HIV-1 Tat Protein Enters Dysfunctional Endothelial Cells via Integrins and Renders Them Permissive to Virus Replication

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
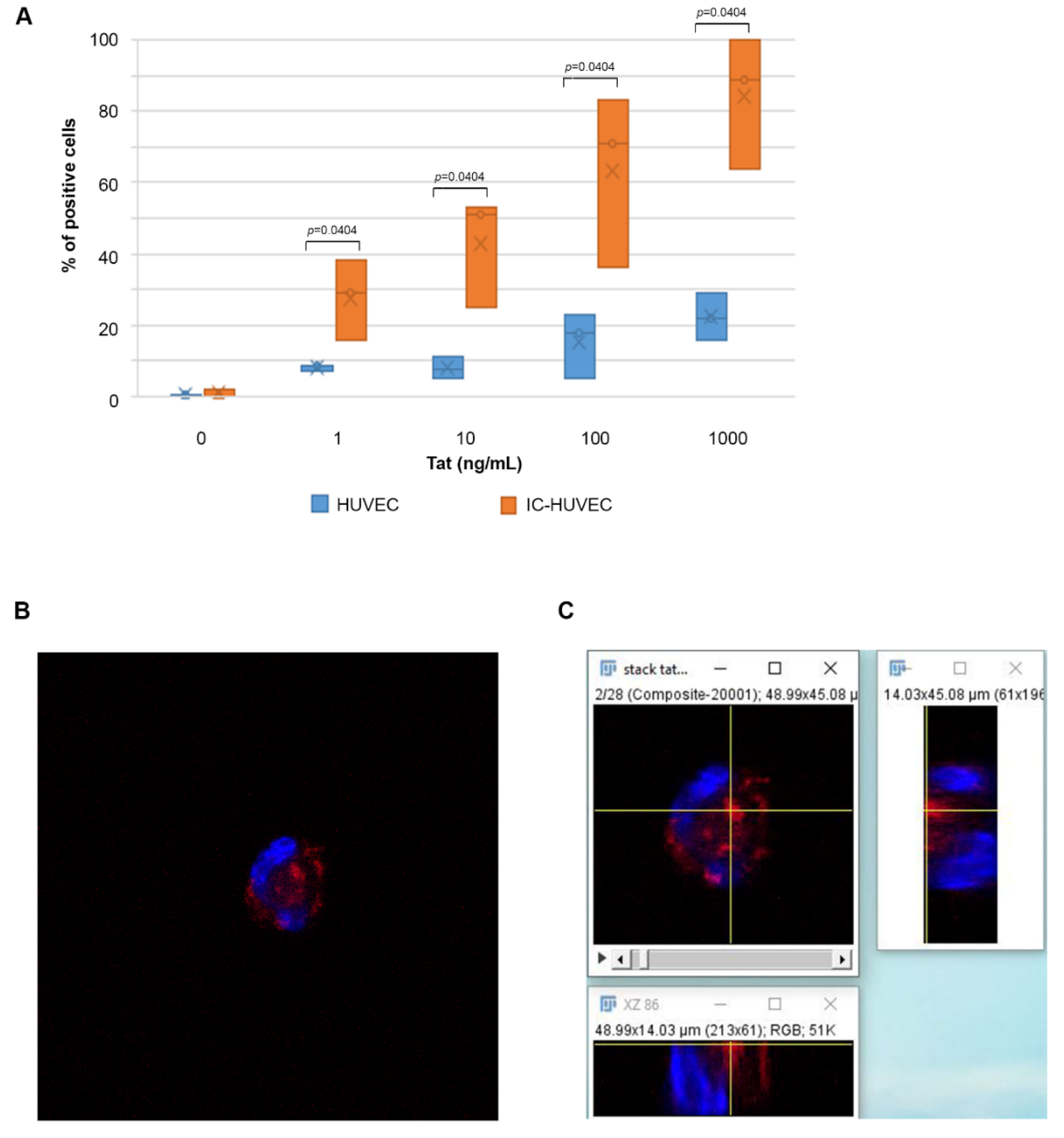
2.1. Biologically Active Tat Enters Primary Endothelial Cells in a Dose-, Time-, and Activation-Dependent Fashion
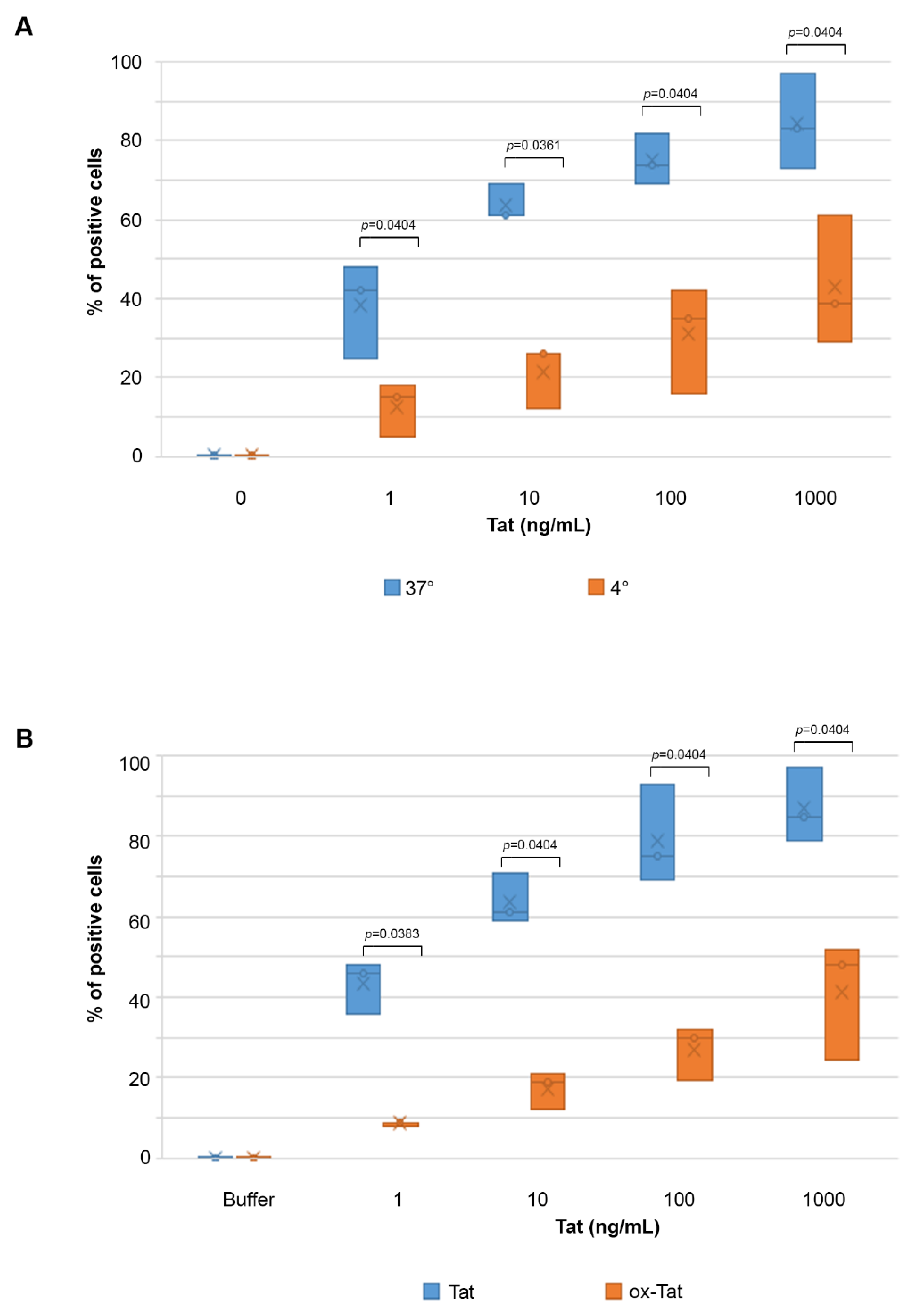
2.2. Tat Entry in IC-HUVEC Is Reduced by Low Temperature or Tat Oxidation
2.3. Uptake of Tat by Activated HUVEC Requires Both the RGD Domain and the Basic Region of Tat, and It Is Competed by Fibronectin and Vitronectin
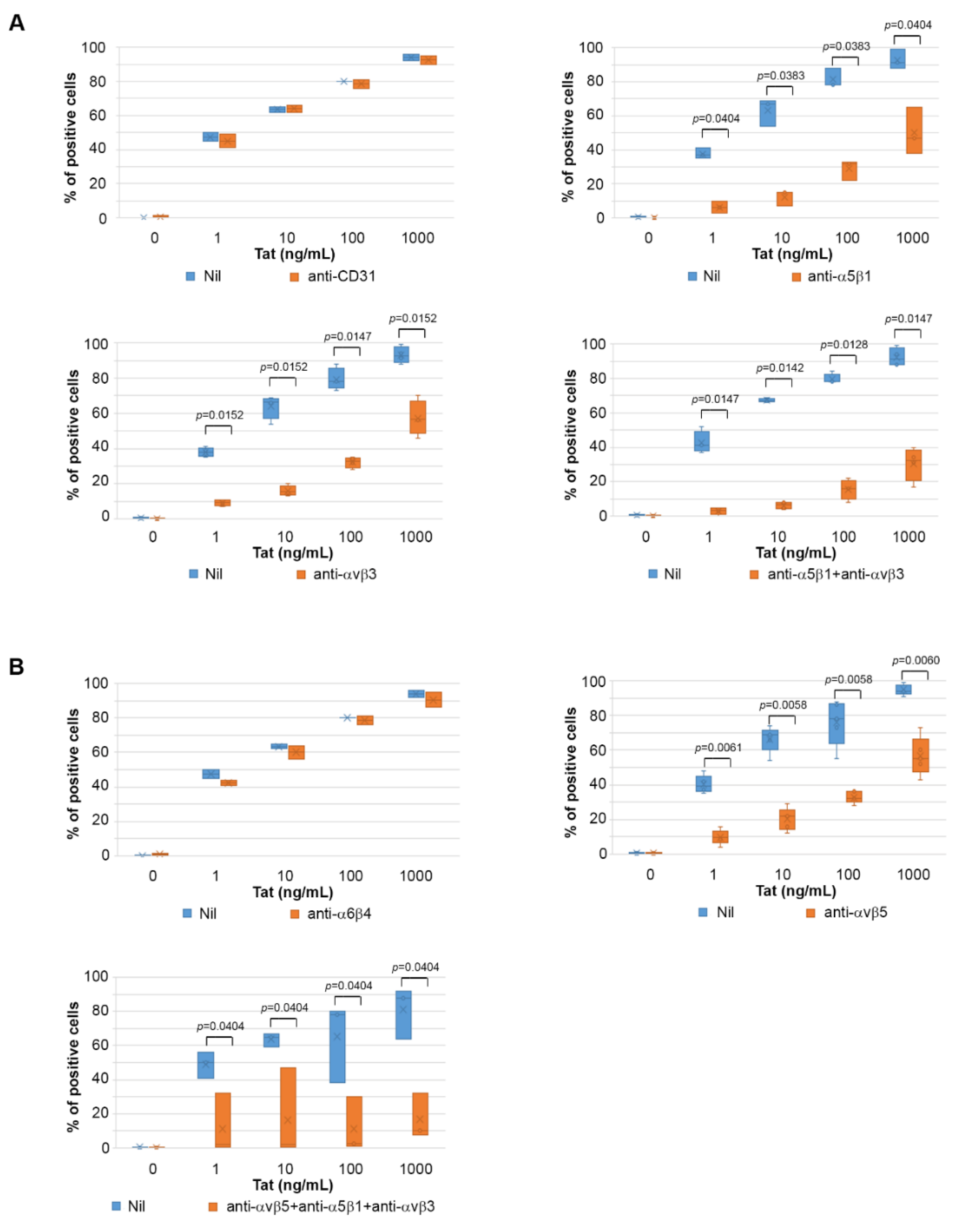
2.4. Tat Entry in IC-HUVEC Is Blocked by Monoclonal Antibodies against the α5β1, αvβ3, and αvβ5 Integrins
2.5. The Tat RGD Domain Is Exposed and Binds αvβ3 in Modeling–Docking of Tat-Integrin Interaction
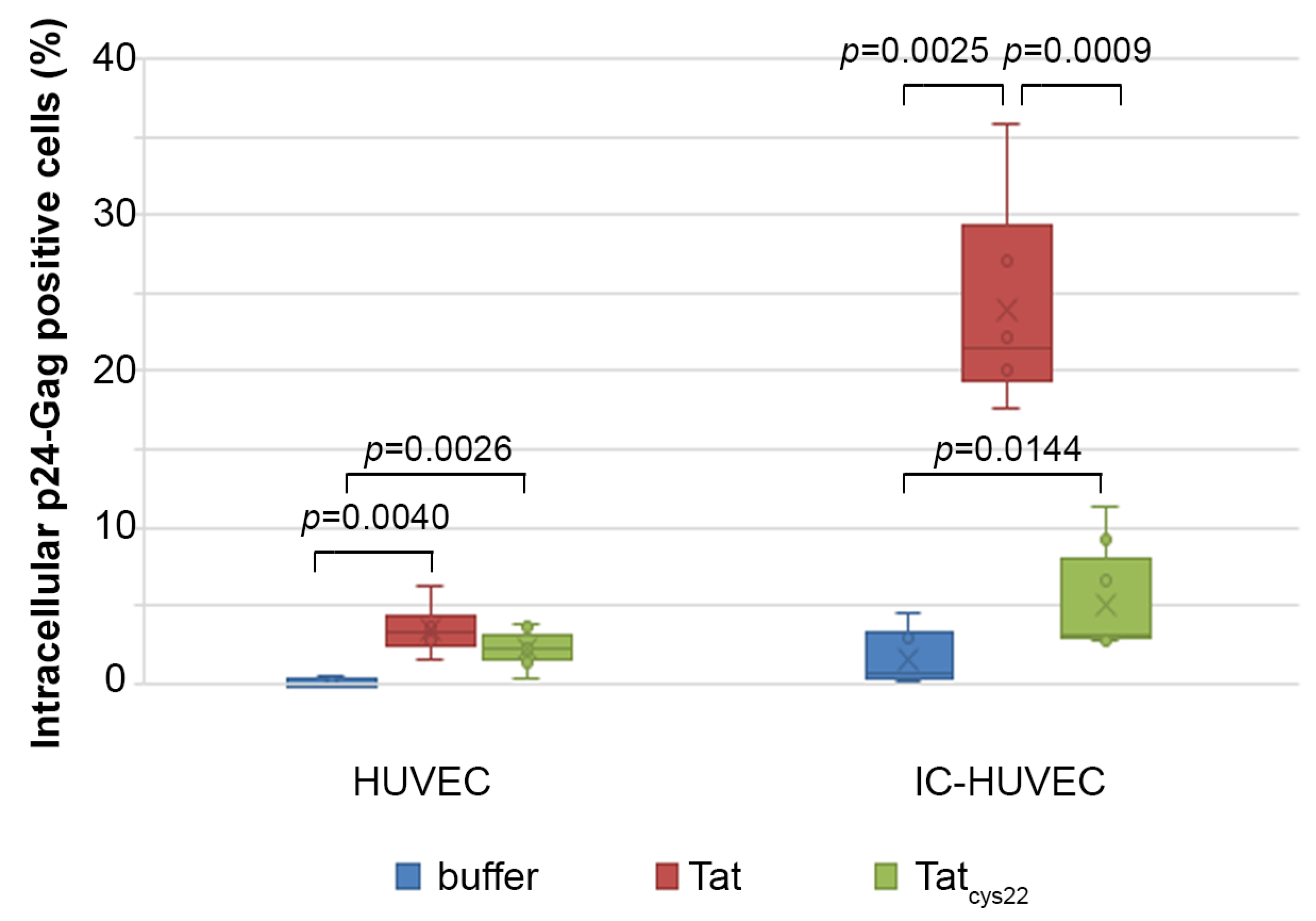
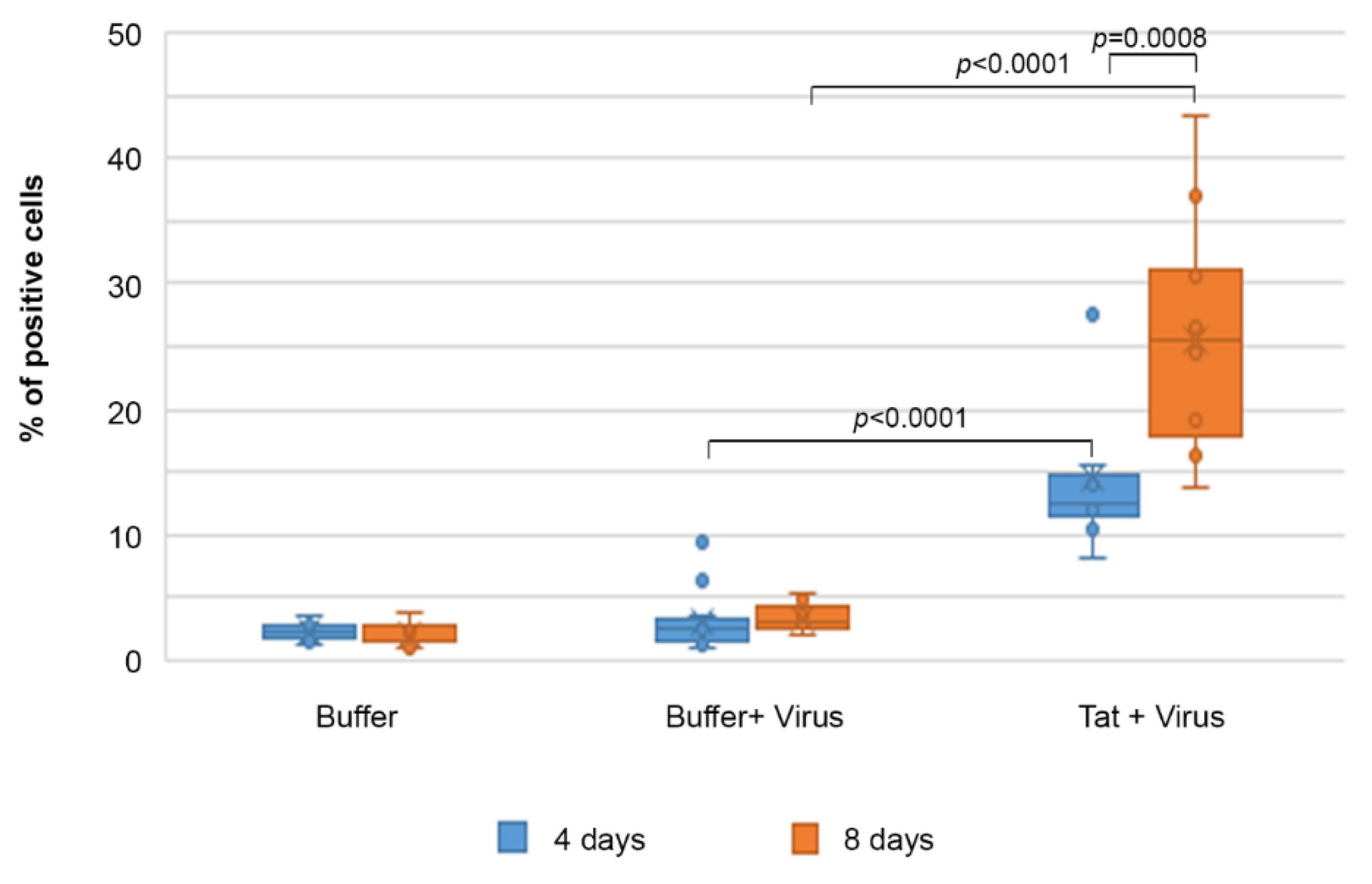
2.6. Tat Mediates Productive HIV-1 Infection of IC-HUVEC
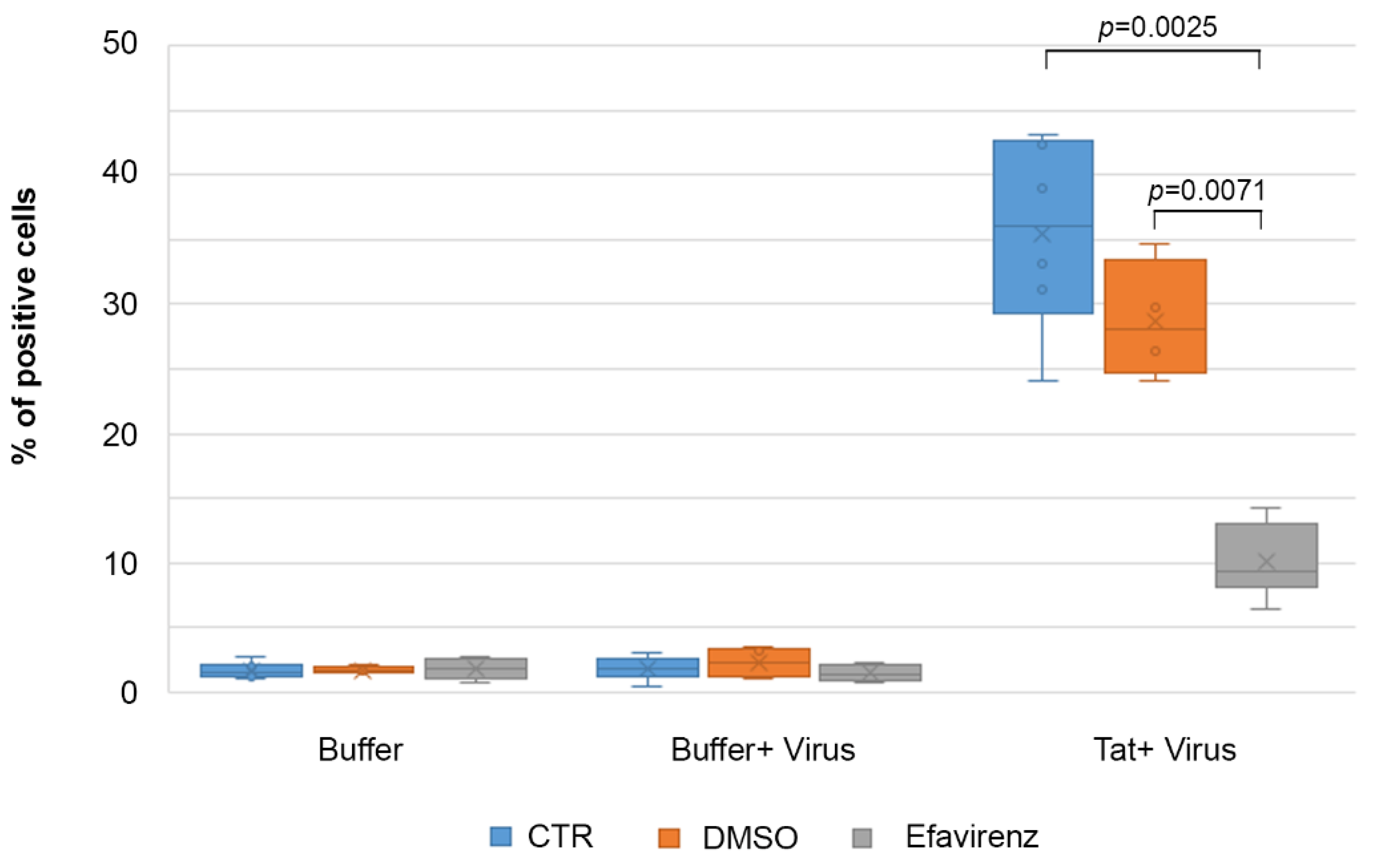
2.7. Efavirenz Suppresses the Tat-Mediated HIV-1 Infection of IC-HUVEC
3. Discussion
4. Materials and Methods
4.1. Tat Protein Production and Purification
4.2. Reagents
4.3. Endothelial Cell Culture and Activation
4.4. Evaluation of Tat Protein Cellular Uptake by Flow Cytometry
4.5. Confocal Microscopy
4.6. Docking Calculations of the Tat-Integrin Complex
4.7. Infection Experiments
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HIV-1 | Human Immunodeficiency Virus 1 |
| HUVEC | human umbilical vein endothelial cells |
| RGD | arginine-glycine-aspartic acid domain |
| cART | combination antiretroviral therapy |
| HSPG | heparan–sulfate proteoglycans |
| IL-1 | interleukin 1 |
| TNF | tumor necrosis factor |
| IFN | interferon |
| ICAM-1 | intercellular adhesion molecule-1 |
| IC-HUVEC | inflammatory cytokines activated HUVEC |
| DMSO | dimethyl sulfoxide |
| PBS | phosphate-buffered saline |
References
- Guerrero, P.A.; McCarty, J.H. Integrins in vascular development and pathology. Adv. Pharmacol. 2018, 81, 129–153. [Google Scholar] [CrossRef] [PubMed]
- Rocha, L.A.; Learmonth, D.A.; Sousa, R.A.; Salgado, A.J. αvβ3 and α5β1 integrin-specific ligands: From tumor angiogenesis inhibitors to vascularization promoters in regenerative medicine? Biotechnol. Adv. 2018, 36, 208–227. [Google Scholar] [CrossRef] [PubMed]
- Shafiei, M.S.; Lui, S.; Rockey, D.C. Integrin-linked kinase regulates endothelial cell nitric oxide synthase expression in hepatic sinusoidal endothelial cells. Liver Int. 2015, 35, 1213–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Lin, J.; Hu, T.; Ren, Z.; Li, L.; Hameed, I.; Zhang, X.; Men, C.; Guo, Y.; Xu, D.; et al. Galectin-3 exacerbates ox-LDL-mediated endothelial injury by inducing inflammation via integrin β1-RhoA-JNK signaling activation. J. Cell. Physiol. 2019, 234, 10990–11000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Li, R.; Ross, R.S.; Manso, A.M. Integrins and integrin-related proteins in cardiac fibrosis. J. Mol. Cell. Cardiol. 2016, 93, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Finney, A.C.; Stokes, K.Y.; Pattillo, C.B.; Orr, A.W. Integrin signaling in atherosclerosis. Cell. Mol. Life Sci. 2017, 74, 2263–2282. [Google Scholar] [CrossRef]
- Edwards, D.N.; Bix, G.J. Roles of blood-brain barrier integrins and extracellular matrix in stroke. Am. J. Physiol. Cell Physiol. 2019, 316, C252–C263. [Google Scholar] [CrossRef]
- Labus, J.; Wöltje, K.; Stolte, K.N.; Häckel, S.; Kim, K.S.; Hildmann, A.; Danker, K. IL-1β promotes transendothelial migration of PBMCs by upregulation of the FN/α5β1 signalling pathway in immortalised human brain microvascular endothelial cells. Exp. Cell Res. 2018, 373, 99–111. [Google Scholar] [CrossRef]
- Natarajan, M.; Udden, M.M.; McIntire, L.V. Adhesion of sickle red blood cells and damage to interleukin-1 beta stimulated endothelial cells under flow in vitro. Blood 1996, 87, 4845–4852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.Y.; Pitson, S.M.; Bonder, C.S. Tumor necrosis factor-induced neutrophil adhesion occurs via sphingosine kinase-1-dependent activation of endothelial α5β1 integrin. Am. J. Pathol. 2010, 177, 436–446. [Google Scholar] [CrossRef]
- Danese, S.; Sans, M.; de la Motte, C.; Graziani, C.; West, G.; Phillips, M.H.; Pola, R.; Rutella, S.; Willis, J.; Gasbarrini, A.; et al. Angiogenesis as a novel component of inflammatory bowel disease pathogenesis. Gastroenterology 2006, 130, 2060–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barillari, G.; Albonici, L.; Incerpi, S.; Bogetto, L.; Pistritto, G.; Volpi, A.; Ensoli, B.; Manzari, V. Inflammatory cytokines stimulate vascular smooth muscle cells locomotion and growth by enhancing alpha5beta1 integrin expression and function. Atherosclerosis 2001, 154, 377–385. [Google Scholar] [CrossRef]
- Zicari, S.; Sessa, L.; Cotugno, N.; Ruggiero, A.; Morrocchi, E.; Concato, C.; Rocca, S.; Zangari, P.; Manno, E.C.; Palma, P. Immune activation, inflammation, and non-AIDS co-morbidities in HIV-infected patients under long-term ART. Viruses 2019, 11, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinh, D.M.; Volpe, G.E.; Duffalo, C.; Bhalchandra, S.; Tai, A.K.; Kane, A.V.; Wanke, C.A.; Ward, H.D. Intestinal microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J. Infect. Dis. 2015, 211, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachdeva, N.; Yoon, H.S.; Oshima, K.; Garcia, D.; Goodkin, K.; Asthana, D. Biochip array-based analysis of plasma cytokines in HIV patients with immunological and virological discordance. Scand. J. Immunol. 2007, 65, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Süsal, C.; Daniel, V.; Naujokat, C.; Zimmermann, R.; Huth-Kühne, A.; Opelz, G. Short communication: Decreasing soluble CD30 and increasing IFN-gamma plasma levels are indicators of effective highly active antiretroviral therapy. AIDS Res. Hum. Retrovir. 2007, 23, 886–890. [Google Scholar] [CrossRef] [PubMed]
- López, M.; San Román, J.; Estrada, V.; Vispo, E.; Blanco, F.; Soriano, V. Endothelial dysfunction in HIV infection—the role of circulating endothelial cells, microparticles, endothelial progenitor cells and macrophages. AIDS Rev. 2012, 14, 223–230. [Google Scholar] [PubMed]
- Marincowitz, C.; Genis, A.; Goswami, N.; De Boever, P.; Nawrot, T.S.; Strijdom, H. Vascular endothelial dysfunction in the wake of HIV and ART. FEBS J. 2019, 286, 1256–1270. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.S.V.; Stelzle, D.; Lee, K.K.; Beck, E.J.; Alam, S.; Clifford, S.; Longenecker, C.T.; Strachan, F.; Bagchi, S.; Whiteley, W.; et al. Global Burden of Atherosclerotic Cardiovascular Disease in People Living With HIV. Circulation 2018, 138, 1100–1112. [Google Scholar] [CrossRef]
- Bush, K.N.V.; Teel, J.L.; Watts, J.A.; Gore, R.S.; Alvarado, G.; Harper, N.L.; Okulicz, J.F. Association of Endothelial Dysfunction and Antiretroviral Therapy in Early HIV Infection. JAMA Netw. Open 2019, 2, e1913615. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Toborek, M.; Lee, Y.W.; Flora, G.; Pu, H.; Andras, I.E.; Wylegala, E.; Hennig, B.; Nath, A. Mechanisms of the blood-brain barrier disruption in HIV-1 infection. Cell. Mol. Neurobiol. 2005, 25, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Marsh, J.W. Selective transcription and modulation of resting T cell activity by preintegrated HIV DNA. Science 2001, 293, 1503–1506. [Google Scholar] [CrossRef] [Green Version]
- Chi, D.; Henry, J.; Kelley, J.; Thorpe, R.; Smith, J.K.; Krishnaswamy, G. The effects of HIV infection on endothelial function. Endothelium 2000, 7, 223–242. [Google Scholar] [CrossRef] [PubMed]
- Matzen, K.; Dirkx, A.E.; oude Egbrink, M.G.; Speth, C.; Gotte, M.; Ascherl, G.; Grimm, T.; Griffioen, A.W.; Sturzl, M. HIV-1 Tat increases the adhesion of monocytes and T-cells to the endothelium in vitro and in vivo: Implications for AIDS-associated vasculopathy. Virus Res. 2004, 104, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Joos, B.; Wong, J.K.; Ott, P.; Opravil, M.; Hirschel, B.; Weber, R.; Huldrych, F.G.; Swiss HIV Cohort Study. Attenuated and nonproductive viral transcription in the lymphatic tissue of HIV-1-infected patients receiving potent antiretroviral therapy. J. Infect. Dis. 2004, 189, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Ferdin, J.; Goričar, K.; Dolžan, V.; Plemenitaš, A.; Martin, J.N.; Peterlin, B.M.; Deeks, S.G.; Lenassi, M. Viral protein Nef is detected in plasma of half of HIV-infected adults with undetectable plasma HIV RNA. PLoS ONE 2018, 13, e0191613. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yi, R.; Green, L.A.; Chelvanambi, S.; Seimetz, M.; Clauss, M. Increased cardiovascular disease risk in the HIV-positive population on ART: Potential role of HIV-Nef and Tat. Cardiovasc. Pathol. 2015, 24, 279–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, L.; Parker, I.; Sutliff, R.L.; Platt, M.O.; Gleason, R.L. Endothelial dysfunction, arterial stiffening, and intima-media thickening in large arteries from HIV-1 transgenic mice. Ann. Biomed. Eng. 2013, 41, 682–693. [Google Scholar] [CrossRef] [Green Version]
- Kline, E.R.; Kleinhenz, D.J.; Liang, B.; Dikalov, S.; Guidot, D.M.; Hart, C.M.; Jones, D.P.; Sutliff, R.L. Vascular oxidative stress and nitric oxide depletion in HIV-1 transgenic rats are reversed by glutathione restoration. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, 2792–2804. [Google Scholar] [CrossRef] [Green Version]
- Bussolino, F.; Mitola, S.; Serini, G.; Barillari, G.; Ensoli, B. Interactions between endothelial cells and HIV-1. Int. J. Biochem. Cell Biol. 2001, 33, 371–390. [Google Scholar] [CrossRef]
- Toschi, E.; Barillari, G.; Sgadari, C.; Bacigalupo, I.; Cereseto, A.; Carlei, D.; Palladino, C.; Zietz, C.; Leone, P.; Stürzl, M.; et al. Activation of Matrix-Metalloproteinase-2 and Membrane-Type-1-Matrix-Metalloproteinase in Endothelial Cells and Induction of Vascular Permeability in vivo by the Human Immunodeficiency Virus-1 Tat protein and Basic Fibroblast Growth Factor. Mol. Biol. Cell 2001, 12, 2934–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ensoli, B.; Buonaguro, L.; Barillari, G.; Fiorelli, V.; Gendelman, R.; Morgan, R.A.; Wingfield, P.; Gallo, R.C. Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. J. Virol. 1993, 67, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.C.; Samaniego, F.; Nair, B.C.; Buonaguro, L.; Ensoli, B. HIV-1 Tat protein exits from cells via a leaderless secretory pathway and binds to extracellular matrix-associated heparan sulfate proteoglycans through its basic region. AIDS 1997, 11, 1421–1431. [Google Scholar] [CrossRef]
- Rayne, F.; Debaisieux, S.; Yezid, H.; Lin, Y.L.; Mettling, C.; Konate, K.; Chazal, N.; Arold, S.T.; Pugnière, M.; Sanchez, F.; et al. Phosphatidylinositol-[4,5]-bisphosphate enables efficient secretion of HIV-1 Tat by infected T-cells. EMBO J. 2010, 29, 1348–1362. [Google Scholar] [CrossRef]
- Zeitler, M.; Steringer, J.P.; Müller, H.M.; Mayer, M.P.; Nickel, W. HIV-Tat Protein Forms Phosphoinositide-dependent Membrane Pores Implicated in Unconventional Protein Secretion. J. Biol. Chem. 2015, 290, 21976–21984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanales-Belasio, E.; Moretti, S.; Nappi, F.; Barillari, G.; Micheletti, F.; Cafaro, A.; Ensoli, B. Native HIV-1 Tat protein targets monocyte-derived dendritic cells and enhances their maturation, function, and antigen-specific T cell responses. J. Immunol. 2002, 168, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanales-Belasio, E.; Moretti, S.; Fiorelli, V.; Tripiciano, A.; Cossut, M.R.P.; Scoglio, A.; Collacchi, B.; Nappi, F.; Macchia, I.; Bellino, S.; et al. HIV-1 Tat Addresses Dendritic Cells to Induce a Predominant Th1-Type Adaptive Immune Response That Appears Prevalent in the Asymptomatic Stage of Infection. J. Immunol. 2009, 182, 2888–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barillari, G.; Ensoli, B. Angiogenic effects of extracellular human immunodeficiency virus type 1 Tat protein and its role in the pathogenesis of AIDS-associated Kaposi’s sarcoma. Clin. Microbiol. Rev. 2002, 15, 310–326. [Google Scholar] [CrossRef] [Green Version]
- Barillari, G.; Gendelman, R.; Gallo, R.C.; Ensoli, B. The Tat protein of human immunodeficiency virus type 1, a growth factor for AIDS Kaposi sarcoma and cytokine-activated vascular cells, induces adhesion of the same cell types by using integrin receptors recognizing the RGD amino acid sequence. Proc. Natl. Acad. Sci. USA 1993, 90, 7941–7945. [Google Scholar] [CrossRef] [Green Version]
- Barillari, G.; Sgadari, C.; Palladino, C.; Gendelman, R.; Caputo, A.; Bohan Morris, C.; Nair, B.C.; Markham, P.; Nel, A.; Stürzl, M.; et al. Inflammatory cytokines synergize with the HIV-1 Tat protein to promote angiogenesis and Kaposi’s sarcoma via induction of basic fibroblast growth factor and avb3 integrin. J. Immunol. 1999, 163, 1929–1935. [Google Scholar] [PubMed]
- Samaniego, F.; Markham, P.D.; Gendelman, R.; Gallo, R.C.; Ensoli, B. Inflammatory cytokines induce endothelial cells to produce and release basic fibroblast growth factor and to promote Kaposi’s sarcoma-like lesions in nude mice. J. Immunol. 1997, 158, 1887–1894. [Google Scholar] [PubMed]
- Dandachi, D.; Morón, F. Effects of HIV on the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1263, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Ensoli, B.; Gendelman, R.; Markham, P.; Fiorelli, V.; Colombini, S.; Raffeld, M.; Cafaro, A.; Chang, H.K.; Brady, J.N.; Gallo, R.C. Synergy between basic fibroblast growth factor and HIV-1 Tat protein in induction of Kaposi’s sarcoma. Nature 1994, 371, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Buonaguro, F.M.; Giraldo, G.; Ensoli, B. The human immunodeficiency virus type 1 Tat protein transactivates tumor necrosis factor beta gene expression through a TAR-like structure. J. Virol. 1994, 68, 2677–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosino, C.; Ruocco, M.R.; Chen, X.; Mallardo, M.; Baudi, F.; Trematerra, S.; Quinto, I.; Venuta, S.; Scala, G. HIV-1 Tat induces the expression of the interleukin-6 (IL6) gene by binding to the IL6 leader RNA and by interacting with CAAT enhancer-binding protein beta (NF-IL6) transcription factors. J. Biol. Chem. 1997, 272, 14883–14892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafrenie, R.M.; Wahl, L.M.; Epstein, J.S.; Hewlett, I.K.; Yamada, K.M.; Dhawan, S. HIV-1-Tat modulates the function of monocytes and alters their interactions with microvessel endothelial cells. A mechanism of HIV pathogenesis. J. Immunol. 1996, 156, 1638–1645. [Google Scholar] [PubMed]
- Lafrenie, R.M.; Wahl, L.M.; Epstein, J.S.; Hewlett, I.K.; Yamada, K.M.; Dhawan, S. HIV-1-Tat protein promotes chemotaxis and invasive behavior by monocytes. J. Immunol. 1996, 157, 974–977. [Google Scholar] [PubMed]
- Lafrenie, R.M.; Wahl, L.M.; Epstein, J.S.; Yamada, K.M.; Dhawan, S. Activation of monocytes by HIV-Tat treatment is mediated by cytokine expression. J. Immunol. 1997, 159, 4077–4083. [Google Scholar] [PubMed]
- Dhawan, S.; Puri, R.K.; Kumar, A.; Duplan, H.; Masson, J.M.; Aggarwal, B.B. Human immunodeficiency virus-1-tat protein induces the cell surface expression of endothelial leukocyte adhesion molecule-1, vascular cell adhesion molecule-1, and intercellular adhesion molecule-1 in human endothelial cells. Blood 1997, 90, 1535–1544. [Google Scholar] [PubMed]
- Westendorp, M.O.; Frank, R.; Ochsenbauer, C.; Stricker, K.; Dhein, J.; Walczak, H.; Debatin, K.M.; Krammer, P.H. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 Tat and gp120. Nature 1995, 375, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Neuveut, C.; Tiffany, H.L.; Benkirane, M.; Rich, E.A.; Murphy, P.M.; Jeang, K.T. Selective CXCR4 antagonism by Tat: Implications for in vivo expansion of coreceptor use by HIV-1. Proc. Natl. Acad. Sci. USA 2000, 97, 11466–11471. [Google Scholar] [CrossRef] [Green Version]
- Poggi, A.; Carosio, R.; Fenoglio, D.; Brenci, S.; Murdaca, G.; Setti, M.; Indiveri, F.; Scabini, S.; Ferrero, E.; Zocchi, M.R. Migration of V delta 1 and V delta 2 T cells in response to CXCR3 and CXCR4 ligands in healthy donors and HIV-1-infected patients: Competition by HIV-1 Tat. Blood 2004, 103, 2205–2213. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.P.; Patel, K.; Johnson, K.R.; Maric, D.; Calabresi, P.A.; Hasbun, R.; Nath, A. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc. Natl. Acad. Sci. USA 2013, 110, 13588–13593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, D.A.; Frankel, A.D. Endocytosis and targeting of exogenous HIV-1 Tat protein. EMBO J. 1991, 10, 1733–1739, PMCID: PMC45284. [Google Scholar] [CrossRef] [PubMed]
- Mitola, S.; Soldi, R.; Zanon, I.; Barra, L.; Gutierrez, M.I.; Berkhout, B.; Giacca, M.; Bussolino, F. Identification of specific molecular structures of human immunodeficiency virus type 1 Tat relevant for its biological effects on vascular endothelial cells. J. Virol. 2000, 74, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Barillari, G.; Sgadari, C.; Fiorelli, V.; Samaniego, F.; Colombini, S.; Manzari, V.; Modesti, A.; Nair, B.C.; Cafaro, A.; Stürzl, M.; et al. The Tat protein of human immunodeficiency virus type-1 promotes vascular cell growth and locomotion by engaging the a5b1 and avb3 integrins by mobilizing sequestered basic fibroblast growth factor. Blood 1999, 94, 663–672. [Google Scholar] [PubMed]
- Vogel, B.E.; Lee, S.J.; Hildebrand, A.; Craig, W.; Pierschbacher, M.D.; Wong-Staal, F.; Ruoslahti, E. A novel integrin specificity exemplified by binding of the alpha v beta 5 integrin to the basic domain of the HIV Tat protein and vitronectin. J. Cell Biol. 1993, 121, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Mathias, P.; Galleno, M.; Nemerow, G.R. Interactions of soluble recombinant integrin alphav beta5 with human adenoviruses. J. Virol. 1998, 72, 8669–8675. [Google Scholar] [CrossRef]
- Zhu, X.; Evans, J.P. Analysis of the roles of RGD-binding integrins, alpha(4)/alpha(9) integrins, alpha(6) integrins, and CD9 in the interaction of the fertilin beta (ADAM2) disintegrin domain with the mouse egg membrane. Biol. Reprod. 2002, 66, 1193–1202. [Google Scholar] [CrossRef]
- Xiong, J.P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science 2002, 296, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prlic, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Di Costanzo, L.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, 271–281. [Google Scholar] [CrossRef]
- Hill, H.R.; Augustine, N.H.; Williams, P.A.; Brown, E.J.; Bohnsack, J.F. Mechanism of fibronectin enhancement of group B streptococcal phagocytosis by human neutrophils and culture-derived macrophages. Infect. Immun. 1993, 61, 2334–2339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Putten, J.P.; Duensing, T.D.; Cole, R.L. Entry of OpaA+ gonococci into HEp-2 cells requires concerted action of glycosaminoglycans, fibronectin and integrin receptors. Mol. Microbiol. 1998, 29, 369–379. [Google Scholar] [CrossRef]
- Erbacher, P.; Remy, J.-S.; Behr, J.-P. Gene transfer with synthetic virus-like particles via the integrin-mediated endocytosis pathway. Gene Ther. 1999, 6, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Lord, R.; Parsons, M.; Kirby, I.; Beavil, A.; Hunt, J.; Sutton, B.; Santis, G. Analysis of the interaction between RGD-expressing adenovirus type 5 fiber knob domains and alphavbeta3 integrin reveals distinct binding profiles and intracellular trafficking. J. Gen. Virol. 2006, 87, 2497–2505. [Google Scholar] [CrossRef]
- Han, X.; Gong, F.; Chi, L.; Feng, C.; Sun, J.; Chen, Y.; Liu, J.; Shen, Y. Cancer-targeted and glutathione-responsive micellar carriers for controlled delivery of cabazitaxel. Nanotechnology 2019, 30, 055601. [Google Scholar] [CrossRef]
- Argyris, E.G.; Kulkosky, J.; Meyer, M.E.; Xu, Y.; Mukhtar, M.; Pomerantz, R.J.; Williams, K.J. The perlecan heparan sulfate proteoglycan mediates cellular uptake of HIV-1 Tat through a pathway responsible for biological activity. Virology 2004, 330, 481–486. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Saredy, J.; Yang, W.Y.; Sun, Y.; Lu, Y.; Saaoud, F.; Drummer, C.; Johnson, C.; Xu, K.; Jiang, X.; et al. Vascular Endothelial Cells and Innate Immunity. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e138–e152. [Google Scholar] [CrossRef]
- Fernandez Pujol, B.; Lucibello, F.C.; Zuzarte, M.; Lütjens, P.; Müller, R.; Havemann, K. Dendritic cells derived from peripheral monocytes express endothelial markers and in the presence of angiogenic growth factors differentiate into endothelial-like cells. Eur. J. Cell Biol. 2001, 80, 99–110. [Google Scholar] [CrossRef]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Arderiu, G.; Espinosa, S.; Peña, E.; Crespo, J.; Aledo, R.; Bogdanov, V.Y.; Badimon, L. Tissue factor variants induce monocyte transformation and transdifferentiation into endothelial cell-like cells. J. Thromb. Haemost. 2017, 15, 1689–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzuca, P.; Caruso, A.; Caccuri, F. HIV-1 infection, microenvironment and endothelial cell dysfunction. New Microbiol. 2016, 39, 163–173. [Google Scholar] [PubMed]
- Wang, D.; Melancon, J.K.; Verbesey, J.; Hu, H.; Liu, C.; Aslam, S.; Young, M.; Wilcox, C.S. Microvascular Endothelial Dysfunction and Enhanced Thromboxane and Endothelial Contractility in Patients with HIV. J. AIDS Clin. Res. 2013, 4, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gresele, P.; Falcinelli, E.; Sebastiano, M.; Baldelli, F. Endothelial and platelet function alterations in HIV-infected patients. Thromb. Res. 2012, 129, 301–308. [Google Scholar] [CrossRef]
- Kobayashi, S.; Hamamoto, Y.; Kobayashi, N.; Yamamoto, N. Serum level of TNF alpha in HIV-infected individuals. AIDS 1990, 4, 169–170. [Google Scholar] [PubMed]
- Lähdevirta, J.; Maury, C.P.; Teppo, A.M.; Repo, H. Elevated levels of circulating cachectin/tumor necrosis factor in patients with acquired immunodeficiency syndrome. Am. J. Med. 1988, 85, 289–291. [Google Scholar] [CrossRef]
- Pugliese, A.; Torre, D.; Saini, A.; Pagliano, G.; Gallo, G.; Pistono, P.G.; Paggi, G.C. Cytokine detection in HIV-1/HHV-8 co-infected subjects. Cell. Biochem. Funct. 2002, 20, 191–194. [Google Scholar] [CrossRef]
- Anand, A.R.; Rachel, G.; Parthasarathy, D. HIV proteins and endothelial dysfunction: Implications in cardiovascular disease. Front. Cardiovasc. Med. 2018, 5, 185. [Google Scholar] [CrossRef]
- Ensoli, B.; Bellino, S.; Tripiciano, A.; Longo, O.; Francavilla, A.; Marcotullio, S.; Cafaro, A.; Picconi, O.; Paniccia, G.; Scoglio, A.; et al. Tat Reduces Immune Activation and Loss of Regulatory T-Cells and Improves Immune Function in Subjects on HAART. PLoS ONE 2010, 5, e13540. [Google Scholar] [CrossRef] [Green Version]
- Mediouni, S.; Darque, A.; Baillat, G.; Ravaux, I.; Dhiver, C.; Tissot-Dupont, H.; Mokhtari, M.; Moreau, H.; Tamalet, C.; Brunet, C.; et al. Antiretroviral therapy does not block the secretion of the human immunodeficiency virus Tat protein. Infect. Disord. Drug Targets 2012, 12, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Langner, J.; Riemann, D.; Machulla, H.K.G. Antigen Processing and Presentation by Vascular Endothelial Cells: Biochemical and Cell Physiological Preconditions. Endothelium 1995, 3, 141–150. [Google Scholar] [CrossRef]
- Savage, C.O.; Brooks, C.J.; Harcourt, G.C.; Picard, J.K.; King, W.; Sansom, D.M.; Willcox, N. Human vascular endothelial cells process and present autoantigen to human T cell lines. Int. Immunol. 1995, 7, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Marelli-Berg, F.M.; Hargreaves, R.E.; Carmichael, P.; Dorling, A.; Lombardi, G.; Lechler, R.I. Major histocompatibility complex class II-expressing endothelial cells induce allospecific nonresponsiveness in naive T cells. J. Exp. Med. 1996, 183, 1603–1612. [Google Scholar] [CrossRef] [Green Version]
- Pober, J.S.; Merola, J.; Liu, R.; Manes, T.D. Antigen Presentation by Vascular Cells. Front. Immunol. 2017, 8, 1907. [Google Scholar] [CrossRef] [Green Version]
- Sigal, A.; Kim, J.T.; Balazs, A.B.; Dekel, E.; Mayo, A.; Milo, R.; Baltimore, D. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 2011, 477, 95–98. [Google Scholar] [CrossRef]
- Baroldi, G.; Corallo, S.; Moroni, M.; Repossini, A.; Mutinelli, M.R.; Lazzarin, A.; Antonacci, C.M.; Cristina, S.; Negri, C. Focal lymphocytic myocarditis in acquired immunodeficiency syndrome (AIDS): A correlative morphologic and clinical study in 26 consecutive fatal cases. J. Am. Coll. Cardiol. 1988, 12, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Birdsall, H.H.; Porter, W.J.; Green, D.M.; Rubio, J.; Trial, J.; Rossen, R.D. Impact of fibronectin fragments on the transendothelial migration of HIV-infected leukocytes and the development of subendothelial foci of infectious leukocytes. J. Immunol. 2004, 173, 2746–2754. [Google Scholar] [CrossRef] [Green Version]
- Kruize, Z.; Kootstra, N.A. The Role of Macrophages in HIV-1 Persistence and Pathogenesis. Front. Microbiol. 2019, 10, 2828. [Google Scholar] [CrossRef] [Green Version]
- Cafaro, A.; Tripiciano, A.; Picconi, O.; Sgadari, C.; Moretti, S.; Buttò, S.; Monini, P.; Ensoli, B. Anti-Tat Immunity in HIV-1 Infection: Effects of Naturally Occurring and Vaccine-Induced Antibodies Against Tat on the Course of the Disease. Vaccines 2019, 7, 99. [Google Scholar] [CrossRef] [Green Version]
- Ensoli, F.; Cafaro, A.; Casabianca, A.; Tripiciano, A.; Bellino, S.; Longo, O.; Francavilla, V.; Picconi, O.; Sgadari, C.; Moretti, S.; et al. HIV-1 Tat immunization restores immune homeostasis and attacks the HAART-resistant blood HIV DNA: Results of a randomized phase II exploratory clinical trial. Retrovirology 2015, 12, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ensoli, B.; Nchabeleng, M.; Ensoli, F.; Tripiciano, A.; Bellino, S.; Picconi, O.; Sgadari, C.; Longo, O.; Tavoschi, L.; Joffe, D.; et al. HIV-Tat immunization induces cross-clade neutralizing antibodies and CD4 [+] T cell increases in antiretroviral-treated South African volunteers: A randomized phase II clinical trial. Retrovirology 2016, 13, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sgadari, C.; Monini, P.; Tripiciano, A.; Picconi, O.; Casabianca, A.; Orlandi, C.; Moretti, S.; Francavilla, V.; Arancio, A.; Paniccia, G.; et al. Continued Decay of HIV Proviral DNA Upon Vaccination With HIV-1 Tat of Subjects on Long-Term ART: An 8-Year Follow-Up Study. Front. Immunol. 2019, 10, 233. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.G.; Feinberg, M.B.; Joseph, S.F.; Harper, M.E.; Marselle, L.M.; Reyes, G.; Gonda, M.A.; Aldovini, A.; Debouk, C.; Gallo, R.C.; et al. The trans-activator gene of HTLV-III is essential for virus replication. Nature 1986, 320, 367–371. [Google Scholar] [CrossRef]
- Pierleoni, R.; Menotta, M.; Antonelli, A.; Sfara, C.; Serafini, G.; Dominici, S.; Laguardia, M.E.; Salis, A.; Damonte, G.; Banci, L.; et al. Effect of the redox state on HIV-1 tat protein multimerization and cell internalization and trafficking. Mol. Cell Biochem. 2010, 345, 105–118. [Google Scholar] [CrossRef]
- Rossi, C.; Balboni, P.G.; Betti, M.; Marconi, P.C.; Bozzini, R.; Grossi, M.P.; Barbanti-Brodano, G.; Caputo, A. Inhibition of HIV-1 replication by a Tat transdominant negative mutant in human peripheral blood lymphocytes from healthy donors and HIV-1-infected patients. Gene Ther. 1997, 4, 1261–1269. [Google Scholar] [CrossRef]
- Monini, P.; Cafaro, A.; Srivastava, I.K.; Moretti, S.; Sharma, V.A.; Andreini, C.; Chiozzini, C.; Ferrantelli, F.; Cossut, M.R.P.; Tripiciano, A.; et al. HIV-1 Tat Promotes Integrin-Mediated HIV Transmission to Dendritic Cells by Binding Env Spikes and Competes Neutralization by Anti-HIV Abs. PLoS ONE 2012, 7, e48781. [Google Scholar] [CrossRef]
- Shi, F.; Sottile, J. Caveolin-1-dependent beta1 integrin endocytosis is a critical regulator of fibronectin turnover. J. Cell Sci. 2008, 121, 2360–2371. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 1–37. [Google Scholar] [CrossRef] [Green Version]
- Péloponèse, J.M.; Collette, Y.; Grégoire, C.; Bailly, C.; Campèse, D.; Meurs, E.F.; Olive, D.; Loret, E.P. Full peptide synthesis, purification, and characterization of six Tat variants. Differences observed between HIV-1 isolates from Africa and other continents. J. Biol. Chem. 1999, 274, 11473–11478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregoire, C.; Péloponèse, J.M.; Esquieu, D.; Opi, S.; Campbell, G.; Solomiac, M.; Lebrun, E.; Lebreton, J.; Loret, E.P. Homonuclear (1)H-NMR assignment and structural characterization of human immunodeficiency virus type 1 Tat Mal protein. Biopolymers 2001, 62, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, S.J.; Thornton, J.M. NACCESS Computer Program; Department of Biochemistry and Molecular Biology, University College London: London, UK, 1993. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tat-Interacting Residues | Integrin-Interacting Residues | Interaction Type |
|---|---|---|
| 17-GLN | 1126-ASP | SC-SC H-bond |
| 56-ARG | 1180-MET | MC-SC H-bond |
| 62-SER | 1214-ARG | SC-SC H-bond |
| 75-SER | 178-TYR | SC-SC H-bond |
| 78-ARG | 150-ASP | SC-SC H-bond |
| 78-ARG * | 218-ASP * | SC-SC H-bond |
| 80-ASP | 1122-TYR | SC-MC H-bond |
| 80-ASP | 4001-MN2+ | SC-Mt bond |
| 80-ASP | 1123-SER | SC-MC H-bond |
| 86-GLU | 248-ARG | SC-SC H-bond |
| 86-GLU | 1253-LYS | SC-SC H-bond |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cafaro, A.; Barillari, G.; Moretti, S.; Palladino, C.; Tripiciano, A.; Falchi, M.; Picconi, O.; Pavone Cossut, M.R.; Campagna, M.; Arancio, A.; et al. HIV-1 Tat Protein Enters Dysfunctional Endothelial Cells via Integrins and Renders Them Permissive to Virus Replication. Int. J. Mol. Sci. 2021, 22, 317. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010317
Cafaro A, Barillari G, Moretti S, Palladino C, Tripiciano A, Falchi M, Picconi O, Pavone Cossut MR, Campagna M, Arancio A, et al. HIV-1 Tat Protein Enters Dysfunctional Endothelial Cells via Integrins and Renders Them Permissive to Virus Replication. International Journal of Molecular Sciences. 2021; 22(1):317. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010317
Chicago/Turabian StyleCafaro, Aurelio, Giovanni Barillari, Sonia Moretti, Clelia Palladino, Antonella Tripiciano, Mario Falchi, Orietta Picconi, Maria Rosaria Pavone Cossut, Massimo Campagna, Angela Arancio, and et al. 2021. "HIV-1 Tat Protein Enters Dysfunctional Endothelial Cells via Integrins and Renders Them Permissive to Virus Replication" International Journal of Molecular Sciences 22, no. 1: 317. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010317