
Role of HMOX1 Promoter Genetic Variants in Chemoresistance and Chemotherapy Induced Neutropenia in Children with Acute Lymphoblastic Leukemia

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
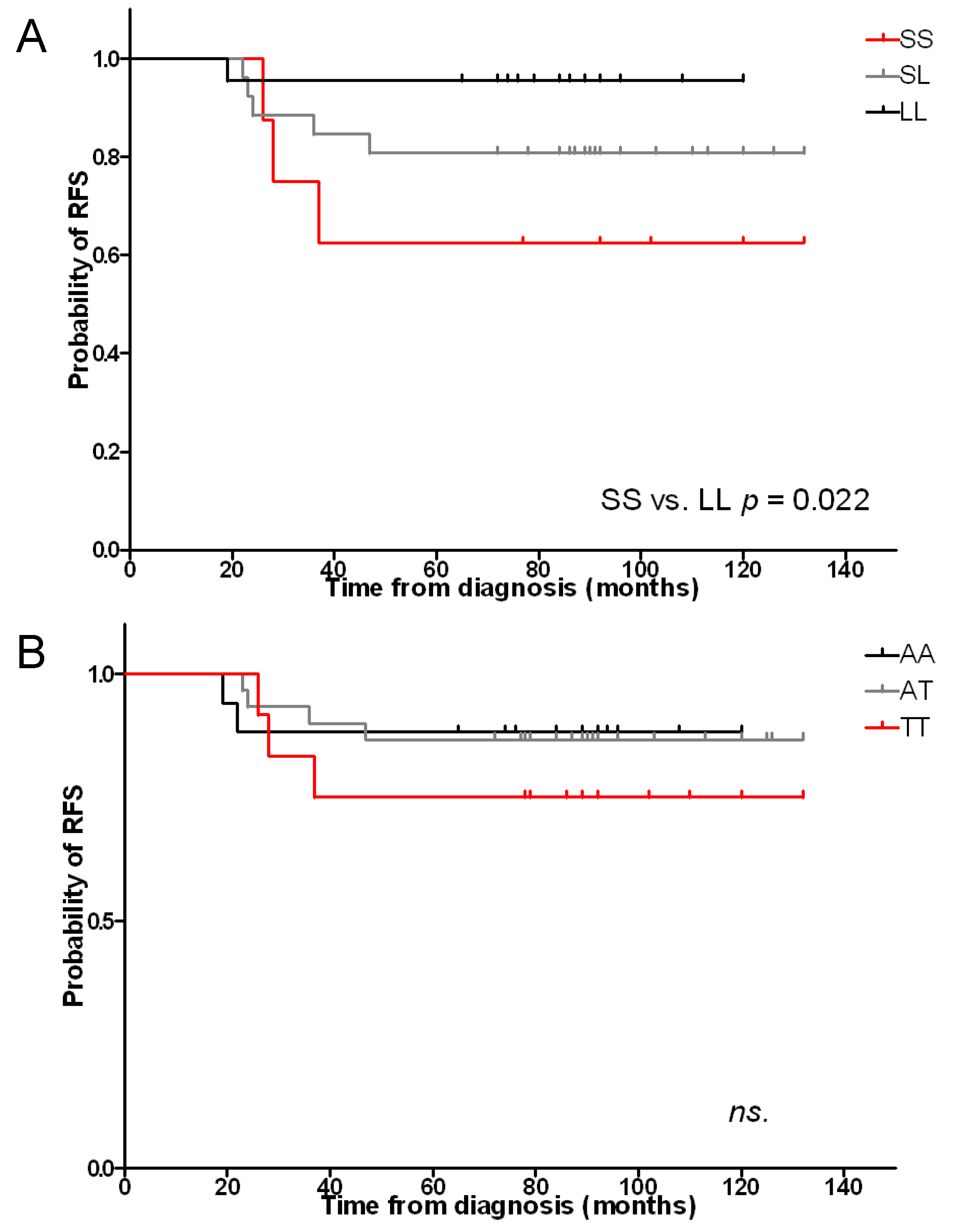
2.1. Length and A(−413)T SNP Polymorphisms and Risk of Relapse
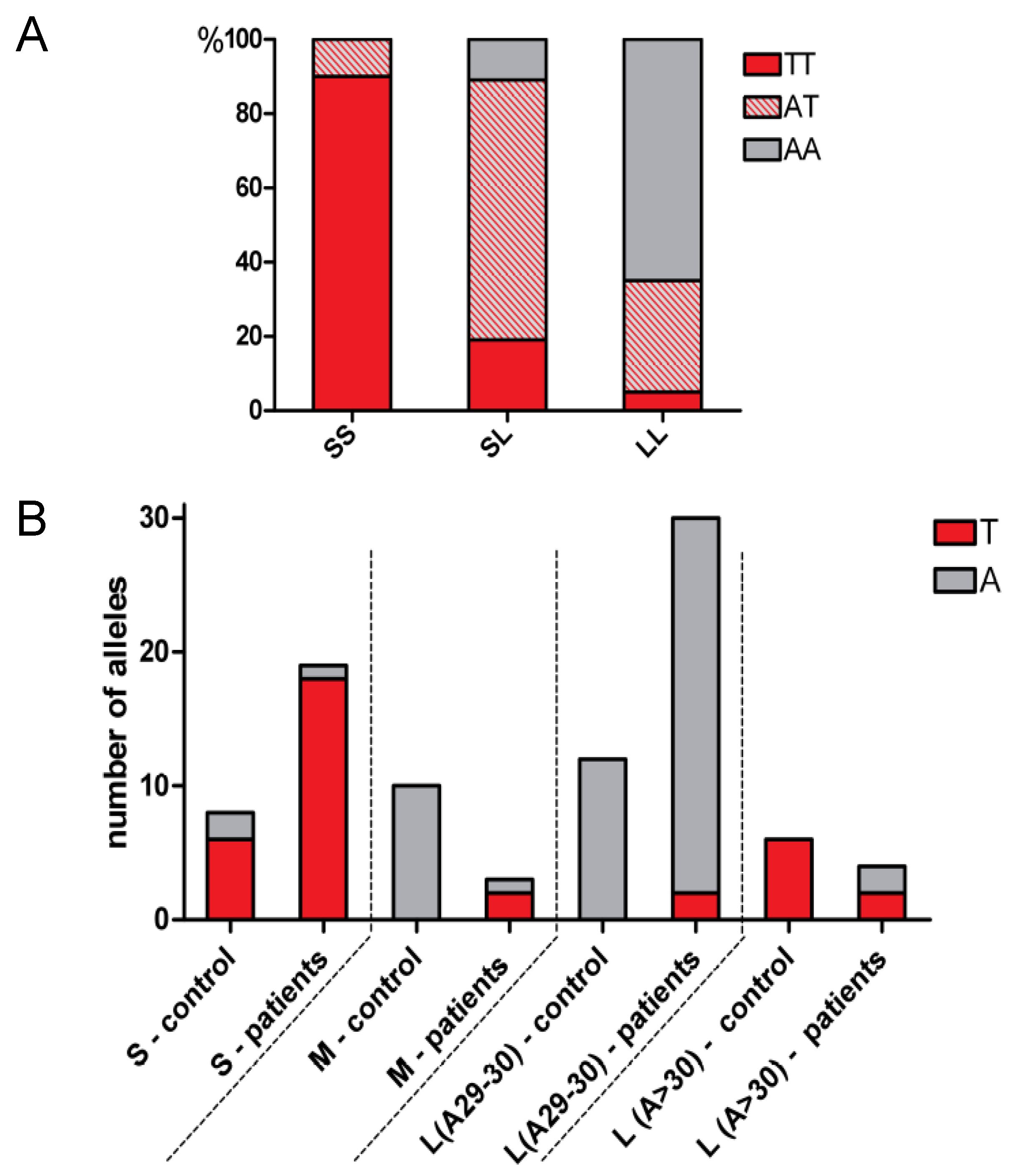
2.2. Co-Segregation of A(−413)T SNP with Length Polymorphisms
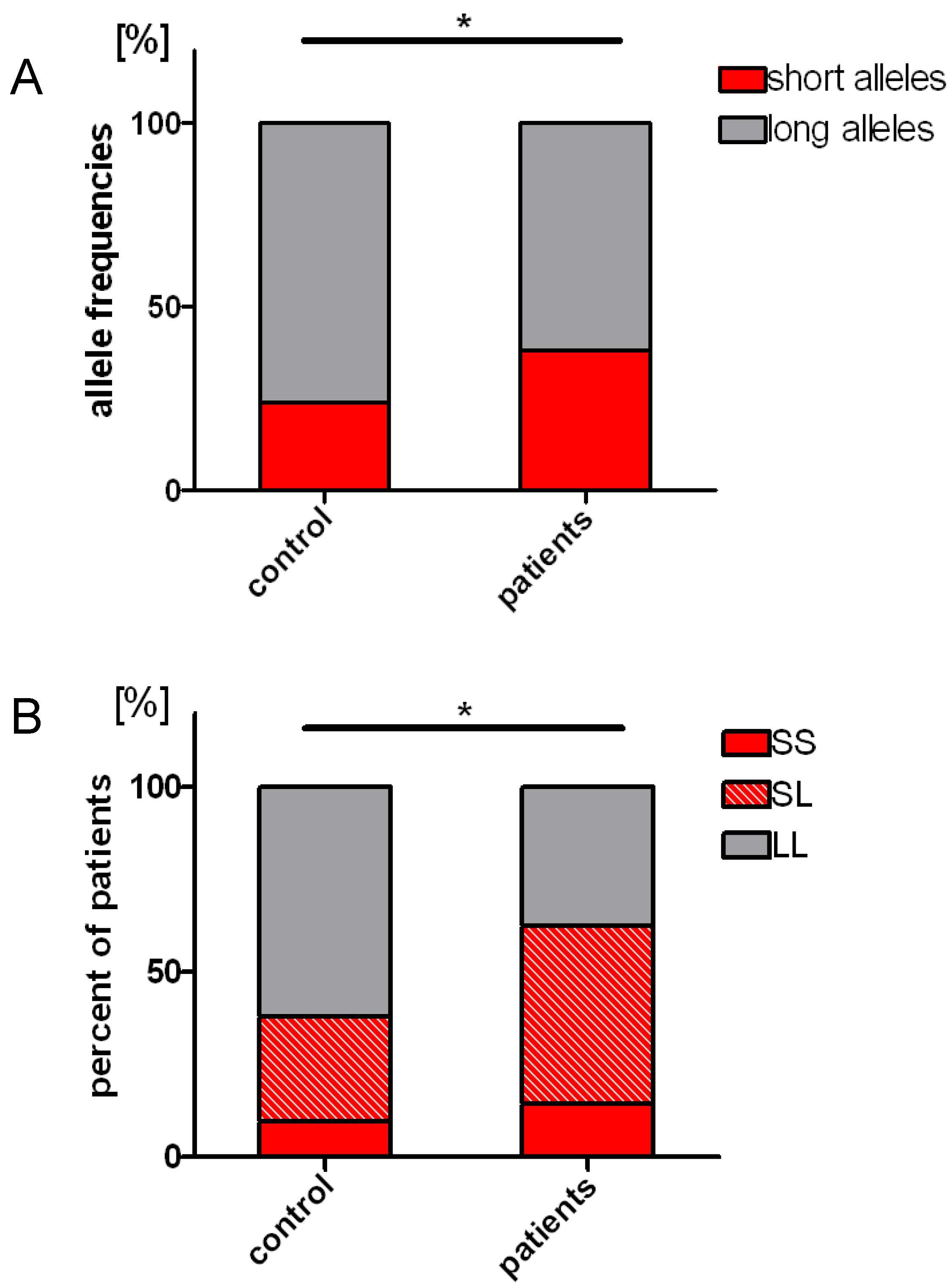
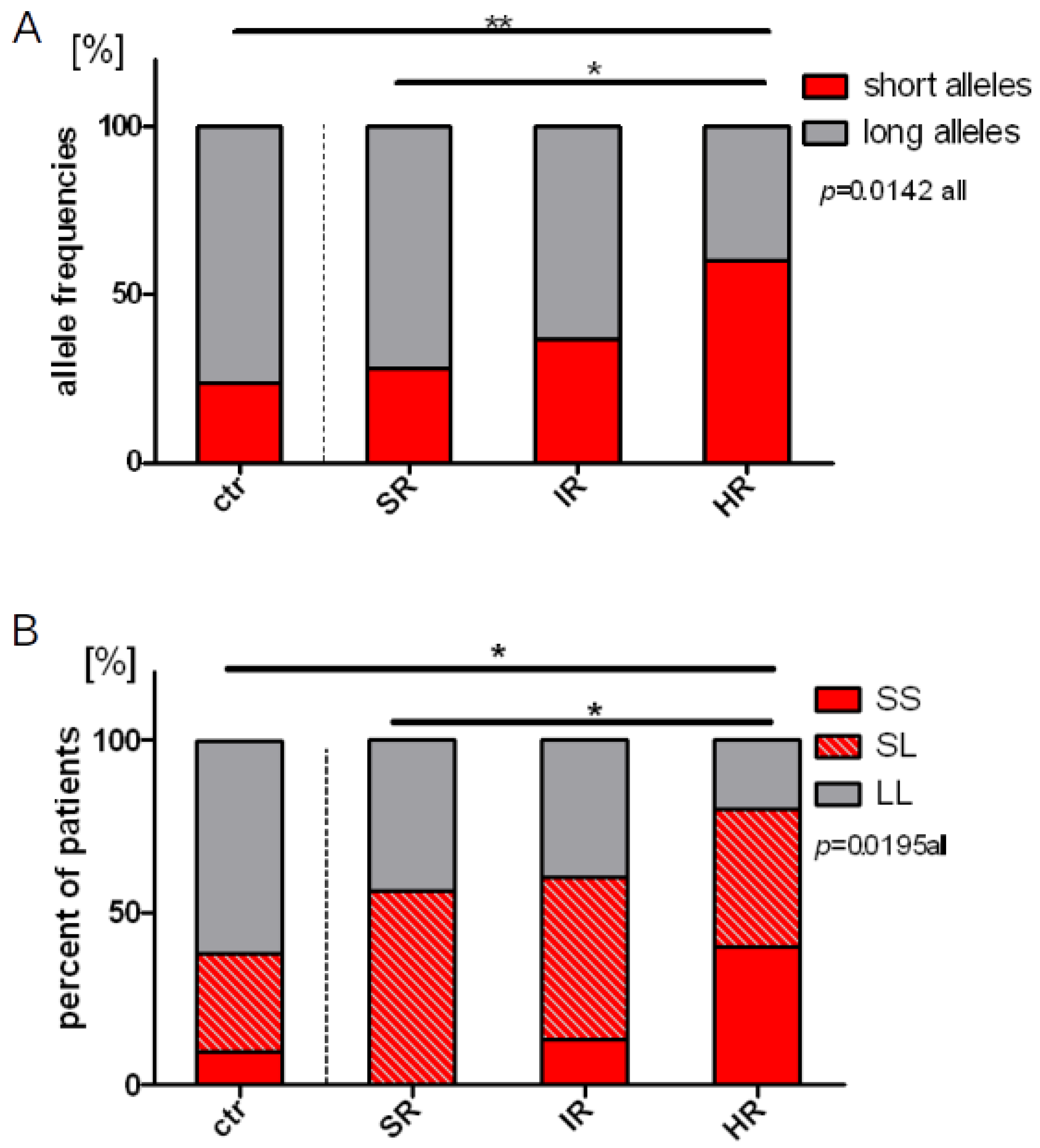
2.3. Distribution of the Length Polymorphisms and Acute Lymphoblastic Leukemia Prevalence
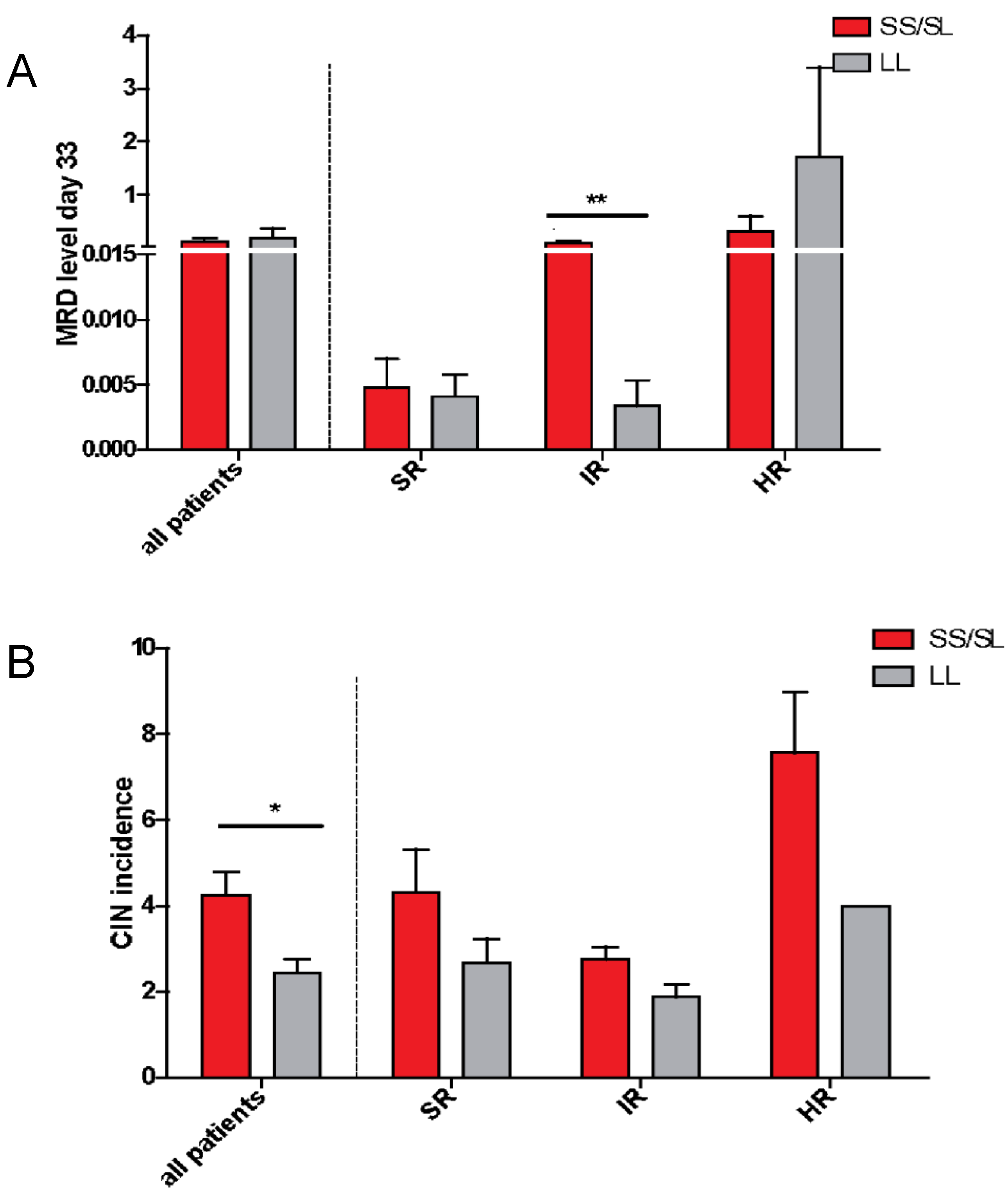
2.4. Length Polymorphisms and Treatment Response
2.5. Chemotherapy-Induced Neutropenia (CIN)
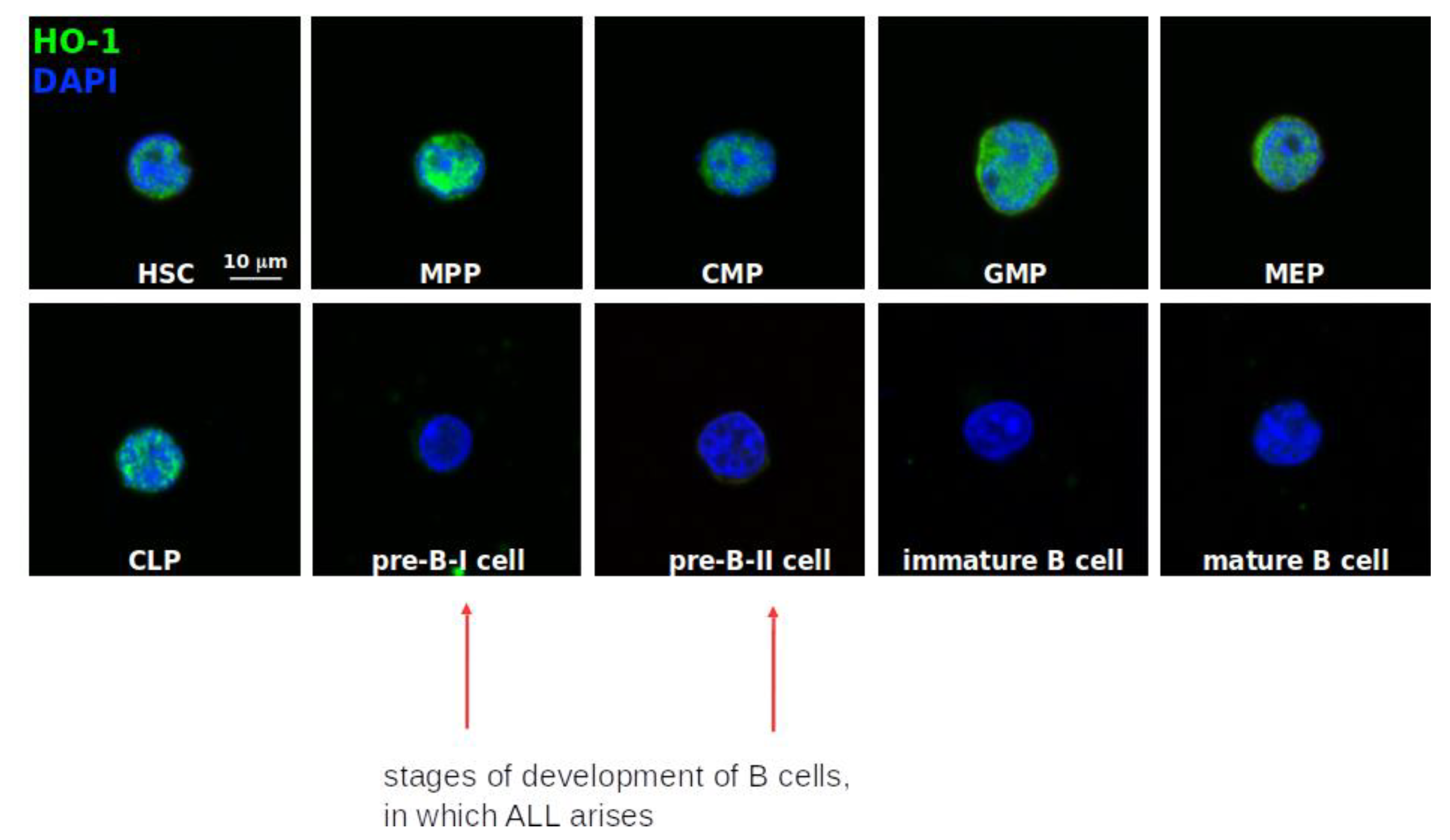
2.6. HO-1 Expression and Its Localization within Normal Human Hematopoietic System
- Hematopoietic stem cells (HSC): Lin−CD34+CD38−CD90+CD45RA−
- Multipotent progenitors (MPP): Lin−CD34+CD38−CD90−CD45RA−
- Common lymphoid progenitors (CLP): Lin−CD34+CD38−CD10+
- Common myeloid progenitors (CMP): Lin−CD34+CD38+CD123+/lowCD45RA−
- Megakaryocyte-erythroid progenitor (MEP): Lin−CD34+CD38+CD123−CD45RA−
- Granulocyte-macrophage progenitor (GMP): Lin−CD34+CD38−CD123+CD45RA+
- Lymphoid maturation steps were established based on:
- -
- preBI: CD19+CD34+CD10+CD20−
- -
- preBII: CD19+CD34−CD10+CD20dim
- -
- Immature B cells: CD19+CD34−CD10+CD20+
- -
- Mature B cells: CD19+CD34−CD10−CD20+
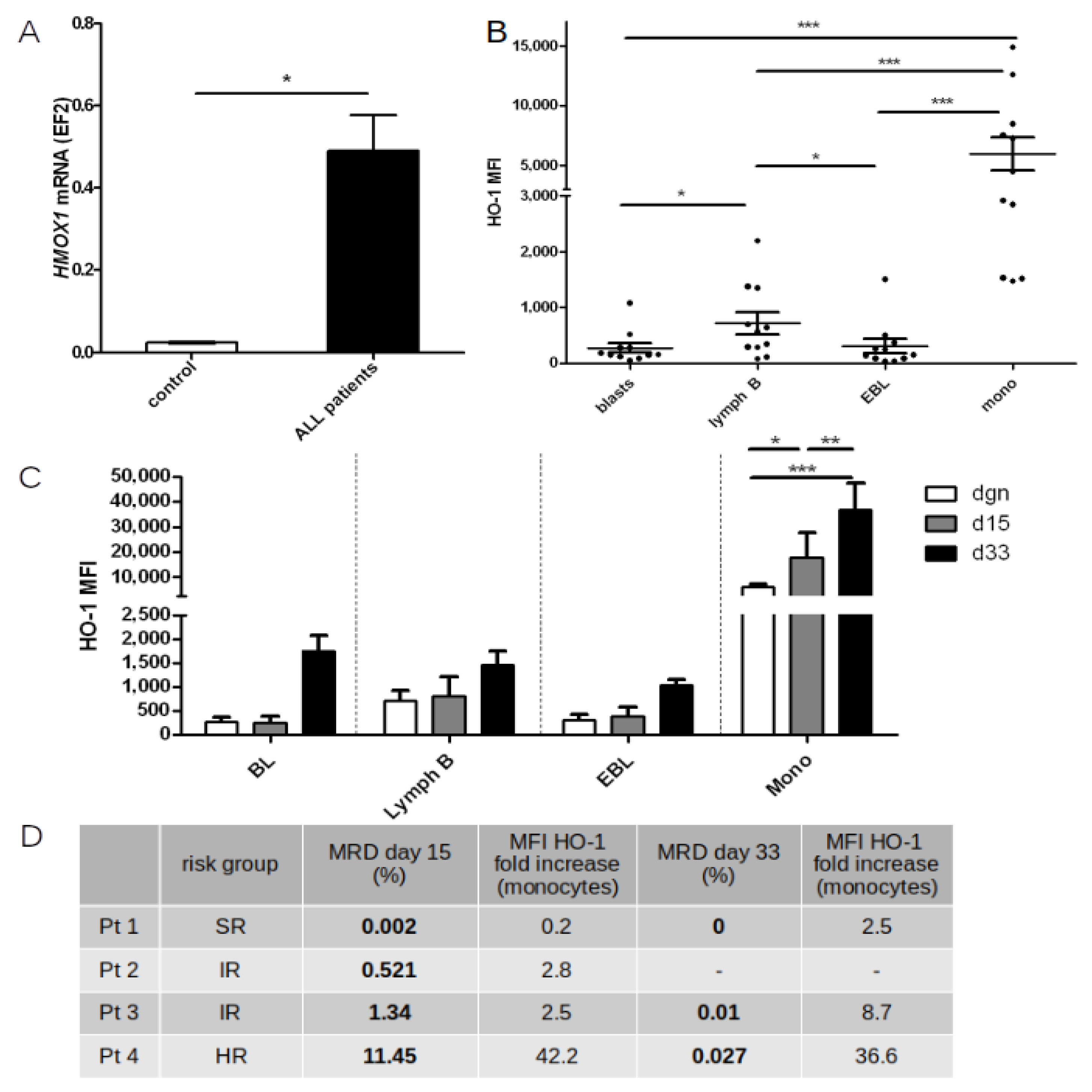
2.7. HO-1 Expression in Peripheral Blood and Bone Marrow Cells before and during Chemotherapy
2.8. Supplementary Information—A-SNP-T Polymorphism
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Patients’ Samples
4.3. DNA Isolation
4.4. Length HMOX1 Gene Promoter Polymorphism
4.5. SNP HMOX1 Gene Promoter Polymorphism
4.6. Cell Sorting, Immunofluorescent Staining and Confocal Analysis
4.7. RNA Isolation, qRT-PCR
4.8. MRD Detection Using Flow Cytometry
4.9. HO-1 Detection Using Flow Cytometry
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [Green Version]
- Campana, D. Minimal residual disease in acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2010, 2010, 7–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curran, E.; Stock, W. How I treat acute lymphoblastic leukemia in older adolescents and young adults. Blood 2015, 125, 3702–3710. [Google Scholar] [CrossRef] [PubMed]
- Flohr, T.; Schrauder, A.; Cazzaniga, G.; Panzer-Grumayer, R.; van der Velden, V.; Fischer, S.; Stanulla, M.; Basso, G.; Niggli, F.K.; Schafer, B.W.; et al. Minimal residual disease-directed risk stratification using real-time quantitative PCR analysis of immunoglobulin and T-cell receptor gene rearrangements in the international multicenter trial AIEOP-BFM ALL 2000 for childhood acute lymphoblastic leukemia. Leukemia 2008, 22, 771–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pui, C.H.; Yang, J.J.; Hunger, S.P.; Pieters, R.; Schrappe, M.; Biondi, A.; Vora, A.; Baruchel, A.; Silverman, L.B.; Schmiegelow, K.; et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J. Clin. Oncol. 2015, 33, 2938–2948. [Google Scholar] [CrossRef]
- Tasian, S.K.; Hunger, S.P. Genomic characterization of paediatric acute lymphoblastic leukaemia: An opportunity for precision medicine therapeutics. Br. J. Haematol. 2017, 176, 867–882. [Google Scholar] [CrossRef]
- Chessells, J.M.; Veys, P.; Kempski, H.; Henley, P.; Leiper, A.; Webb, D.; Hann, I.M. Long-term follow-up of relapsed childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2003, 123, 396–405. [Google Scholar] [CrossRef]
- Pui, C.H.; Evans, W.E. Treatment of acute lymphoblastic leukemia. N. Engl. J. Med. 2006, 354, 166–178. [Google Scholar] [CrossRef]
- Nguyen, K.; Devidas, M.; Cheng, S.C.; La, M.; Raetz, E.A.; Carroll, W.L.; Winick, N.J.; Hunger, S.P.; Gaynon, P.S.; Loh, M.L. Factors influencing survival after relapse from acute lymphoblastic leukemia: A Children’s Oncology Group study. Leukemia 2008, 22, 2142–2150. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Umerez, M.; Garcia-Obregon, S.; Martin-Guerrero, I.; Astigarraga, I.; Gutierrez-Camino, A.; Garcia-Orad, A. Role of miRNAs in treatment response and toxicity of childhood acute lymphoblastic leukemia. Pharmacogenomics 2018, 19, 361–373. [Google Scholar] [CrossRef]
- Lyman, G.H. Chemotherapy dose intensity and quality cancer care. Oncology 2006, 20, 16–25. [Google Scholar] [PubMed]
- Beveridge, R.A.; Miller, J.A.; Kales, A.N.; Binder, R.A.; Robert, N.J.; Harvey, J.H.; Windsor, K.; Gore, I.; Cantrell, J.; Thompson, K.A.; et al. A comparison of efficacy of sargramostim (yeast-derived RhuGM-CSF) and filgrastim (bacteria-derived RhuG-CSF) in the therapeutic setting of chemotherapy-induced myelosuppression. Cancer Investig. 1998, 16, 366–373. [Google Scholar] [CrossRef]
- Hassan, B.A.; Yusoff, Z.B.; Othman, S.B. Filgrastim and antibiotics treatment reduces neutropenia severity in solid cancer patients. Asian Pac. J. Cancer Prev. 2009, 10, 641–644. [Google Scholar] [PubMed]
- Lyman, G.H.; Lyman, C.H.; Agboola, O. Risk models for predicting chemotherapy-induced neutropenia. Oncology 2005, 10, 427–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Slot, A.J.; Zuurmond, A.M.; Bardoel, A.F.; Wijmenga, C.; Pruijs, H.E.; Sillence, D.O.; Brinckmann, J.; Abraham, D.J.; Black, C.M.; Verzijl, N.; et al. Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J. Biol. Chem. 2003, 278, 40967–40972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innocenti, R.; Rigacci, L.; Restelli, U.; Scappini, B.; Gianfaldoni, G.; Fanci, R.; Mannelli, F.; Scolari, F.; Croce, D.; Bonizzoni, E.; et al. Lenograstim and filgrastim in the febrile neutropenia prophylaxis of hospitalized patients: Efficacy and cost of the prophylaxis in a retrospective survey. J. Blood Med. 2019, 10, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Low, S.K.; Chung, S.; Takahashi, A.; Zembutsu, H.; Mushiroda, T.; Kubo, M.; Nakamura, Y. Genome-wide association study of chemotherapeutic agent-induced severe neutropenia/leucopenia for patients in Biobank Japan. Cancer Sci. 2013, 104, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Gatti, D.M.; Weber, S.N.; Goodwin, N.C.; Lammert, F.; Churchill, G.A. Genetic background influences susceptibility to chemotherapy-induced hematotoxicity. Pharm. J. 2018, 18, 319–330. [Google Scholar] [CrossRef]
- Fu, J.; Si, L.; Zhuang, Y.; Zhang, A.; Sun, N.; Li, D.; Hao, B.; Ju, X. Wnt/betacatenin inhibition reverses multidrug resistance in pediatric acute lymphoblastic leukemia. Oncol. Rep. 2019, 41, 1387–1394. [Google Scholar] [CrossRef] [Green Version]
- Simizu, S.; Takada, M.; Umezawa, K.; Imoto, M. Requirement of caspase-3(-like) protease-mediated hydrogen peroxide production for apoptosis induced by various anticancer drugs. J. Biol. Chem. 1998, 273, 26900–26907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsura, T.; Kai, M.; Fujii, Y.; Ito, H.; Yamada, K. Hydrogen peroxide-induced apoptosis in HL-60 cells requires caspase-3 activation. Free Radic. Res. 1999, 30, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, H.; Ito, Y.; Naganawa, T.; Banno, Y.; Nakashima, S.; Yoshimura, S.; Sawada, M.; Nishimura, Y.; Nozawa, Y.; Sakai, N. Activation of caspase-9 and -3 during H2O2-induced apoptosis of PC12 cells independent of ceramide formation. Neurol. Res. 2000, 22, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.G.; Xia, H.L.; Just, T.; Dai, Y.R. Hydroxyl radical-induced apoptosis in human tumor cells is associated with telomere shortening but not telomerase inhibition and caspase activation. FEBS Lett. 2001, 488, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Skonieczna, M.; Hejmo, T.; Poterala-Hejmo, A.; Cieslar-Pobuda, A.; Buldak, R.J. NADPH Oxidases: Insights into Selected Functions and Mechanisms of Action in Cancer and Stem Cells. Oxidative Med. Cell. Longev. 2017, 2017, 9420539. [Google Scholar] [CrossRef]
- Berberat, P.O.; Dambrauskas, Z.; Gulbinas, A.; Giese, T.; Giese, N.; Kunzli, B.; Autschbach, F.; Meuer, S.; Buchler, M.W.; Friess, H. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin. Cancer Res. 2005, 11, 3790–3798. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wang, J.; Fang, Q.; Gao, R.; Shi, Q.; Zhang, H.; Zhao, J. Upregulated heme oxygenase-1 expression of mouse mesenchymal stem cells resists to chemotherapy-induced bone marrow suppression. Chin. Med. J. 2014, 127, 1310–1316. [Google Scholar]
- Yang, Y.C.; Huang, Y.T.; Hsieh, C.W.; Yang, P.M.; Wung, B.S. Carbon monoxide induces heme oxygenase-1 to modulate STAT3 activation in endothelial cells via S-glutathionylation. PLoS ONE 2014, 9, e100677. [Google Scholar] [CrossRef]
- Zhe, N.; Wang, J.; Chen, S.; Lin, X.; Chai, Q.; Zhang, Y.; Zhao, J.; Fang, Q. Heme oxygenase-1 plays a crucial role in chemoresistance in acute myeloid leukemia. Hematology 2015, 20, 384–391. [Google Scholar] [CrossRef]
- Kuwano, A.; Ikeda, H.; Takeda, K.; Nakai, H.; Kondo, I.; Shibahara, S. Mapping of the human gene for inducible heme oxygenase to chromosome 22q12. Tohoku J. Exp. Med. 1994, 172, 389–392. [Google Scholar] [CrossRef] [Green Version]
- Grochot-Przeczek, A.; Dulak, J.; Jozkowicz, A. Haem oxygenase-1: Non-canonical roles in physiology and pathology. Clin. Sci. 2012, 122, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshane, J.; Chen, S.; Caballero, S.; Grochot-Przeczek, A.; Was, H.; Li Calzi, S.; Lach, R.; Hock, T.D.; Chen, B.; Hill-Kapturczak, N.; et al. Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J. Exp. Med. 2007, 204, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Jozkowicz, A.; Was, H.; Dulak, J. Heme oxygenase-1 in tumors: Is it a false friend? Antioxid. Redox Signal. 2007, 9, 2099–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Was, H.; Sokolowska, M.; Sierpniowska, A.; Dominik, P.; Skrzypek, K.; Lackowska, B.; Pratnicki, A.; Grochot-Przeczek, A.; Taha, H.; Kotlinowski, J.; et al. Effects of heme oxygenase-1 on induction and development of chemically induced squamous cell carcinoma in mice. Free Radic. Biol. Med. 2011, 51, 1717–1726. [Google Scholar] [CrossRef] [Green Version]
- Na, H.K.; Surh, Y.J. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Radic. Biol. Med. 2014, 67, 353–365. [Google Scholar] [CrossRef]
- Nowis, D.; Legat, M.; Grzela, T.; Niderla, J.; Wilczek, E.; Wilczynski, G.M.; Glodkowska, E.; Mrowka, P.; Issat, T.; Dulak, J.; et al. Heme oxygenase-1 protects tumor cells against photodynamic therapy-mediated cytotoxicity. Oncogene 2006, 25, 3365–3374. [Google Scholar] [CrossRef] [Green Version]
- Furchgott, R.F.; Jothianandan, D. Endothelium-dependent and -independent vasodilation involving cyclic GMP: Relaxation induced by nitric oxide, carbon monoxide and light. Blood Vessel. 1991, 28, 52–61. [Google Scholar] [CrossRef]
- Morita, T.; Kourembanas, S. Endothelial cell expression of vasoconstrictors and growth factors is regulated by smooth muscle cell-derived carbon monoxide. J. Clin. Investig. 1995, 96, 2676–2682. [Google Scholar] [CrossRef]
- Morita, T.; Mitsialis, S.A.; Koike, H.; Liu, Y.; Kourembanas, S. Carbon monoxide controls the proliferation of hypoxic vascular smooth muscle cells. J. Biol. Chem. 1997, 272, 32804–32809. [Google Scholar] [CrossRef] [Green Version]
- Morita, T.; Perrella, M.A.; Lee, M.E.; Kourembanas, S. Smooth muscle cell-derived carbon monoxide is a regulator of vascular cGMP. Proc. Natl. Acad. Sci. USA 1995, 92, 1475–1479. [Google Scholar] [CrossRef] [Green Version]
- Durante, W.; Schafer, A.I. Carbon monoxide and vascular cell function (review). Int. J. Mol. Med. 1998, 2, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Otterbein, L.E.; Morse, D.; Choi, A.M. Heme oxygenase/carbon monoxide signaling pathways: Regulation and functional significance. Mol. Cell. Biochem. 2002, 234-235, 249–263. [Google Scholar] [CrossRef]
- Bilban, M.; Haschemi, A.; Wegiel, B.; Chin, B.Y.; Wagner, O.; Otterbein, L.E. Heme oxygenase and carbon monoxide initiate homeostatic signaling. J. Mol. Med. 2008, 86, 267–279. [Google Scholar] [CrossRef]
- Kozakowska, M.; Szade, K.; Dulak, J.; Jozkowicz, A. Role of heme oxygenase-1 in postnatal differentiation of stem cells: A possible cross-talk with microRNAs. Antioxid. Redox Signal. 2014, 20, 1827–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukowska-Strakova, K.; Ciesla, M.; Szade, K.; Nowak, W.N.; Straka, R.; Szade, A.; Tyszka-Czochara, M.; Najder, K.; Konturek, A.; Siedlar, M.; et al. Heme oxygenase 1 affects granulopoiesis in mice through control of myelocyte proliferation. Immunobiology 2017, 222, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Exner, M.; Minar, E.; Wagner, O.; Schillinger, M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radic. Biol. Med. 2004, 37, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Yamaya, M.; Okinaga, S.; Nakayama, K.; Sekizawa, K.; Shibahara, S.; Sasaki, H. Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to emphysema. Am. J. Hum. Genet. 2000, 66, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, M.; Wong, R.J.; Stevenson, D.K. Heme oxygenase-1 promoter polymorphisms: Do they modulate neonatal hyperbilirubinemia? J. Perinatol. Off. J. Calif. Perinat. Assoc. 2017, 37, 901–905. [Google Scholar] [CrossRef]
- Taha, H.; Skrzypek, K.; Guevara, I.; Nigisch, A.; Mustafa, S.; Grochot-Przeczek, A.; Ferdek, P.; Was, H.; Kotlinowski, J.; Kozakowska, M.; et al. Role of heme oxygenase-1 in human endothelial cells: Lesson from the promoter allelic variants. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1634–1641. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Lin, S.J.; Lin, M.W.; Tsai, H.L.; Kuo, S.S.; Chen, J.W.; Charng, M.J.; Wu, T.C.; Chen, L.C.; Ding, Y.A.; et al. Microsatellite polymorphism in promoter of heme oxygenase-1 gene is associated with susceptibility to coronary artery disease in type 2 diabetic patients. Hum. Genet. 2002, 111, 1–8. [Google Scholar] [CrossRef]
- Hirai, H.; Kubo, H.; Yamaya, M.; Nakayama, K.; Numasaki, M.; Kobayashi, S.; Suzuki, S.; Shibahara, S.; Sasaki, H. Microsatellite polymorphism in heme oxygenase-1 gene promoter is associated with susceptibility to oxidant-induced apoptosis in lymphoblastoid cell lines. Blood 2003, 102, 1619–1621. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, A.; Yamaya, M.; Suzuki, S.; Yasuda, H.; Kubo, H.; Nakayama, K.; Handa, M.; Sasaki, T.; Shibahara, S.; Sekizawa, K.; et al. Association of susceptibility to the development of lung adenocarcinoma with the heme oxygenase-1 gene promoter polymorphism. Hum. Genet. 2005, 116, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Andersen, V.; Christensen, J.; Overvad, K.; Tjonneland, A.; Vogel, U. Heme oxygenase-1 polymorphism is not associated with risk of colorectal cancer: A Danish prospective study. Eur. J. Gastroenterol. Hepatol. 2011, 23, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Murakami, A.; Fujimori, Y.; Yoshikawa, Y.; Yamada, S.; Tamura, K.; Hirayama, N.; Terada, T.; Kuribayashi, K.; Tabata, C.; Fukuoka, K.; et al. Heme oxygenase-1 promoter polymorphism is associated with risk of malignant mesothelioma. Lung 2012, 190, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Daenen, K.E.; Martens, P.; Bammens, B. Association of HO-1 (GT)n Promoter Polymorphism and Cardiovascular Disease: A Reanalysis of the Literature. Can. J. Cardiol. 2016, 32, 160–168. [Google Scholar] [CrossRef]
- Wang, R.; Shen, J.; Yang, R.; Wang, W.G.; Yuan, Y.; Guo, Z.H. Association between heme oxygenase-1 gene promoter polymorphisms and cancer susceptibility: A meta-analysis. Biomed. Rep. 2018, 8, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.W.; Lee, T.C.; Yeh, W.I.; Chung, M.Y.; Liu, C.J.; Chi, L.Y.; Lin, S.C. Polymorphism in heme oxygenase-1 (HO-1) promoter is related to the risk of oral squamous cell carcinoma occurring on male areca chewers. Br. J. Cancer 2004, 91, 1551–1555. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.C.; Ambrosone, C.B.; Ahn, J.; Choi, J.Y.; McCullough, M.L.; Stevens, V.L.; Rodriguez, C.; Thun, M.J.; Calle, E.E. Genetic variability in iron-related oxidative stress pathways (Nrf2, NQ01, NOS3, and HO-1), iron intake, and risk of postmenopausal breast cancer. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1784–1794. [Google Scholar] [CrossRef] [Green Version]
- Lo, S.S.; Lin, S.C.; Wu, C.W.; Chen, J.H.; Yeh, W.I.; Chung, M.Y.; Lui, W.Y. Heme oxygenase-1 gene promoter polymorphism is associated with risk of gastric adenocarcinoma and lymphovascular tumor invasion. Ann. Surg. Oncol. 2007, 14, 2250–2256. [Google Scholar] [CrossRef]
- Hu, J.L.; Li, Z.Y.; Liu, W.; Zhang, R.G.; Li, G.L.; Wang, T.; Ren, J.H.; Wu, G. Polymorphism in heme oxygenase-1 (HO-1) promoter and alcohol are related to the risk of esophageal squamous cell carcinoma on Chinese males. Neoplasma 2010, 57, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, I.; Krogler, J.; Endler, G.; Kaufmann, S.; Mustafa, S.; Exner, M.; Mannhalter, C.; Wagner, O.; Pehamberger, H. A microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with risk for melanoma. Int. J. Cancer 2006, 119, 1312–1315. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Goto, Y.; Takagi, S.; Baba, S.; Tago, N.; Nonogi, H.; Iwai, N. A promoter variant of the heme oxygenase-1 gene may reduce the incidence of ischemic heart disease in Japanese. Atherosclerosis 2004, 173, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Buis, C.I.; van der Steege, G.; Visser, D.S.; Nolte, I.M.; Hepkema, B.G.; Nijsten, M.; Slooff, M.J.; Porte, R.J. Heme oxygenase-1 genotype of the donor is associated with graft survival after liver transplantation. Am. J. Transplant. 2008, 8, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Shao, Y.; Yao, N.; Chen, X.; Hu, L.; He, T. Association of heme oxygenase-1 polymorphisms with cancer risk: A systematic review and meta-analysis. J. Balk. Union Oncol. 2015, 20, 1142–1153. [Google Scholar]
- Bean, C.J.; Boulet, S.L.; Ellingsen, D.; Pyle, M.E.; Barron-Casella, E.A.; Casella, J.F.; Payne, A.B.; Driggers, J.; Trau, H.A.; Yang, G.; et al. Heme oxygenase-1 gene promoter polymorphism is associated with reduced incidence of acute chest syndrome among children with sickle cell disease. Blood 2012, 120, 3822–3828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.Y.; Guan, J.; Li, H.; Zhou, Z.Q.; Zhou, G.W. Heme Oxygenase-1 Promoter Polymorphism Protects Liver Allograft. Indian J. Surg. 2016, 78, 14–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueda, B.; Oliver, J.; Robledo, G.; Lopez-Nevot, M.A.; Balsa, A.; Pascual-Salcedo, D.; Gonzalez-Gay, M.A.; Gonzalez-Escribano, M.F.; Martin, J. HO-1 promoter polymorphism associated with rheumatoid arthritis. Arthritis Rheum. 2007, 56, 3953–3958. [Google Scholar] [CrossRef]
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef] [Green Version]
- Vanella, L.; Barbagallo, I.; Tibullo, D.; Forte, S.; Zappala, A.; Li Volti, G. The non-canonical functions of the heme oxygenases. Oncotarget 2016, 7, 69075–69086. [Google Scholar] [CrossRef] [Green Version]
- Good, Z.; Sarno, J.; Jager, A.; Samusik, N.; Aghaeepour, N.; Simonds, E.F.; White, L.; Lacayo, N.J.; Fantl, W.J.; Fazio, G.; et al. Single-cell developmental classification of B cell precursor acute lymphoblastic leukemia at diagnosis reveals predictors of relapse. Nat. Med. 2018, 24, 474–483. [Google Scholar] [CrossRef]
- Swaminathan, S.; Duy, C.; Muschen, M. BACH2-BCL6 balance regulates selection at the pre-B cell receptor checkpoint. Trends Immunol. 2014, 35, 131–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seita, J.; Sahoo, D.; Rossi, D.J.; Bhattacharya, D.; Serwold, T.; Inlay, M.A.; Ehrlich, L.I.; Fathman, J.W.; Dill, D.L.; Weissman, I.L. Gene Expression Commons: An open platform for absolute gene expression profiling. PLoS ONE 2012, 7, e40321. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. HO-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. Heme oxygenase: Function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988, 2, 2557–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, J.; Cook, J.L. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am. J. Respir. Cell Mol. Biol. 2007, 36, 166–174. [Google Scholar] [CrossRef]
- Zhang, M.M.; Zheng, Y.Y.; Gao, Y.; Zhang, J.Z.; Liu, F.; Yang, Y.N.; Li, X.M.; Ma, Y.T.; Xie, X. Heme oxygenase-1 gene promoter polymorphisms are associated with coronary heart disease and restenosis after percutaneous coronary intervention: A meta-analysis. Oncotarget 2016, 7, 83437–83450. [Google Scholar] [CrossRef] [Green Version]
- Sawa, T.; Mounawar, M.; Tatemichi, M.; Gilibert, I.; Katoh, T.; Ohshima, H. Increased risk of gastric cancer in Japanese subjects is associated with microsatellite polymorphisms in the heme oxygenase-1 and the inducible nitric oxide synthase gene promoters. Cancer Lett. 2008, 269, 78–84. [Google Scholar] [CrossRef]
- Vashist, Y.K.; Uzungolu, G.; Kutup, A.; Gebauer, F.; Koenig, A.; Deutsch, L.; Zehler, O.; Busch, P.; Kalinin, V.; Izbicki, J.R.; et al. Heme oxygenase-1 germ line GTn promoter polymorphism is an independent prognosticator of tumor recurrence and survival in pancreatic cancer. J. Surg. Oncol. 2011, 104, 305–311. [Google Scholar] [CrossRef]
- Chau, L.Y. Heme oxygenase-1: Emerging target of cancer therapy. J. Biomed. Sci. 2015, 22, 22. [Google Scholar] [CrossRef] [Green Version]
- Ciesla, M.; Marona, P.; Kozakowska, M.; Jez, M.; Seczynska, M.; Loboda, A.; Bukowska-Strakova, K.; Szade, A.; Walawender, M.; Kusior, M.; et al. Heme Oxygenase-1 Controls an HDAC4-miR-206 Pathway of Oxidative Stress in Rhabdomyosarcoma. Cancer Res. 2016, 76, 5707–5718. [Google Scholar] [CrossRef] [Green Version]
- Szade, K.; Zukowska, M.; Szade, A.; Nowak, W.; Skulimowska, I.; Ciesla, M.; Bukowska-Strakova, K.; Gulati, G.S.; Kachamakova-Trojanowska, N.; Kusienicka, A.; et al. Heme oxygenase-1 deficiency triggers exhaustion of hematopoietic stem cells. EMBO Rep. 2020, 21, e47895. [Google Scholar] [CrossRef] [PubMed]
- Kapturczak, M.H.; Wasserfall, C.; Brusko, T.; Campbell-Thompson, M.; Ellis, T.M.; Atkinson, M.A.; Agarwal, A. Heme oxygenase-1 modulates early inflammatory responses: Evidence from the heme oxygenase-1-deficient mouse. Am. J. Pathol. 2004, 165, 1045–1053. [Google Scholar] [CrossRef]
- Swaminathan, S.; Huang, C.; Geng, H.; Chen, Z.; Harvey, R.; Kang, H.; Ng, C.; Titz, B.; Hurtz, C.; Sadiyah, M.F.; et al. BACH2 mediates negative selection and p53-dependent tumor suppression at the pre-B cell receptor checkpoint. Nat. Med. 2013, 19, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Watanabe-Matsui, M.; Muto, A.; Matsui, T.; Itoh-Nakadai, A.; Nakajima, O.; Murayama, K.; Yamamoto, M.; Ikeda-Saito, M.; Igarashi, K. Heme regulates B-cell differentiation, antibody class switch, and heme oxygenase-1 expression in B cells as a ligand of Bach2. Blood 2011, 117, 5438–5448. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Li, C.; Wu, Q.; An, P.; Huang, L.; Wang, J.; Chen, C.; Chen, X.; Zhang, F.; Ma, L.; et al. Iron-dependent histone 3 lysine 9 demethylation controls B cell proliferation and humoral immune responses. Nat. Commun. 2019, 10, 2935. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, D.B.; Anselmino, N.; Brandani, J.N.; Jaworski, F.M.; Paez, A.V.; Mazaira, G.; Meiss, R.P.; Nunez, M.; Nemirovsky, S.I.; Giudice, J.; et al. Heme Oxygenase 1 Impairs Glucocorticoid Receptor Activity in Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 1006. [Google Scholar] [CrossRef] [Green Version]
- Inaba, H.; Pui, C.H. Glucocorticoid use in acute lymphoblastic leukaemia. Lancet. Oncol. 2010, 11, 1096–1106. [Google Scholar] [CrossRef] [Green Version]
- Allen, A.; Vaninov, N.; Li, M.; Nguyen, S.; Singh, M.; Igo, P.; Tilles, A.W.; O’Rourke, B.; Miller, B.L.K.; Parekkadan, B.; et al. Mesenchymal Stromal Cell Bioreactor for Ex Vivo Reprogramming of Human Immune Cells. Sci. Rep. 2020, 10, 10142. [Google Scholar] [CrossRef]
- Goodman, A.I.; Choudhury, M.; da Silva, J.L.; Schwartzman, M.L.; Abraham, N.G. Overexpression of the heme oxygenase gene in renal cell carcinoma. Proc. Soc. Exp. Biol. Med. 1997, 214, 54–61. [Google Scholar] [CrossRef]
- Mayerhofer, M.; Florian, S.; Krauth, M.T.; Aichberger, K.J.; Bilban, M.; Marculescu, R.; Printz, D.; Fritsch, G.; Wagner, O.; Selzer, E.; et al. Identification of heme oxygenase-1 as a novel BCR/ABL-dependent survival factor in chronic myeloid leukemia. Cancer Res. 2004, 64, 3148–3154. [Google Scholar] [CrossRef] [Green Version]
- Was, H.; Cichon, T.; Smolarczyk, R.; Rudnicka, D.; Stopa, M.; Chevalier, C.; Leger, J.J.; Lackowska, B.; Grochot, A.; Bojkowska, K.; et al. Overexpression of heme oxygenase-1 in murine melanoma: Increased proliferation and viability of tumor cells, decreased survival of mice. Am. J. Pathol. 2006, 169, 2181–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degese, M.S.; Mendizabal, J.E.; Gandini, N.A.; Gutkind, J.S.; Molinolo, A.; Hewitt, S.M.; Curino, A.C.; Coso, O.A.; Facchinetti, M.M. Expression of heme oxygenase-1 in non-small cell lung cancer (NSCLC) and its correlation with clinical data. Lung Cancer 2012, 77, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Noh, S.J.; Bae, J.S.; Jamiyandorj, U.; Park, H.S.; Kwon, K.S.; Jung, S.H.; Youn, H.J.; Lee, H.; Park, B.H.; Chung, M.J.; et al. Expression of nerve growth factor and heme oxygenase-1 predict poor survival of breast carcinoma patients. BMC Cancer 2013, 13, 516. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.Y.; Liu, C.L.; Chen, M.J.; Lee, J.J.; Pun, P.C.; Cheng, S.P. Expression of haem oxygenase-1 correlates with tumour aggressiveness and BRAF V600E expression in thyroid cancer. Histopathology 2015, 66, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Nishie, A.; Ono, M.; Shono, T.; Fukushi, J.; Otsubo, M.; Onoue, H.; Ito, Y.; Inamura, T.; Ikezaki, K.; Fukui, M.; et al. Macrophage infiltration and heme oxygenase-1 expression correlate with angiogenesis in human gliomas. Clin. Cancer Res. 1999, 5, 1107–1113. [Google Scholar] [PubMed]
- Torisu-Itakura, H.; Furue, M.; Kuwano, M.; Ono, M. Co-expression of thymidine phosphorylase and heme oxygenase-1 in macrophages in human malignant vertical growth melanomas. Jpn. J. Cancer Res. Gann 2000, 91, 906–910. [Google Scholar] [CrossRef] [PubMed]
- Boschetto, P.; Zeni, E.; Mazzetti, L.; Miotto, D.; Lo Cascio, N.; Maestrelli, P.; Marian, E.; Querzoli, P.; Pedriali, M.; Murer, B.; et al. Decreased heme-oxygenase (HO)-1 in the macrophages of non-small cell lung cancer. Lung Cancer 2008, 59, 192–197. [Google Scholar] [CrossRef]
- Lavrovsky, Y.; Schwartzman, M.L.; Levere, R.D.; Kappas, A.; Abraham, N.G. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc. Natl. Acad. Sci. USA 1994, 91, 5987–5991. [Google Scholar] [CrossRef] [Green Version]
- Chow, A.; Lucas, D.; Hidalgo, A.; Mendez-Ferrer, S.; Hashimoto, D.; Scheiermann, C.; Battista, M.; Leboeuf, M.; Prophete, C.; van Rooijen, N.; et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J. Exp. Med. 2011, 208, 261–271. [Google Scholar] [CrossRef]
- Szade, K.; Gulati, G.S.; Chan, C.K.F.; Kao, K.S.; Miyanishi, M.; Marjon, K.D.; Sinha, R.; George, B.M.; Chen, J.Y.; Weissman, I.L. Where Hematopoietic Stem Cells Live: The Bone Marrow Niche. Antioxid. Redox Signal. 2018, 29, 191–204. [Google Scholar] [CrossRef]
- Cunnington, A.J.; de Souza, J.B.; Walther, M.; Riley, E.M. Malaria impairs resistance to Salmonella through heme- and heme oxygenase-dependent dysfunctional granulocyte mobilization. Nat. Med. 2011, 18, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Crawford, J.; Dale, D.C.; Lyman, G.H. Chemotherapy-induced neutropenia: Risks, consequences, and new directions for its management. Cancer 2004, 100, 228–237. [Google Scholar] [CrossRef]
- Zhang, H.; Nguyen-Jackson, H.; Panopoulos, A.D.; Li, H.S.; Murray, P.J.; Watowich, S.S. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood 2010, 116, 2462–2471. [Google Scholar] [CrossRef] [Green Version]
- Manz, M.G.; Boettcher, S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar] [CrossRef]
- Kawakami, M.; Tsutsumi, H.; Kumakawa, T.; Abe, H.; Hirai, M.; Kurosawa, S.; Mori, M.; Fukushima, M. Levels of serum granulocyte colony-stimulating factor in patients with infections. Blood 1990, 76, 1962–1964. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, H.E.; Maitland, M.L.; Dolan, M.E.; Cox, N.J.; Ratain, M.J. Cancer pharmacogenomics: Strategies and challenges. Nat. Rev. Genet. 2013, 14, 23–34. [Google Scholar] [CrossRef]
- Szade, K.; Bukowska-Strakova, K.; Nowak, W.N.; Szade, A.; Kachamakova-Trojanowska, N.; Zukowska, M.; Jozkowicz, A.; Dulak, J. Murine bone marrow Lin(-)Sca(-)1(+)CD45(-) very small embryonic-like (VSEL) cells are heterogeneous population lacking Oct-4A expression. PLoS ONE 2013, 8, e63329. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bukowska-Strakova, K.; Włodek, J.; Pitera, E.; Kozakowska, M.; Konturek-Cieśla, A.; Cieśla, M.; Gońka, M.; Nowak, W.; Wieczorek, A.; Pawińska-Wąsikowska, K.; et al. Role of HMOX1 Promoter Genetic Variants in Chemoresistance and Chemotherapy Induced Neutropenia in Children with Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2021, 22, 988. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22030988
Bukowska-Strakova K, Włodek J, Pitera E, Kozakowska M, Konturek-Cieśla A, Cieśla M, Gońka M, Nowak W, Wieczorek A, Pawińska-Wąsikowska K, et al. Role of HMOX1 Promoter Genetic Variants in Chemoresistance and Chemotherapy Induced Neutropenia in Children with Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences. 2021; 22(3):988. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22030988
Chicago/Turabian StyleBukowska-Strakova, Karolina, Joanna Włodek, Ewelina Pitera, Magdalena Kozakowska, Anna Konturek-Cieśla, Maciej Cieśla, Monika Gońka, Witold Nowak, Aleksandra Wieczorek, Katarzyna Pawińska-Wąsikowska, and et al. 2021. "Role of HMOX1 Promoter Genetic Variants in Chemoresistance and Chemotherapy Induced Neutropenia in Children with Acute Lymphoblastic Leukemia" International Journal of Molecular Sciences 22, no. 3: 988. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22030988