Cell-Permeable Succinate Rescues Mitochondrial Respiration in Cellular Models of Statin Toxicity

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
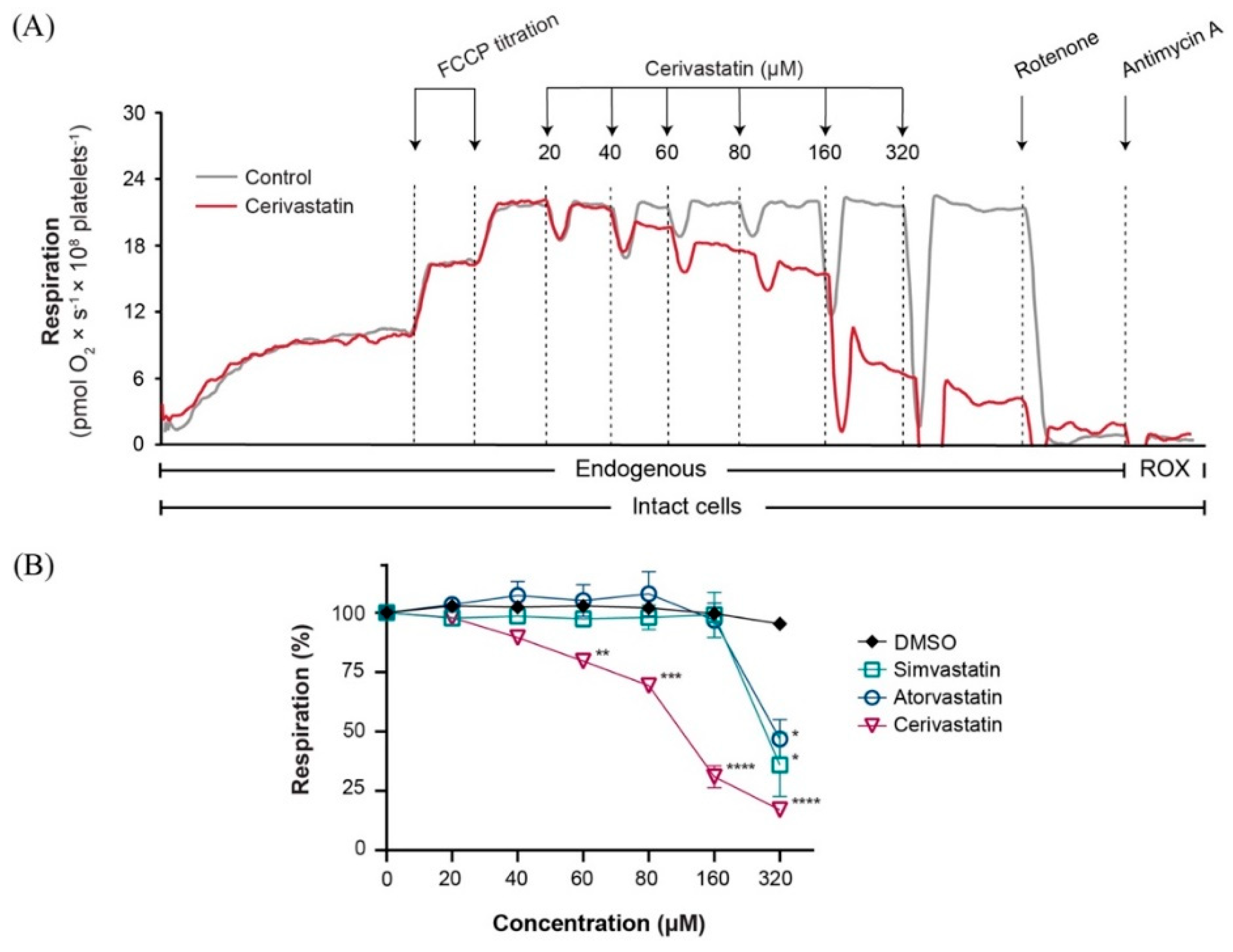
2.1. Statins Decreased Mitochondrial Respiration in Intact Platelets in a Concentration-Dependent Manner
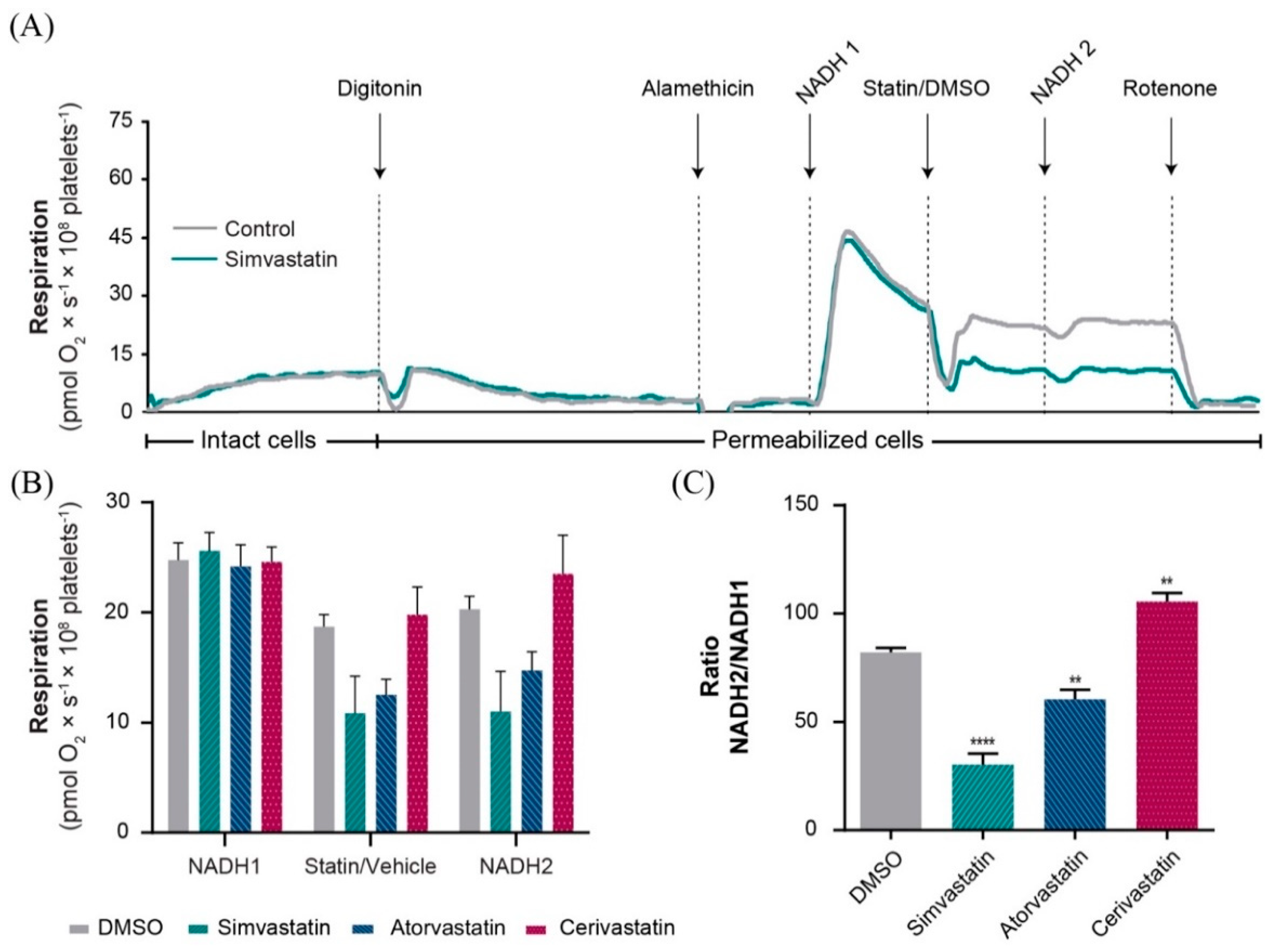
2.2. Statins Induced Mitochondrial Dysfunction in Permeabilized Platelets via Several Distinct Mechanisms
2.3. Simvastatin and Atorvastatin (but Not Cerivastatin) Directly Inhibited Complex I-Linked Respiration
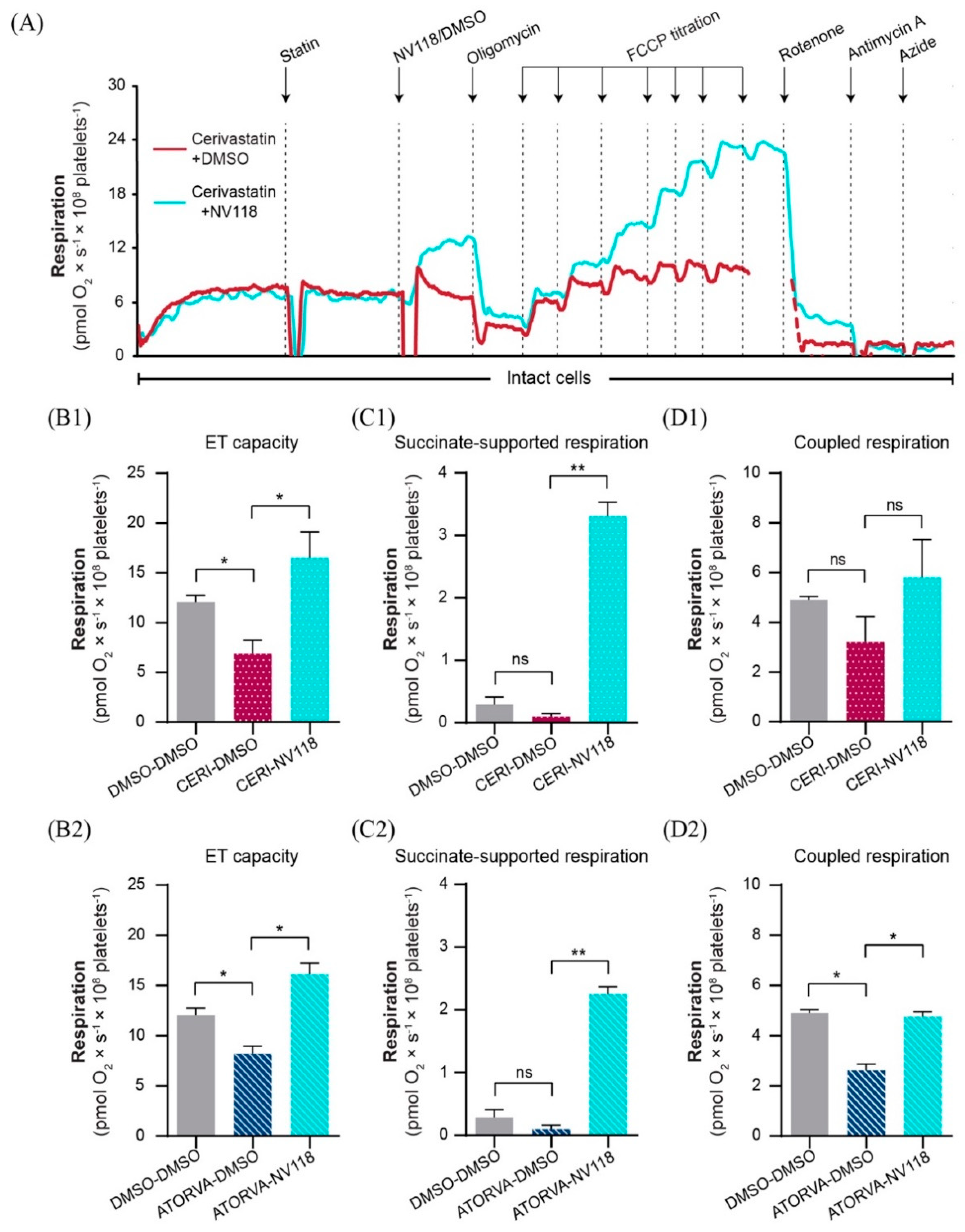
2.4. Cell-Permeable Succinate Bypassed the NADH-Linked Mitochondrial Dysfunction Induced by Statins in Human Platelets
2.5. Cell-Permeable Succinate Bypassed Mitochondrial Dysfunction Induced by Statins in HepG2 Cells
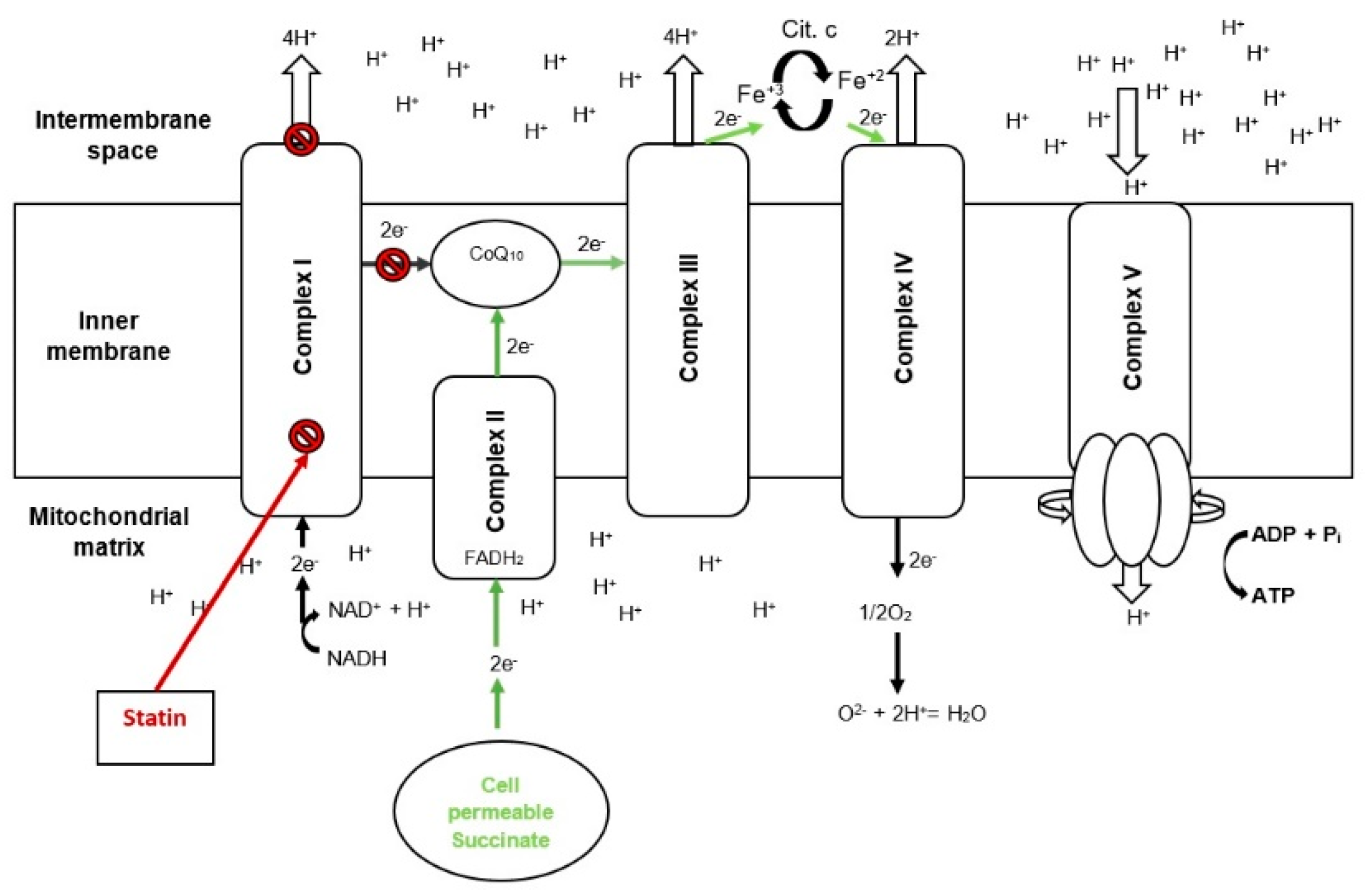
3. Discussion
4. Materials and Methods
4.1. Chemicals and Human Samples
4.2. Mitochondrial Respiration
4.3. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADP | Adenosine diphosphate |
| ATP | Adenosine triphosphate |
| Cit. c | Cytochrome c |
| DMSO | Dimethyl sulfoxide |
| ET | Electron transport |
| ETS | Electron transport system |
| FCCP | Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone |
| LDL | Low-density lipoprotein |
| LEAK | Non-phosphorylating respiration |
| OXPHOS | Oxidative phosphorylation |
| NADH | Nicotinamide adenine dinucleotide reduced form |
| ROS | Reactive oxygen species |
| ROX | Residual oxygen consumption |
| SAMS | Statin-associated muscle symptoms |
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics—2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2019. [Google Scholar] [CrossRef]
- Ference, B.A.; Ginsberg, H.N.; Graham, I.; Ray, K.K.; Packard, C.J.; Bruckert, E.; Hegele, R.A.; Krauss, R.M.; Raal, F.J.; Schunkert, H.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2017, 38, 2459–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björkhem-Bergman, L.; Lindh, J.D.; Bergman, P. What is a relevant statin concentration in cell experiments claiming pleiotropic effects? Br. J. Clin. Pharm. 2011, 72, 164–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroes, E.S.; Thompson, P.D.; Corsini, A.; Vladutiu, G.D.; Raal, F.J.; Ray, K.K.; Roden, M.; Stein, E.; Tokgözoğlu, L.; Nordestgaard, B.G.; et al. Statin-associated muscle symptoms: Impact on statin therapy-European Atherosclerosis Society Consensus Panel Statement on Assessment, Aetiology and Management. Eur. Heart J. 2015, 36, 1012–1022. [Google Scholar] [CrossRef]
- Mach, F.; Ray, K.K.; Wiklund, O.; Corsini, A.; Catapano, A.L.; Bruckert, E.; De Backer, G.; Hegele, R.A.; Hovingh, G.K.; Jacobson, T.A.; et al. Adverse effects of statin therapy: Perception vs. the evidence-focus on glucose homeostasis, cognitive, renal and hepatic function, haemorrhagic stroke and cataract. Eur. Heart J. 2018, 39, 2526–2539. [Google Scholar] [CrossRef]
- Apostolopoulou, M.; Corsini, A.; Roden, M. The role of mitochondria in statin-induced myopathy. Eur. J. Clin. Investig. 2015, 45, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, R.; Wierzbicki, A.S. Statins, Muscle Disease and Mitochondria. J. Clin. Med. 2017, 6, 75. [Google Scholar] [CrossRef] [Green Version]
- Bouitbir, J.; Sanvee, G.M.; Panajatovic, M.V.; Singh, F.; Krahenbuhl, S. Mechanisms of statin-associated skeletal muscle-associated symptoms. Pharmacol. Res. 2019, 104201. [Google Scholar] [CrossRef]
- Muntean, D.M.; Thompson, P.D.; Catapano, A.L.; Stasiolek, M.; Fabis, J.; Muntner, P.; Serban, M.C.; Banach, M. Statin-associated myopathy and the quest for biomarkers: Can we effectively predict statin-associated muscle symptoms? Drug Discov. Today 2017, 22, 85–96. [Google Scholar] [CrossRef]
- Vevera, J.; Fišar, Z.; Nekovářová, T.; Vrablík, M.; Zlatohlávek, L.; Hroudová, J.; Singh, N.; Raboch, J.; Valeš, K. Statin-Induced Changes in Mitochondrial Respiration in Blood Platelets in Rats and Human With Dyslipidemia. Physiol. Res. 2016, 65, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Allard, N.A.E.; Schirris, T.J.J.; Verheggen, R.J.; Russel, F.G.M.; Rodenburg, R.J.; Smeitink, J.A.M.; Thompson, P.D.; Hopman, M.T.E.; Timmers, S. Statins Affect Skeletal Muscle Performance: Evidence for Disturbances in Energy Metabolism. J. Clin. Endocrinol. Metab. 2018, 103, 75–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durhuus, J.A.; Hansson, S.; Morville, T.; Kuhlman, A.B.; Dohlmann, T.L.; Larsen, S.; Helge, J.W.; Angleys, M.; Muniesa-Vargas, A.; Bundgaard, J.R.; et al. Simvastatin improves mitochondrial respiration in peripheral blood cells. Sci. Rep. 2020, 10, 17012. [Google Scholar] [CrossRef] [PubMed]
- Petrus, A.T.; Lighezan, D.L.; Danila, M.D.; Duicu, O.M.; Sturza, A.; Muntean, D.M.; Ionita, I. Assessment of platelet respiration as emerging biomarker of disease. Physiol. Res. 2019, 68, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Sjövall, F.; Ehinger, J.K.H.; Marelsson, S.E.; Morota, S.; Åsander Frostner, E.; Uchino, H.; Lundgren, J.; Arnbjörnsson, E.; Hansson, M.J.; Fellman, V.; et al. Mitochondrial respiration in human viable platelets—Methodology and influence of gender, age and storage. Mitochondrion 2013, 13, 7–14. [Google Scholar] [CrossRef]
- Braganza, A.; Corey, C.G.; Santanasto, A.J.; Distefano, G.; Coen, P.M.; Glynn, N.W.; Nouraie, S.-M.; Goodpaster, B.H.; Newman, A.B.; Shiva, S. Platelet bioenergetics correlate with muscle energetics and are altered in older adults. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Sirvent, P.; Mercier, J.; Vassort, G.; Lacampagne, A. Simvastatin triggers mitochondria-induced Ca2+ signaling alteration in skeletal muscle. Biochem. Biophys. Res. Commun. 2005, 329, 1067–1075. [Google Scholar] [CrossRef]
- Ehinger, J.K.; Piel, S.; Ford, R.; Karlsson, M.; Sjövall, F.; Frostner, E.Å.; Morota, S.; Taylor, R.W.; Turnbull, D.M.; Cornell, C.; et al. Cell-permeable succinate prodrugs bypass mitochondrial complex I deficiency. Nat. Commun. 2016, 7, 12317. [Google Scholar] [CrossRef] [Green Version]
- Piel, S.; Ehinger, J.K.; Chamkha, I.; Frostner, E.Å.; Sjövall, F.; Elmér, E.; Hansson, M.J. Bioenergetic bypass using cell-permeable succinate, but not methylene blue, attenuates metformin-induced lactate production. Intensive Care Med. Exp. 2018, 6, 22. [Google Scholar] [CrossRef]
- Piel, S.; Chamkha, I.; Dehlin, A.K.; Ehinger, J.K.; Sjövall, F.; Elmér, E.; Hansson, M.J. Cell-permeable succinate prodrugs rescue mitochondrial respiration in cellular models of acute acetaminophen overdose. PLoS ONE 2020, 15, e0231173. [Google Scholar] [CrossRef]
- Furberg, C.D.; Pitt, B. Withdrawal of cerivastatin from the world market. Curr. Control. Trials Cardiovasc. Med. 2001, 2, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control an Introduction to OXPHOS Analysis, 4th ed.; OROBOROS MiPNet Publications: Innsbruck, Austria, 2014; p. 81. [Google Scholar]
- Kaufmann, P.; Török, M.; Zahno, A.; Waldhauser, K.M.; Brecht, K.; Krähenbühl, S. Toxicity of statins on rat skeletal muscle mitochondria. Cell. Mol. Life Sci. CMLS 2006, 63, 2415–2425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broniarek, I.; Jarmuszkiewicz, W. Atorvastatin affects negatively respiratory function of isolated endothelial mitochondria. Arch. Biochem. Biophys. 2018, 637, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Broniarek, I.; Dominiak, K.; Galganski, L.; Jarmuszkiewicz, W. The Influence of Statins on the Aerobic Metabolism of Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gvozdjakova, A.; Sumbalova, Z.; Kucharska, J.; Chladekova, A.; Rausova, Z.; Vancova, O.; Komlosi, M.; Ulicna, O.; Mojto, V. Platelet mitochondrial bioenergetic analysis in patients with nephropathies and non-communicable diseases: A new method. Bratisl. Lek. Listy 2019, 120, 630–635. [Google Scholar] [CrossRef] [Green Version]
- Mück, W. Clinical Pharmacokinetics of Cerivastatin. Clin. Pharmacokinet. 2000, 39, 99–116. [Google Scholar] [CrossRef]
- Lennernäs, H. Clinical Pharmacokinetics of Atorvastatin. Clin. Pharmacokinet. 2003, 42, 1141–1160. [Google Scholar] [CrossRef]
- Kashani, A.; Phillips, C.O.; Foody, J.M.; Wang, Y.; Mangalmurti, S.; Ko, D.T.; Krumholz, H.M. Risks Associated With Statin Therapy. Circulation 2006, 114, 2788–2797. [Google Scholar] [CrossRef] [Green Version]
- Hirota, T.; Ieiri, I. Drug–drug interactions that interfere with statin metabolism. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1435–1447. [Google Scholar] [CrossRef]
- Wooten, J.M. A Brief Drug Class Review: Considerations for Statin Use, Toxicity, and Drug Interactions. South Med. J. 2018, 111, 39–44. [Google Scholar] [CrossRef] [Green Version]
- Duncan, A.J.; Hargreaves, I.P.; Damian, M.S.; Land, J.M.; Heales, S.J.R. Decreased Ubiquinone Availability and Impaired Mitochondrial Cytochrome Oxidase Activity Associated With Statin Treatment. Toxicol. Mech. Methods 2009, 19, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Wiklund, O.; Pirazzi, C.; Romeo, S. Monitoring of lipids, enzymes, and creatine kinase in patients on lipid-lowering drug therapy. Curr. Cardiol. Rep. 2013, 15, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Task Force, M.; Perk, J.; De Backer, G.; Gohlke, H.; Graham, I.; Reiner, Ž.; Verschuren, M.; Albus, C.; Benlian, P.; Boysen, G.; et al. European Guidelines on cardiovascular disease prevention in clinical practice (version 2012): The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts) Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR)†. Eur. Heart J. 2012, 33, 1635–1701. [Google Scholar] [CrossRef] [Green Version]
- Azie, N.E.; Brater, D.C.; Becker, P.A.; Jones, D.R.; Hall, S.D. The interaction of diltiazem with lovastatin and pravastatin. Clin. Pharmacol. Ther. 1998, 64, 369–377. [Google Scholar] [CrossRef]
- Bellosta, S.; Paoletti, R.; Corsini, A. Safety of Statins. Circulation 2004, 109, III-50–III-57. [Google Scholar] [CrossRef] [Green Version]
- Herminghaus, A.; Laser, E.; Schulz, J.; Truse, R.; Vollmer, C.; Bauer, I.; Picker, O. Pravastatin and Gemfibrozil Modulate Differently Hepatic and Colonic Mitochondrial Respiration in Tissue Homogenates from Healthy Rats. Cells 2019, 8, 983. [Google Scholar] [CrossRef] [Green Version]
- Fromenty, B.; Fisch, C.; Berson, A.; Letteron, P.; Larrey, D.; Pessayre, D. Dual effect of amiodarone on mitochondrial respiration. Initial protonophoric uncoupling effect followed by inhibition of the respiratory chain at the levels of complex I and complex II. J. Pharmacol. Exp. Ther. 1990, 255, 1377. [Google Scholar]
- Rodriguez, R.J.; Acosta, D., Jr. Inhibition of mitochondrial function in isolated rat liver mitochondria by azole antifungals. J. Biochem. Toxicol. 1996, 11, 127–131. [Google Scholar] [CrossRef]
- Salimi, A.; Eybagi, S.; Seydi, E.; Naserzadeh, P.; Kazerouni, N.P.; Pourahmad, J. Toxicity of macrolide antibiotics on isolated heart mitochondria: A justification for their cardiotoxic adverse effect. Xenobiotica 2016, 46, 82–93. [Google Scholar] [CrossRef]
- Wagner, B.K.; Kitami, T.; Gilbert, T.J.; Peck, D.; Ramanathan, A.; Schreiber, S.L.; Golub, T.R.; Mootha, V.K. Large-scale chemical dissection of mitochondrial function. Nat. Biotechnol. 2008, 26, 343–351. [Google Scholar] [CrossRef] [Green Version]
- von Hafe, M.; Neves, J.S.; Vale, C.; Borges-Canha, M.; Leite-Moreira, A. The impact of thyroid hormone dysfunction on ischemic heart disease. Endocr. Connect. 2019, 8, R76–R90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- du Souich, P.; Roederer, G.; Dufour, R. Myotoxicity of statins: Mechanism of action. Pharmacol. Ther. 2017, 175, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hoek, J.B.; Cahill, A.; Pastorino, J.G. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology 2002, 122, 2049–2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Sobenin, I.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. Mitochondrial Aging and Age-Related Dysfunction of Mitochondria. BioMed Res. Int. 2014, 2014, 7. [Google Scholar] [CrossRef] [Green Version]
- Nadanaciva, S.; Dykens, J.A.; Bernal, A.; Capaldi, R.A.; Will, Y. Mitochondrial impairment by PPAR agonists and statins identified via immunocaptured OXPHOS complex activities and respiration. Toxicol. Appl. Pharmacol. 2007, 223, 277–287. [Google Scholar] [CrossRef]
- Ji, S.; You, Y.; Kerner, J.; Hoppel, C.L.; Schoeb, T.R.; Chick, W.S.H.; Hamm, D.A.; Sharer, J.D.; Wood, P.A. Homozygous carnitine palmitoyltransferase 1b (muscle isoform) deficiency is lethal in the mouse. Mol. Genet. Metab. 2008, 93, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Keung, W.; Ussher, J.R.; Jaswal, J.S.; Raubenheimer, M.; Lam, V.H.M.; Wagg, C.S.; Lopaschuk, G.D. Inhibition of carnitine palmitoyltransferase-1 activity alleviates insulin resistance in diet-induced obese mice. Diabetes 2013, 62, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Kimura, J. Mechanism of Statin-Induced Rhabdomyolysis. J. Pharmacol. Sci. 2013, 123, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Michalska-Kasiczak, M.; Sahebkar, A.; Mikhailidis, D.P.; Rysz, J.; Muntner, P.; Toth, P.P.; Jones, S.R.; Rizzo, M.; Kees Hovingh, G.; Farnier, M.; et al. Analysis of vitamin D levels in patients with and without statin-associated myalgia—A systematic review and meta-analysis of 7 studies with 2420 patients. Int. J. Cardiol. 2015, 178, 111–116. [Google Scholar] [CrossRef]
- Turner, R.M.; Pirmohamed, M. Statin-Related Myotoxicity: A Comprehensive Review of Pharmacokinetic, Pharmacogenomic and Muscle Components. J. Clin. Med. 2020, 9, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riche, K.D.; Arnall, J.; Rieser, K.; East, H.E.; Riche, D.M. Impact of vitamin D status on statin-induced myopathy. J. Clin. Transl. Endocrinol. 2016, 6, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Dohlmann, T.L.; Morville, T.; Kuhlman, A.B.; Chrøis, K.M.; Helge, J.W.; Dela, F.; Larsen, S. Statin Treatment Decreases Mitochondrial Respiration But Muscle Coenzyme Q10 Levels Are Unaltered: The LIFESTAT Study. J. Clin. Endocrinol. Metab. 2018, 104, 2501–2508. [Google Scholar] [CrossRef] [PubMed]
- Päivä, H.; Thelen, K.M.; Van Coster, R.; Smet, J.; De Paepe, B.; Mattila, K.M.; Laakso, J.; Lehtimäki, T.; von Bergmann, K.; Lütjohann, D.; et al. High-dose statins and skeletal muscle metabolism in humans: A randomized, controlled trial. Clin. Pharmacol. Ther. 2005, 78, 60–68. [Google Scholar] [CrossRef]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim. Et Biophys. Acta Bioenerg. 2016, 1857, 1086–1101. [Google Scholar] [CrossRef]
- Lin, C.S.; Sharpley, M.S.; Fan, W.; Waymire, K.G.; Sadun, A.A.; Carelli, V.; Ross-Cisneros, F.N.; Baciu, P.; Sung, E.; McManus, M.J.; et al. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc. Natl. Acad. Sci. USA 2012, 109, 20065–20070. [Google Scholar] [CrossRef] [Green Version]
- Gnaiger, E.; Kuznetsov, A.V.; Schneeberger, S.; Seiler, R.; Brandacher, G.; Steurer, W.; Margreiter, R. Mitochondria in the Cold. In Proceedings of Life in the Cold; Springer: Berlin/Heidelberg, Germany, 2000; pp. 431–442. [Google Scholar]
- Zhang, H.; Plutzky, J.; Skentzos, S.; Morrison, F.; Mar, P.; Shubina, M.; Turchin, A. Discontinuation of statins in routine care settings: A cohort study. Ann. Intern. Med. 2013, 158, 526–534. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effect | Simvastatin | Atorvastatin | Cerivastatin |
|---|---|---|---|
| OXPHOS coupling efficiency reduction | YES | YES | YES |
| ET capacity inhibition | YES | YES | YES |
| NADH-linked ETS inhibition | YES | YES | YES |
| Direct inhibition of NADH-dehydrogenase | YES | YES | NO |
| Succinate-linked ETS inhibition | YES | NO | NO |
| Uncoupling (increased LEAK respiration) | NO | YES | YES |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avram, V.F.; Chamkha, I.; Åsander-Frostner, E.; Ehinger, J.K.; Timar, R.Z.; Hansson, M.J.; Muntean, D.M.; Elmér, E. Cell-Permeable Succinate Rescues Mitochondrial Respiration in Cellular Models of Statin Toxicity. Int. J. Mol. Sci. 2021, 22, 424. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010424
Avram VF, Chamkha I, Åsander-Frostner E, Ehinger JK, Timar RZ, Hansson MJ, Muntean DM, Elmér E. Cell-Permeable Succinate Rescues Mitochondrial Respiration in Cellular Models of Statin Toxicity. International Journal of Molecular Sciences. 2021; 22(1):424. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010424
Chicago/Turabian StyleAvram, Vlad F., Imen Chamkha, Eleonor Åsander-Frostner, Johannes K. Ehinger, Romulus Z. Timar, Magnus J. Hansson, Danina M. Muntean, and Eskil Elmér. 2021. "Cell-Permeable Succinate Rescues Mitochondrial Respiration in Cellular Models of Statin Toxicity" International Journal of Molecular Sciences 22, no. 1: 424. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22010424