activates p38 and AMPK in skeletal muscle in vivo. J. Appl. Physiol. 2008, 104, 178–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobrzyn, P.; Dobrzyn, A.; Miyazaki, M.; Cohen, P.; Asilmaz, E.; Hardie, D.G.; Friedman, J.M.; Ntambi, J.M. Stearoyl-CoA desaturase 1 deficiency increases fatty acid oxidation by activating AMP-activated protein kinase in liver. Proc. Natl. Acad. Sci. USA 2004, 101, 6409–6414. [Google Scholar] [CrossRef] [Green Version]
- Dobrzyn, A.; Dobrzyn, P.; Lee, S.H.; Miyazaki, M.; Cohen, P.; Asilmaz, E.; Hardie, D.G.; Friedman, J.M.; Ntambi, J.M. Stearoyl-CoA desaturase-1 deficiency reduces ceramide synthesis by downregulating serine palmitoyltransferase and increasing beta-oxidation in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E599–E607. [Google Scholar] [CrossRef] [Green Version]
- López, M.; Varela, L.; Vázquez, M.J.; Rodríguez-Cuenca, S.; González, C.R.; Velagapudi, V.R.; Morgan, D.A.; Schoenmakers, E.; Agassandian, K.; Lage, R.; et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat. Med. 2010, 16, 1001–1008. [Google Scholar] [CrossRef]
- Gluvic, Z.; Obradovic, M.M.; Sudar-Milovanovic, E.M.; Zafirovic, S.S.; Radak, D.J.; Essack, M.M.; Bajic, V.B.; Takashi, G.; Isenovic, C. Regulation of nitric oxide production in hypothyroidism. Biomed. Pharmacother. 2020, 124, 109881. [Google Scholar] [CrossRef]
- Sampath, H.; Ntambi, J.M. The role of stearoyl-CoA desaturase in obesity, insulin resistance, and inflammation. Ann. N. Y. Acad. Sci. 2011, 1243, 47–53. [Google Scholar] [CrossRef]
- Peter, A.; Weigert, C.; Staiger, H.; Machicao, F.; Schick, F.; Machann, J.; Stefan, N.; Thamer, C.; Häring, H.U.; Schleicher, E. Individual stearoyl-coa desaturase 1 expression modulates endoplasmic reticulum stress and inflammation in human myotubes and is associated with skeletal muscle lipid storage and insulin sensitivity in vivo. Diabetes 2009, 58, 1757–1765. [Google Scholar] [CrossRef] [Green Version]
- Peter, A.; Weigert, C.; Staiger, H.; Rittig, K.; Cegan, A.; Lutz, P.; Machicao, F.; Häring, H.U.; Schleicher, E. Induction of stearoyl-CoA desaturase protects human arterial endothelial cells against lipotoxicity. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E339–E349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinnamaneni, S.K.; Southgate, R.J.; Febbraio, M.A.; Watt, M.J. Stearoyl CoA desaturase 1 is elevated in obesity but protects against fatty acid-induced skeletal muscle insulin resistance in vitro. Diabetologia 2006, 49, 3027–3037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crispino, M.; Trinchese, G.; Penna, E.; Cimmino, F.; Catapano, A.; Villano, I.; Perrone-Capano, C.; Mollica, M.P. Interplay between peripheral and central inflammation in obesity-promoted disorders: The impact on synaptic mitochondrial functions. Int. J. Mol. Sci. 2020, 21, 5964. [Google Scholar] [CrossRef]
- Shi, J.; Fan, J.; Su, Q.; Yang, Z. Cytokines and abnormal glucose and lipid metabolism. Front. Endocrinol. 2019, 10, 703. [Google Scholar] [CrossRef]
- Hogan, J.C.; Stephens, J.M. Effects of leukemia inhibitory factor on 3T3-L1 adipocytes. J. Endocrinol. 2005, 185, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Hardy, E.; Hardy-Sosa, A.; Fernandez-Patron, C. MMP-2: Is too low as bad as too high in the cardiovascular system? Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1332–H1340. [Google Scholar] [CrossRef]
- Haukeland, J.W.; Dahl, T.B.; Yndestad, A.; Gladhaug, I.P.; Løberg, E.M.; Haaland, T.; Konopski, Z.; Wium, C.; Taasheim, E.; Johansen, E.O.; et al. Fetuin A in nonalcoholic fatty liver disease: In vivo and in vitro studies. Eur. J. Endocrinol. 2012, 166, 503–510. [Google Scholar] [CrossRef]
- Castillo, M.; Hall, J.A.; Correa-Medina, M.; Ueta, C.; Kang, H.W.; Cohen, D.E.; Bianco, A.C. Disruption of thyroid hormone activation in type 2 deiodinase knockout mice causes obesity with glucose intolerance and liver steatosis only at thermoneutrality. Diabetes 2011, 60, 1082–1089. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, M.; Man, W.C.; Ntambi, J.M. Targeted disruption of stearoyl-CoA desaturase1 gene in mice causes atrophy of sebaceous and meibomian glands and depletion of wax esters in the eyelid. J. Nutr. 2001, 131, 2260–2268. [Google Scholar] [CrossRef]
- Costa-e-Sousa, R.H.; Astapova, I.; Ye, F.; Wondisford, F.E.; Hollenberg, A.N. The thyroid axis is regulated by NCoR1 via its actions in the pituitary. Endocrinology 2012, 153, 5049–5057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claycomb, W.C.; Lanson, N.A.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biol. Chem. 1959, 37, 911–917. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | WT Hypo | SCD1−/− | SCD1−/− Hypo | |
|---|---|---|---|---|
| WAT/BW (mg/g) | 11.6 ± 0.7 | 15.0 ± 0.7 * | 6.6 ± 0.6 *,# | 10.8 ± 0.8 #,& |
| HW/BW (mg/g) | 6.0 ± 0.2 | 4.5 ± 0.1 * | 6.3 ± 0.3 # | 4.7 ± 0.1 *,& |
| TG (mg/dl) | 85.5 ± 7.7 | 44.5 ± 3.4 * | 55.5 ± 7.2 * | 54.8 ± 7.8 * |
| FFA (mg/dl) | 23.8 ± 2.2 | 18.7 ± 1.8 * | 21.7 ± 2.2 | 23.4 ± 3.1 |
| Glucose (mg/dl) | 118.9 ± 14.9 | 159.5 ± 19.7 * | 121.4 ± 4.7 | 163.2 ± 12.5 *,& |
| TSH (pg/mL) | 0.104 ± 0.005 | 0.21 ± 0.07 * | 0.066 ± 0.02 *,# | 0.948 ± 0.61 *,#,& |
| fT3 (pMol/dl) | 6.8 ± 0.5 | 6.9 ± 0.5 | 6.3 ± 0.8 | 6.9 ± 0.7 |
| T4 (mg/dl) | 2.1 ± 0.2 | 0.4 ± 0.1 * | 1.4 ± 0.2 *,# | 0.5 ± 0.2 *,& |
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Abhd5 | TGGTGTCCCACATCTACATCA | CAGCGTCCATATTCTGTTTCCA |
| Dio1 | GCTGAAGCGGCTTGTGATATT | GTTGTCAGGGGCGAATCGG |
| Dio2 | AATTATGCCTCGGAGAAGACCG | GGCAGTTGCCTAGTGAAAGGT |
| Dio3 | CACGGCCTTCATGCTCTGG | CGGTTGTCGTCTGATACGCA |
| Lipe | TTCTCCAAAGCACCTAGCCAA | TGTGGAAAACTAAGGGCTTGTTG |
| Pnpla2 | CAACGCCACTCACATCTACGG | GGACACCTCAATAATGTTGGCAC |
| Rpl32 | AGTTCCTGGTCCACAATGTCA | GCACACAAGCCATCTACTCATT |
| Slc16a2 | GTGCTCTTGGTGTGCATTGG | CCGAAGTCCCCGGCATAGG |
| Thra | GGTCACCAGATGGAAAGCGAA | CCTTGTCCCCACACACGAC |
| Thrb | ACACCAGCAATTACCAGAGTG | GCAGCTCGAAGGGACATGA |
| β-actin | TTCTTGGGTATGGAATCCTGT | AGCACTGTGTTGGCATAGAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olichwier, A.; Balatskyi, V.V.; Wolosiewicz, M.; Ntambi, J.M.; Dobrzyn, P. Interplay between Thyroid Hormones and Stearoyl-CoA Desaturase 1 in the Regulation of Lipid Metabolism in the Heart. Int. J. Mol. Sci. 2021, 22, 109. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010109
Olichwier A, Balatskyi VV, Wolosiewicz M, Ntambi JM, Dobrzyn P. Interplay between Thyroid Hormones and Stearoyl-CoA Desaturase 1 in the Regulation of Lipid Metabolism in the Heart. International Journal of Molecular Sciences. 2021; 22(1):109. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010109
Chicago/Turabian StyleOlichwier, Adam, Volodymyr V. Balatskyi, Marcin Wolosiewicz, James M. Ntambi, and Pawel Dobrzyn. 2021. "Interplay between Thyroid Hormones and Stearoyl-CoA Desaturase 1 in the Regulation of Lipid Metabolism in the Heart" International Journal of Molecular Sciences 22, no. 1: 109. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010109