Ingested Ketone Ester Leads to a Rapid Rise of Acetyl-CoA and Competes with Glucose Metabolism in the Brain of Non-Fasted Mice

 ,
,  , , and
, , and 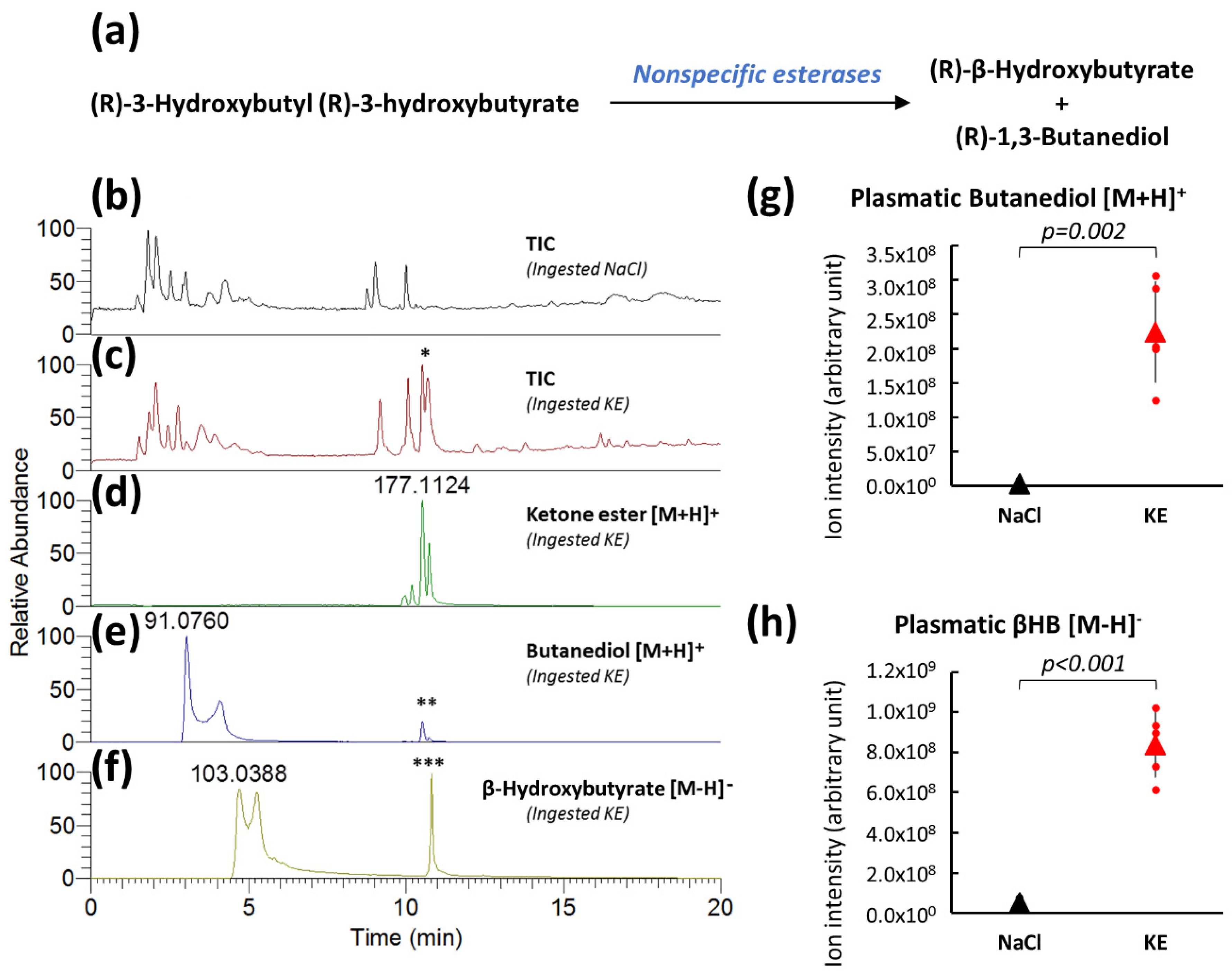{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
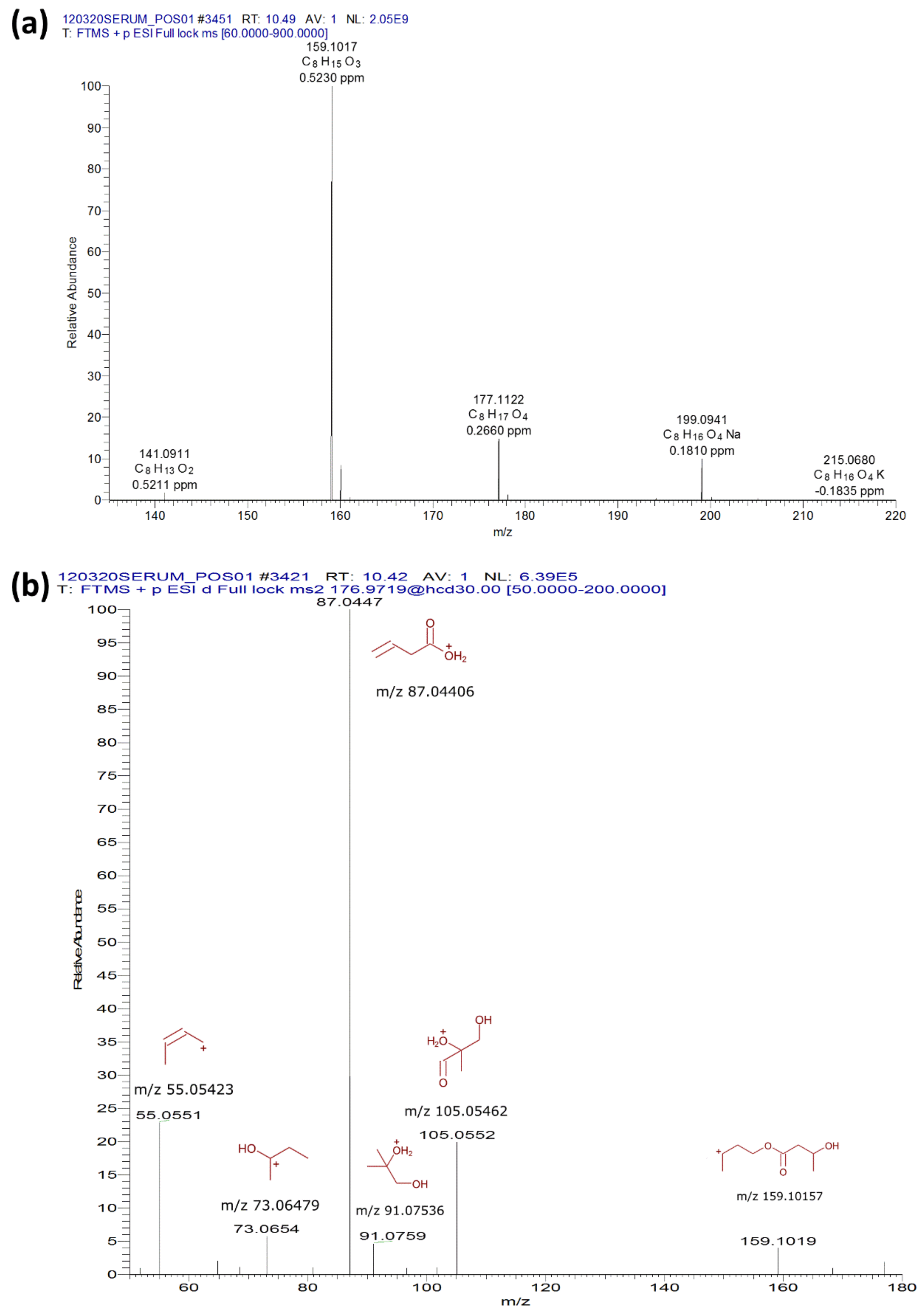
2.1. Plasma Analysis of (R)-3-Hydroxybutyl (R)-3-Hydroxybutyrate-Gavaged Mice
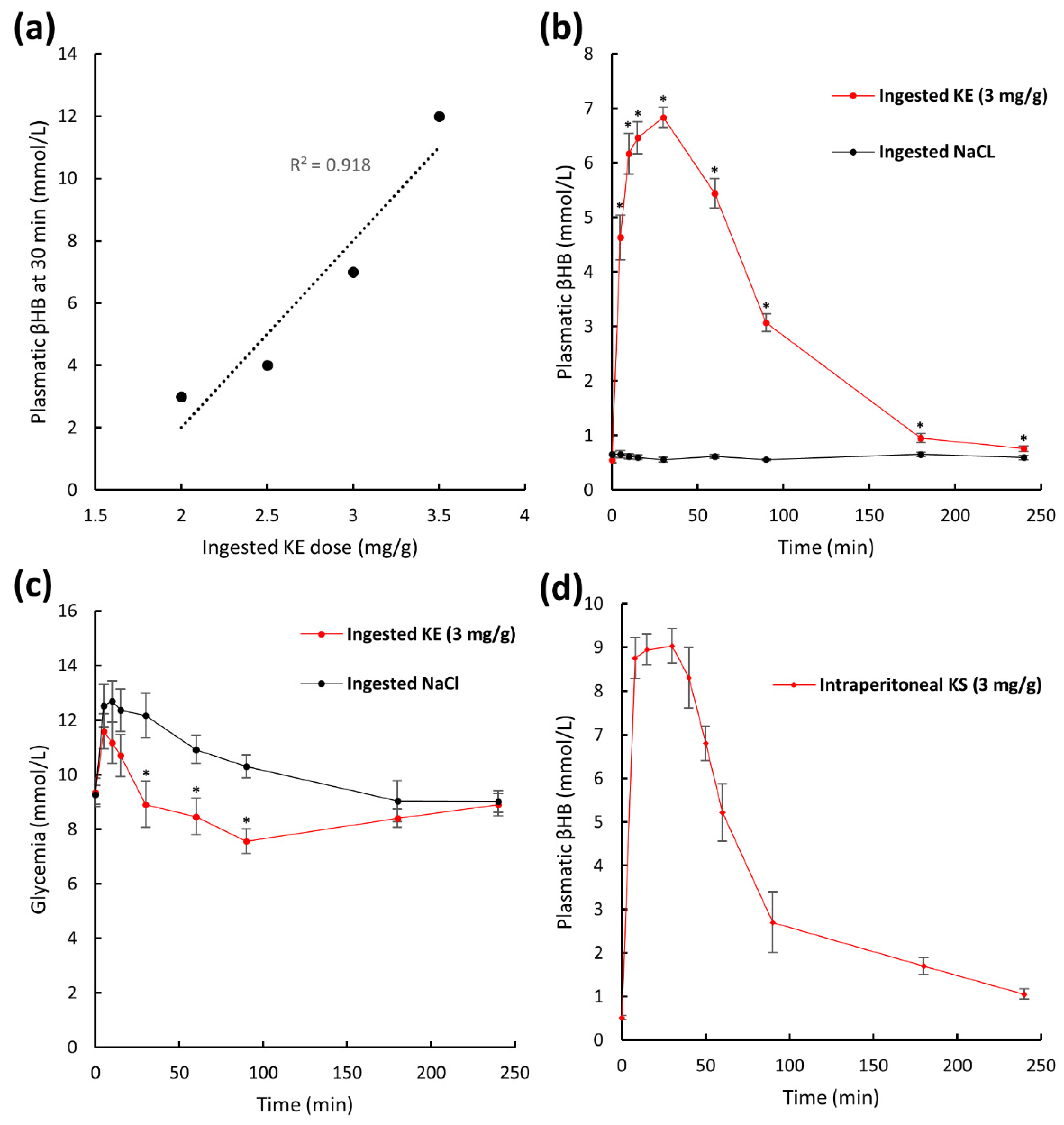
2.2. Plasma Pharmacokinetics of β-Hydroxybutyrate in Non-Fasted Mice after KE Ingestion
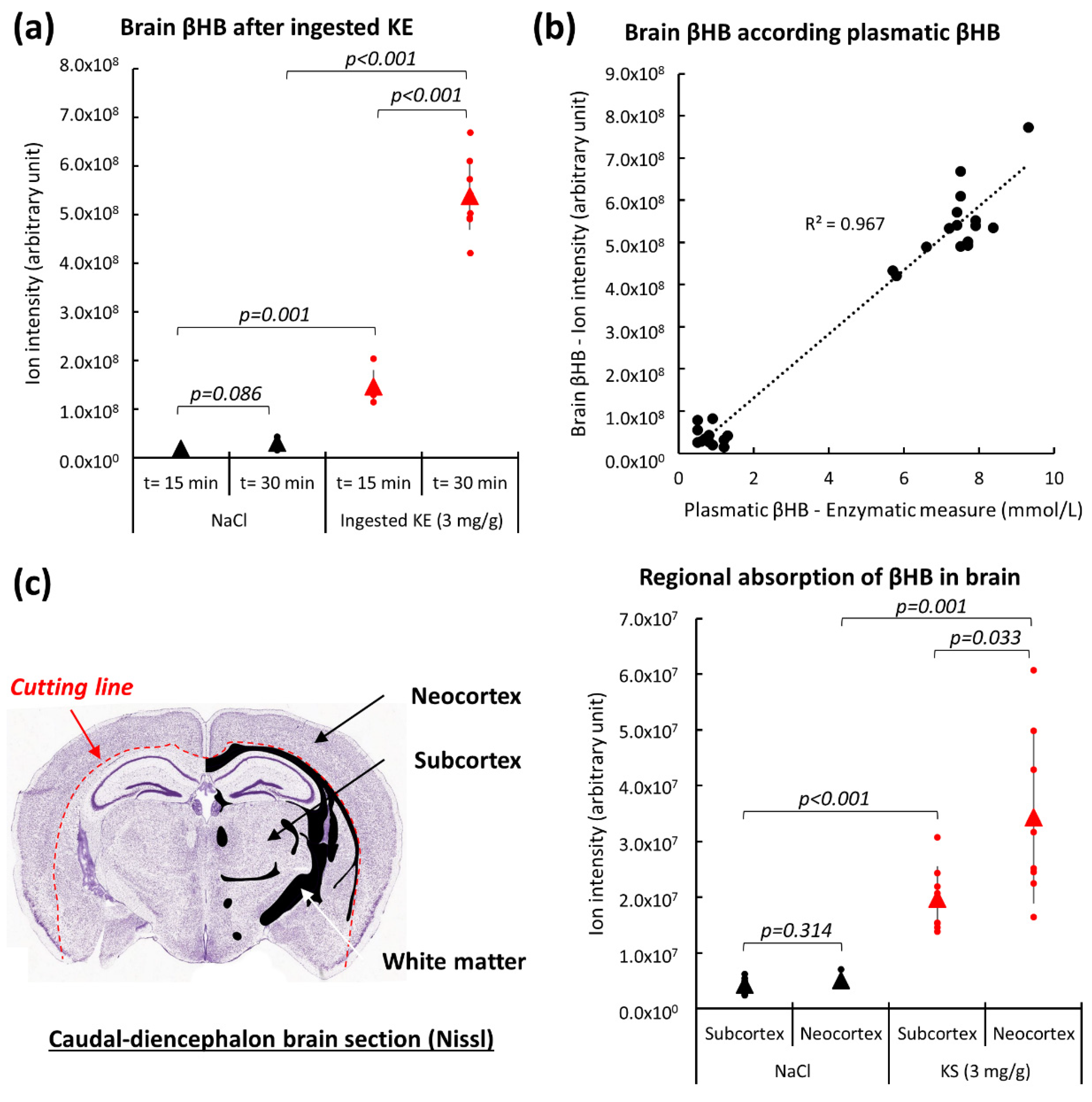
2.3. Brain β-Hydroxybutyrate Levels in KE-Gavaged Mice
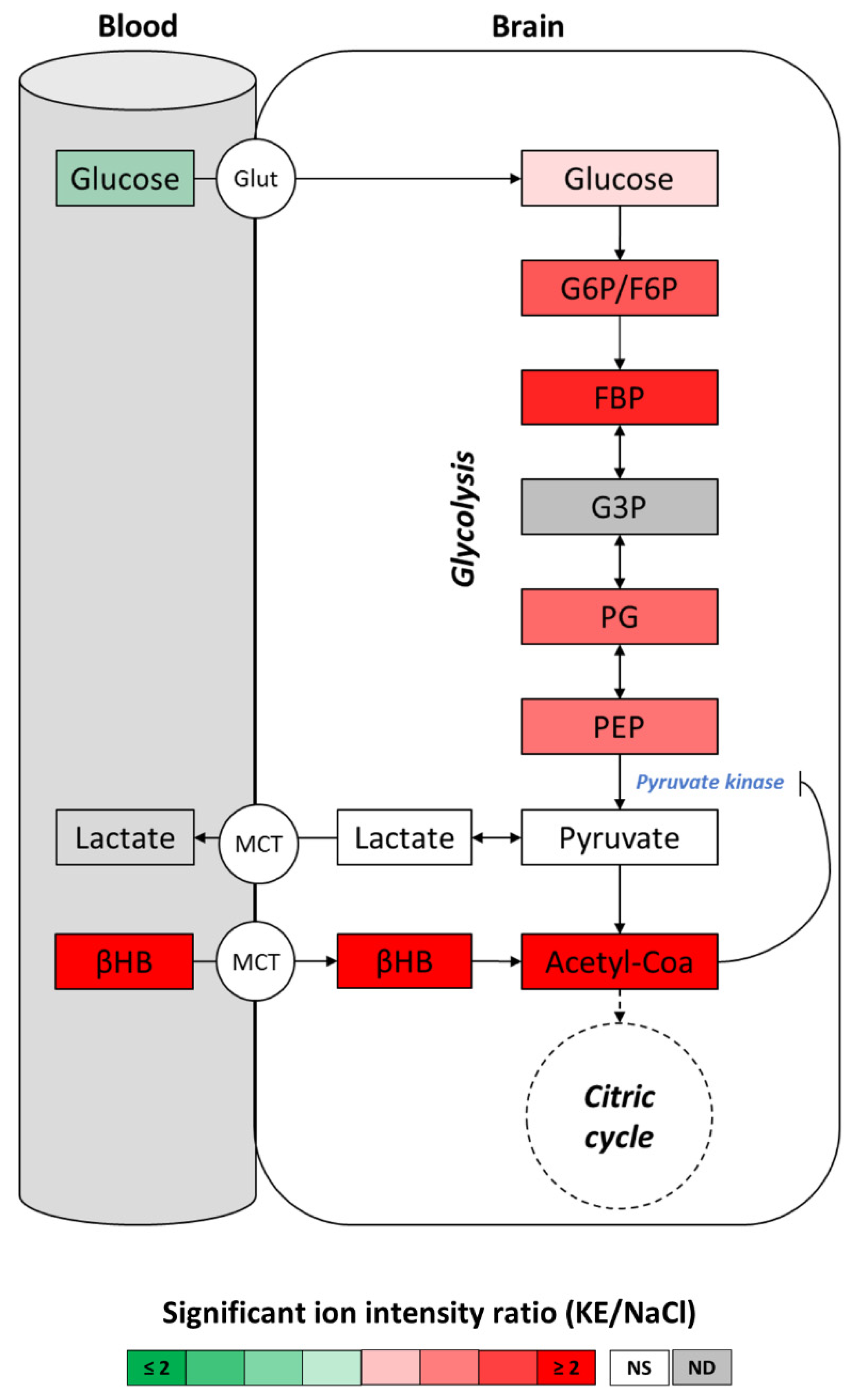
2.4. Energy Metabolism in the Brain of KE-Gavaged Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Ketone Administration
4.3. Blood Collection and Processing for LC-MS Analysis
4.4. Brain Collection and Processing for LC-MS Analysis
4.5. LC-MS Analysis
4.6. Glucose and β-Hydroxybutyrate Enzymatic Measurements
4.7. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| KE | Ketone ester |
| βHB | Β-Hydroxybutyrate |
| Acetyl-CoA | Acetyl-Coenzyme A |
| LC-MS | Liquid chromatography-Mass spectrometry |
References
- Robinson, A.M.; Williamson, D.H. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [CrossRef]
- Morris, A.A.M. Cerebral ketone body metabolism. J. Inherit. Metab. Dis. 2005, 28, 109–121. [Google Scholar] [CrossRef]
- Wood, T.R.; Stubbs, B.J.; Juul, S.E. Exogenous ketone bodies as promising neuroprotective agents for developmental brain injury. Dev. Neurosci. 2018, 40, 451–462. [Google Scholar] [CrossRef]
- Yang, H.; Shan, W.; Zhu, F.; Wu, J.; Wang, Q. Ketone bodies in neurological diseases: Focus on neuroprotection and underlying mechanisms. Front Neurol. 2019, 10, 585. [Google Scholar] [CrossRef] [Green Version]
- White, H.; Venkatesh, K.; Venkatesh, B. Systematic review of the use of ketones in the management of acute and chronic neurological disorders. J. Neurol. Neurosci. 2017, 8, 2. [Google Scholar] [CrossRef]
- White, H.; Venkatesh, B. Clinical review: Ketones and brain injury. Crit. Care. 2011, 15, 219. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Lamanna, J.C.; Puchowicz, M.A. Neuroprotective properties of ketone bodies. Adv. Exp. Med. Biol. 2012, 737, 97–102. [Google Scholar]
- Suzuki, M.; Suzuki, M.; Kitamura, Y.; Mori, S.; Sato, K.; Dohi, S.; Hiraide, A. Beta-hydroxybutyrate, a cerebral function improving agent, protects rat brain against ischemic damage caused by permanent and transient focal cerebral ischemia. Jpn. J. Pharmacol. 2002, 89, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Suzuki, M.; Sato, K.; Dohi, S.; Sato, T.; Matsuura, A.; Hiraide, A. Effect of beta-hydroxybutyrate, a cerebral function improving agent, on cerebral hypoxia, anoxia and ischemia in mice and rats. Jpn. J. Pharmacol. 2001, 87, 143–150. [Google Scholar] [CrossRef]
- Puchowicz, M.A.; Zechel, J.L.; Valerio, J.; Emancipator, D.S.; Xu, K.; Pundik, S.; Lust, W.D. Neuroprotection in diet-induced ketotic rat brain after focal ischemia. J. Cereb. Blood Flow. Metab. 2008, 28, 1907–1916. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Ye, L.; Sharma, K.; Jin, Y.; Harrison, M.M.; Caldwell, T.; Berthiaume, J.M.; Luo, Y.; LaManna, J.C.; Puchowicz, M.A. Diet-induced ketosis protects against focal cerebral ischemia in mouse. Adv. Exp. Med. Biol. 2017, 977, 205–213. [Google Scholar] [PubMed]
- Yin, J.; Han, P.; Tang, Z.; Liu, Q.; Shi, J. Sirtuin 3 mediates neuroprotection of ketones against ischemic stroke. J. Cereb. Blood Flow. Metab. 2015, 35, 1783–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.S.; Woo, D.-C.; Woo, C.-W.; Kim, K.-S. Exogenous β-hydroxybutyrate treatment and neuroprotection in a suckling rat mdel of hypoxic-ischemic encephalopathy. Dev. Neurosci. 2018, 40, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L. The therapeutic implications of ketone bodies: The effects of ketone bodies in pathological conditions: Ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 309–319. [Google Scholar] [CrossRef]
- Clarke, K.; Tchabanenko, K.; Pawlosky, R.; Carter, E.; Todd King, M.; Musa-Veloso, K.; Ho, M.; Roberts, A.; Robertson, J.; Vanitallie, T.B.; et al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul. Toxicol. Pharmacol. 2012, 63, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Taketani, M.; Shii, M.; Ohura, K.; Ninomiya, S.; Imai, T. Carboxylesterase in the liver and small intestine of experimental animals and human. Life Sci. 2007, 81, 924–932. [Google Scholar] [CrossRef]
- Tate, R.L.; Mehlman, M.A.; Tobin, R.B. Metabolie fate of 1,3-butanediol in the rat: Conversion to β-hydroxybutyrate. J. Nutr. 1971, 101, 1719–1726. [Google Scholar] [CrossRef] [Green Version]
- Desrochers, S.; Quinze, K.; Dugas, H.; Dubreuil, P.; Bomont, C.; David, F.; Kamlesh, C.A.; Kumar, A.; Soloviev, V.M.; Powers, L.; et al. R,S-1,3-butanediol acetoacetate esters, potential alternates to lipid emulsions for total parenteral nutrition. J. Nutr. Biochem. 1995, 6, 111–118. [Google Scholar] [CrossRef]
- Kesl, S.L.; Poff, A.M.; Ward, N.P.; Fiorelli, T.N.; Ari, C.; van Putten, A.J.; Sherwood, W.J.; Arnold, P.; D’Agostino, P.D. Effects of exogenous ketone supplementation on blood ketone, glucose, triglyceride, and lipoprotein levels in Sprague–Dawley rats. Nutr. Metab. 2016, 13, 9. [Google Scholar] [CrossRef] [Green Version]
- Amiel, S.A.; Archibald, H.R.; Chusney, G.; Williams, A.J.K.; Gale, E.A.M. Ketone infusion lowers hormonal responses to hypoglycaemia: Evidence for acute cerebral utilization of a non-glucose fuel. Clin. Sci. 1991, 81, 189–194. [Google Scholar] [CrossRef]
- Sato, K.; Kashiwaya, Y.; Keon, C.A.; Tsuchiya, N.; King, M.T.; Radda, G.K.; Chance, B.; Clarke, K.; Veech, R.L. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. 1995, 9, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.; Ross, B.; Arun, P.; Madhavarao, C.; Namboodiri, A. N-Acetylaspartate in the CNS: From neurodiagnostics to neurobiology. Prog. Neurobiol. 2007, 81, 89–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselbalch, S.G.; Madsen, P.L.; Hageman, L.P.; Olsen, K.S.; Justesen, N.; Holm, S.; Paulson, O.B. Changes in cerebral blood flow and carbohydrate metabolism during acute hyperketonemia. Am. J. Physiol. 1996, 270, E746–E751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courchesne-Loyer, A.; Croteau, E.; Castellano, C.-A.; St-Pierre, V.; Hennebelle, M.; Cunnane, S.C. Inverse relationship between brain glucose and ketone metabolism in adults during short-term moderate dietary ketosis: A dual tracer quantitative positron emission tomography study. J. Cereb. Blood Flow. Metab. 2017, 37, 2485–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlosky, R.J.; Kemper, M.F.; Kashiwaya, Y.; King, M.T.; Mattson, M.P.; Veech, R.L. Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 2017, 141, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Valente-Silva, P.; Lemos, C.; Köfalvi, A.; Cunha, R.A.; Jones, J.G. Ketone bodies effectively compete with glucose for neuronal acetyl-CoA generation in rat hippocampal slices: Ketone body oxidation in brain slices. NMR Biomed. 2015, 28, 1111–1116. [Google Scholar] [CrossRef]
- Weber, G.; Lea, M.A.; Stamm, N.B. Inhibition of pyruvate kinase and glucokinase by acetyl-CoA and inhibition of glucokinase by phosphoenolpyruvate. Life Sci. 1967, 6, 2441–2452. [Google Scholar] [CrossRef]
- Murray, A.J.; Knight, N.S.; Cole, M.A.; Cochlin, L.E.; Carter, E.; Tchabanenko, K.; Pichulik, T.; Gulston, M.K.; Atherton, H.J.; Schroeder, M.A.; et al. Novel ketone diet enhances physical and cognitive performance. FASEB J. 2016, 30, 4021–4032. [Google Scholar] [CrossRef] [Green Version]
- Shivva, V.; Cox, P.J.; Clarke, K.; Veech, R.L.; Tucker, I.G.; Duffull, S.B. The population pharmacokinetics of D-β-hydroxybutyrate following administration of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate. AAPS J. 2016, 18, 678–688. [Google Scholar]
- Cox, P.J.; Kirk, T.; Ashmore, T.; Willerton, K.; Evans, R.; Smith, A.; Murray, A.J.; Stubbs, B.; West, J.; McLure, W.S.; et al. Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab. 2016, 24, 256–268. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; Price, N.T. The proton-linked monocarboxylate transporter (MCT) family: Structure, function and regulation. Biochem. J. 1999, 2, 281–299. [Google Scholar] [CrossRef]
- Hasselbalch, S.G.; Knudsen, G.M.; Jakobsen, J.; Hageman, L.P.; Holm, S.; Paulson, O.B. Blood-brain barrier permeability of glucose and ketone bodies during short-term starvation in humans. Am. J. Physiol. Endocrinol. Metab. 1995, 268, E1161–E1166. [Google Scholar] [CrossRef] [PubMed]
- Raskin, N.H.; Sokoloff, L. Alcohol dehydrogenase activity in rat brain and liver. J. Neurochem. 1970, 17, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.A.; Mans, A.M.; Davis, D.W. Regional ketone body utilization by rat brain in starvation and diabetes. Am. J. Physiol. 1986, 250, E169–E178. [Google Scholar] [CrossRef]
- Hawkins, R.; Biebuyck, J. Ketone bodies are selectively used by individual brain regions. Science 1979, 205, 325–327. [Google Scholar] [CrossRef]
- Koehler-Stec, E.M.; Simpson, I.A.; Vannucci, S.J.; Landschulz, K.T.; Landschulz, W.H. Monocarboxylate transporter expression in mouse brain. Am. J. Physiol. Endocrinol. Metab. 1998, 275, E516–E524. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, L.; Bergersen, L.H.; Halestrap, A.P.; Pierre, K. Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J. Neurosci. Res. 2005, 79, 55–64. [Google Scholar] [CrossRef]
- Patel, M.S.; Johnson, C.A.; Rajan, R.; Owen, O.E. The metabolism of ketone bodies in developing human brain: Development of ketone-body-utilizing enzymes and ketone bodies as precursors for lipid synthesis. J. Neurochem. 1975, 25, 905–908. [Google Scholar] [CrossRef]
- Soeters, M.R.; Serlie, M.J.; Sauerwein, H.P.; Duran, M.; Ruiter, J.P.; Kulik, W.; Ackermans, M.T.; Minkler, P.E.; Hoppel, C.L.; Wanders, J.A.R.; et al. Characterization of D-3-hydroxybutyrylcarnitine (ketocarnitine): An identified ketosis-induced metabolite. Metabolism 2012, 61, 966–973. [Google Scholar] [CrossRef]
- Suissa, L.; Panicucci, E.; Perot, C.; Romero, G.; Gazzola, S.; Laksiri, N.; Ackermans, M.T.; Minkler, E.P.; Hoppel, C.L.; Wanders, J.A.R.; et al. Effect of hyperglycemia on stroke outcome is not homogeneous to all patients treated with mechanical thrombectomy. Clin. Neurol. Neurosurg. 2020, 194, 105750. [Google Scholar] [CrossRef]
- Chamorro, Á.; Brown, S.; Amaro, S.; Hill, M.D.; Muir, K.W.; Dippel, D.W.J.; van Zwam, W.; Butcher, K.; Ford, G.A.; den Hertog, M.H. Glucose modifies the effect of endovascular thrombectomy in patients with acute stroke. Stroke 2019, 50, 690–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, M.W.; Barber, P.A.; Desmond, P.M.; Baird, T.A.; Darby, D.G.; Byrnes, G.; Brian, M.; Tress, M.D.; Stephen, M.; Davis, M.D.; et al. Acute hyperglycemia adversely affects stroke outcome: A magnetic resonance imaging and spectroscopy study. Ann. Neurol. 2002, 52, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, R.M.; Stead, L.G. The role of hyperglycemia in acute ischemic stroke. Neurocrit. Care 2006, 5, 153–158. [Google Scholar] [CrossRef]
- Suissa, L.; Flachon, V.; Guigonis, J.-M.; Olivieri, C.-V.; Burel-Vandenbos, F.; Guglielmi, J.; Ambrosetti, D.; Gérard, M.; Franken, P.; Darcourt, J.; et al. Urinary ketone body loss leads to degeneration of brain white matter in elderly SLC5A8-deficient mice. J. Cereb. Blood Flow. Metab. 2020, 40, 1709–1723. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic. Acids. Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suissa, L.; Kotchetkov, P.; Guigonis, J.-M.; Doche, E.; Osman, O.; Pourcher, T.; Lindenthal, S. Ingested Ketone Ester Leads to a Rapid Rise of Acetyl-CoA and Competes with Glucose Metabolism in the Brain of Non-Fasted Mice. Int. J. Mol. Sci. 2021, 22, 524. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020524
Suissa L, Kotchetkov P, Guigonis J-M, Doche E, Osman O, Pourcher T, Lindenthal S. Ingested Ketone Ester Leads to a Rapid Rise of Acetyl-CoA and Competes with Glucose Metabolism in the Brain of Non-Fasted Mice. International Journal of Molecular Sciences. 2021; 22(2):524. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020524
Chicago/Turabian StyleSuissa, Laurent, Pavel Kotchetkov, Jean-Marie Guigonis, Emilie Doche, Ophélie Osman, Thierry Pourcher, and Sabine Lindenthal. 2021. "Ingested Ketone Ester Leads to a Rapid Rise of Acetyl-CoA and Competes with Glucose Metabolism in the Brain of Non-Fasted Mice" International Journal of Molecular Sciences 22, no. 2: 524. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22020524