
Relevance of Notch Signaling for Bone Metabolism and Regeneration

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Bone Remodeling
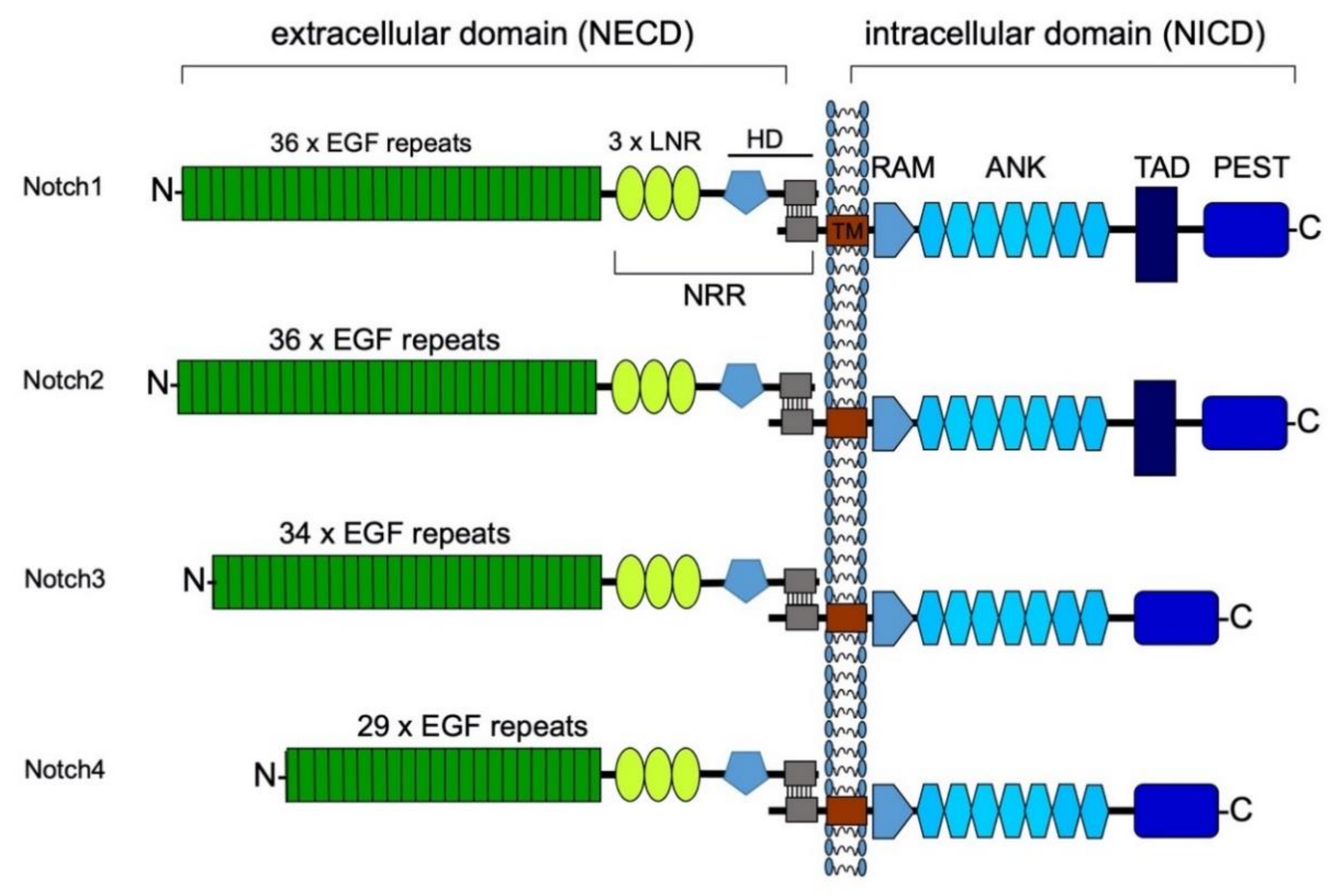
3. Structure of the Notch Receptors and Their Ligands
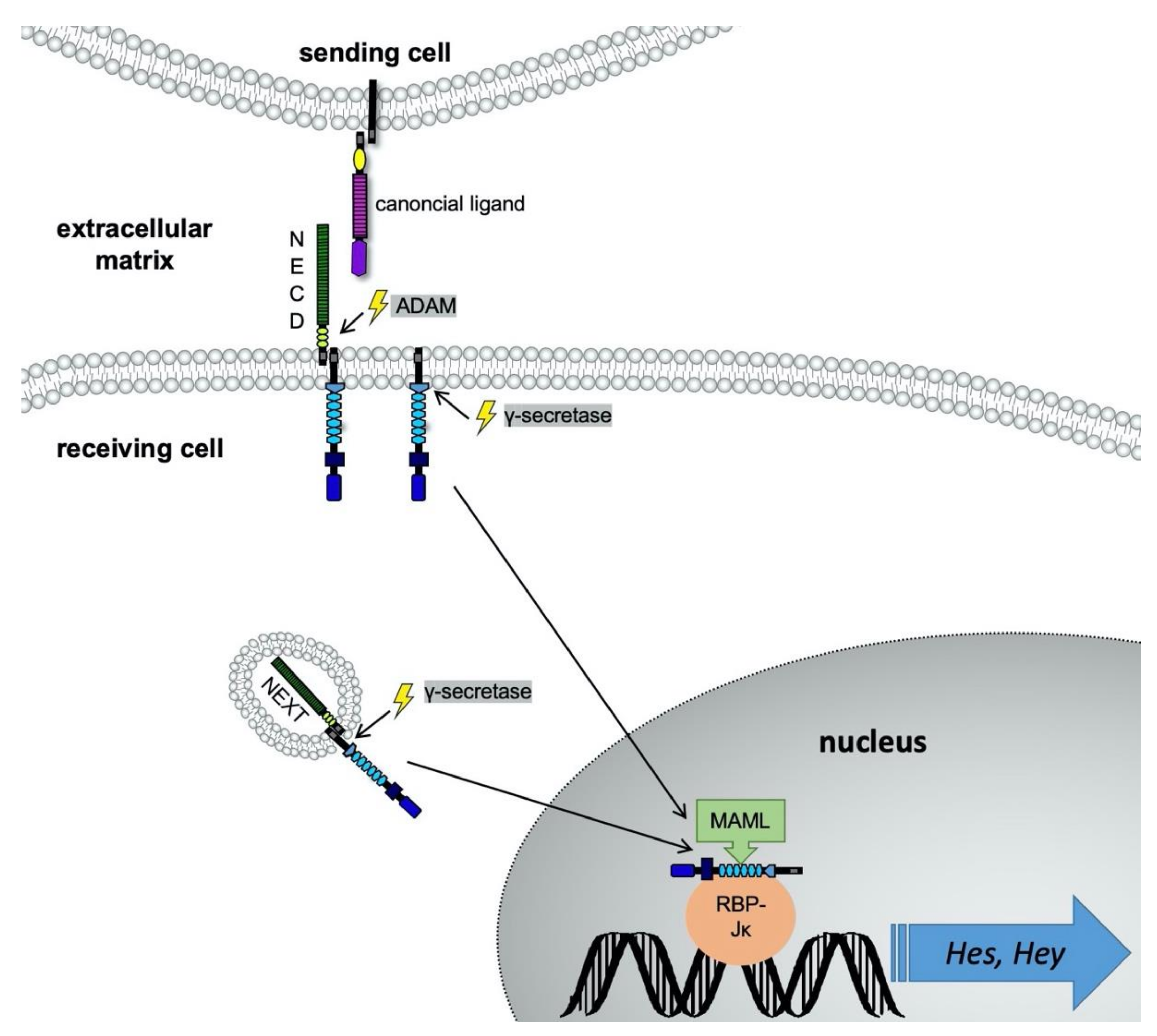
4. Notch Signaling Pathway
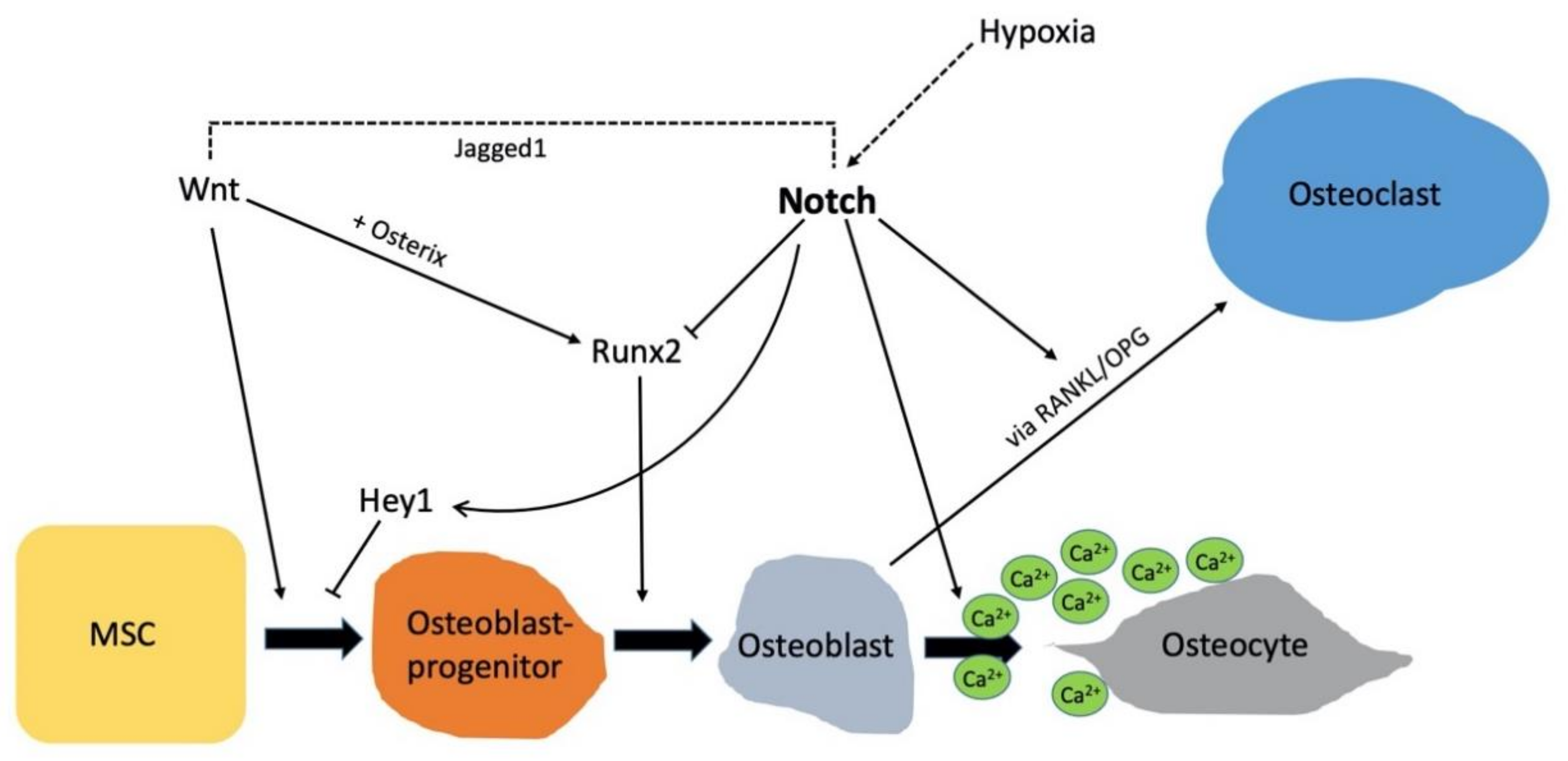
5. The Role of Notch Signaling in Adult Bone Metabolism
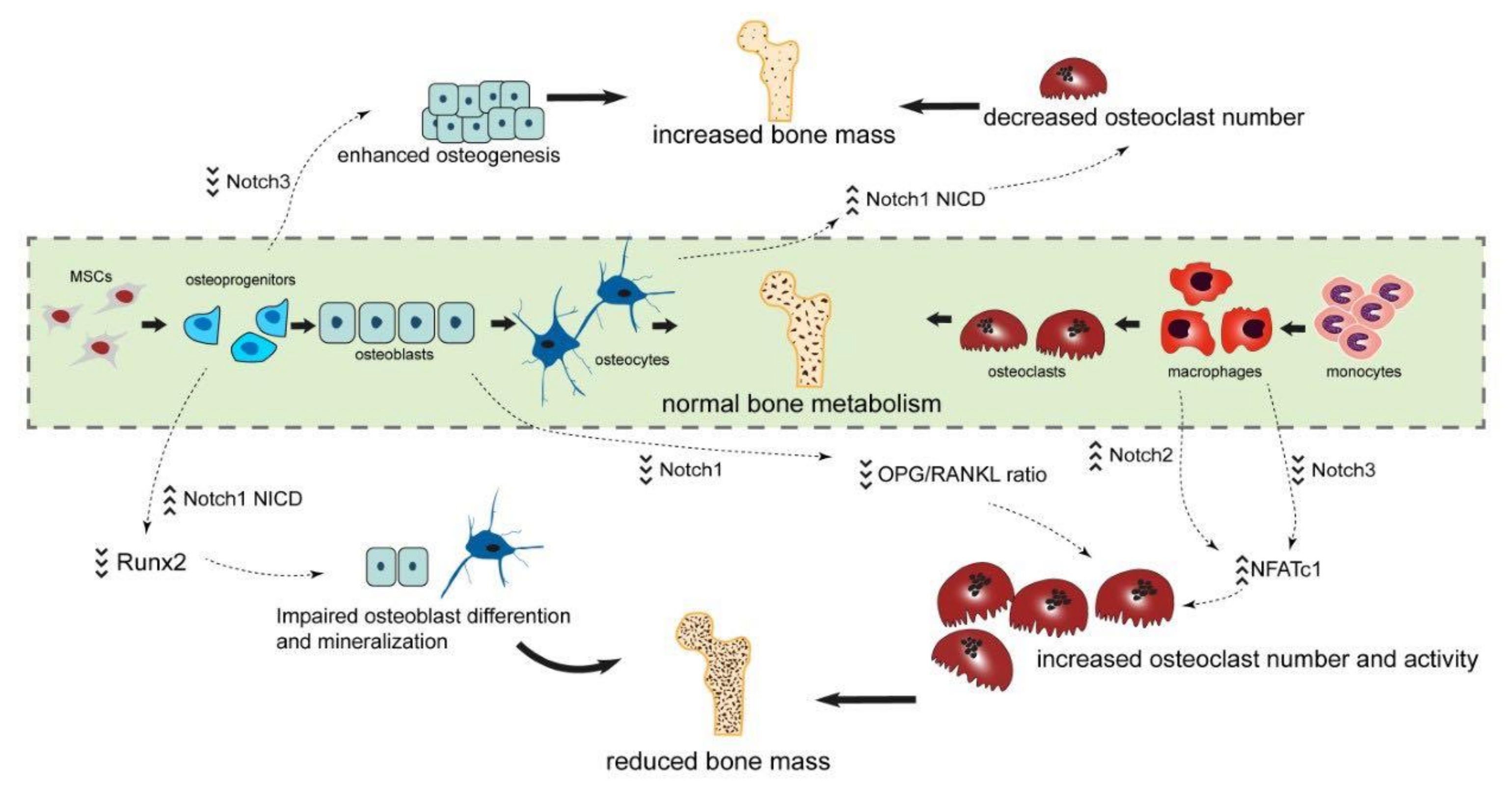
5.1. Function of Receptor Notch1 for Bone Metabolism
5.2. Function of Receptor Notch2 for Bone Metabolism
5.3. Function of Receptor Notch3 and -4 for Bone Metabolism
6. The Role of Notch Signaling in Bone Regeneration
7. Crosstalk of Notch Signaling with Other Pathways
8. Pathologic Mutations in Notch Signaling
8.1. Adams-Oliver Syndrome
8.2. Alagille Syndrome
8.3. Hajdu Cheney Syndrome (HCS)
8.4. Lateral Meningocele Syndrome (LMS)
8.5. CADASIL Syndrome
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANK | ankyrin |
| ALP | alkaline phosphatase |
| ADAM | a disintegrin and metalloprotease |
| AOS | Adams-Oliver syndrome |
| ATF4 | Activating Transcription Factor 4 |
| Bglap | Bone γ-carboxyglutamate protein |
| BMP | Bone Morphogenetic Protein |
| BMSC | bone marrow stem cell |
| CADASIL | cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy |
| Cre | cyclization recombinase |
| Col | collagen |
| DAPT | N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester |
| Dll | Delt-like ligand |
| EGF | Epidermal Growth Factor |
| FBXW7 | F-box/WD repeat-containing protein 7 |
| FGF/-R | fibroblast growth factor/-receptor |
| GSK-3β | Glycogen synthase kinase-3β |
| Hes | hairy enhancer of the split |
| Hey | hairy ears, Y-linked |
| HCS | Hajdu Cheney syndrome |
| Hif | Hypoxia-inducible factor |
| LMS | lateral meningocele syndrome |
| LNR | Lin-12-Notch repeat |
| loxP | locus of X-over P1 |
| MAML | Mastermind-like |
| M-CSF | macrophage-colony stimulating factor |
| MSC | mesenchymal stem cell |
| NECD | Notch extracellular domain |
| NFATc1 | Nuclear Factor of Activated T-cell, cytoplasmic 1 |
| NEXT | Notch extracellular truncation |
| NICD | Notch intracellular domain |
| NRR | negative regulator region |
| OPG | osteoprotegerin |
| Perx1 | Peroxiredoxin-1 |
| PEST | proline, glutamine, serin and threonine |
| RAM | recombination signal binding protein kappa-J region associated module |
| RANK(L) | receptor activator of nuclear factor kappa-B (ligand) |
| RBP-J-κ | Recombination Signal Binding Protein for Immunoglobulin Kappa J Region |
| Runx2 | Runt-related transcription factor 2 |
| TAD | transcriptional activation domain |
| TGF-β | tumor growth factor-β |
| VEGFR1 | vascular endothelial growth factor receptor 1 |
References
- Dexter, J.S. The analysis of a case of continuous variation in Drosophila by a study of its linkage relationships. Am. Nat. 1914, 48, 712–758. [Google Scholar] [CrossRef]
- Morgan, T. The Theory of the Gene. Am. Nat. 1917, 51, 513–544. [Google Scholar] [CrossRef]
- Muller, H.J. Genetic Variability, Twin Hybrids and Constant Hybrids, in a Case of Balanced Lethal Factors. Genetics 1918, 3, 422–499. [Google Scholar] [PubMed]
- Wang, M.M. Notch signaling and Notch signaling modifiers. Int. J. Biochem. Cell Biol. 2011, 43, 1550–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [Green Version]
- Djudjaj, S.; Chatziantoniou, C.; Raffetseder, U.; Guerrot, D.; Dussaule, J.C.; Boor, P.; Kerroch, M.; Hanssen, L.; Brandt, S.; Dittrich, A.; et al. Notch-3 receptor activation drives inflammation and fibrosis following tubulointerstitial kidney injury. J. Pathol. 2012, 228, 286–299. [Google Scholar] [CrossRef]
- Brandt, S.; Ballhause, T.M.; Bernhardt, A.; Becker, A.; Salaru, D.; Le-Deffge, H.M.; Fehr, A.; Fu, Y.; Philipsen, L.; Djudjaj, S.; et al. Fibrosis and Immune Cell Infiltration Are Separate Events Regulated by Cell-Specific Receptor Notch3 Expression. J. Am. Soc. Nephrol. 2020, 31, 2589–2608. [Google Scholar] [CrossRef]
- Canalis, E. Notch in skeletal physiology and disease. Osteoporos Int. 2018, 29, 2611–2621. [Google Scholar] [CrossRef]
- Wagley, Y.; Chesi, A.; Acevedo, P.K.; Lu, S.; Wells, A.D.; Johnson, M.E.; Grant, S.F.A.; Hankenson, K.D. Canonical Notch signaling is required for bone morphogenetic protein-mediated human osteoblast differentiation. Stem Cells 2020, 38, 1332–1347. [Google Scholar] [CrossRef]
- Novak, S.; Roeder, E.; Sinder, B.P.; Adams, D.J.; Siebel, C.W.; Grcevic, D.; Hankenson, K.D.; Matthews, B.G.; Kalajzic, I. Modulation of Notch1 signaling regulates bone fracture healing. J. Orthop. Res. 2020, 38, 2350–2361. [Google Scholar] [CrossRef]
- Brylka, L.J.; Schinke, T. Chemokines in Physiological and Pathological Bone Remodeling. Front. Immunol. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryser, M.D.; Murgas, K.A. Bone remodeling as a spatial evolutionary game. J. Theor. Biol. 2017, 418, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahi, M.; Peymani, A.; Sahmani, M. Regulation of Bone Metabolism. Rep. Biochem. Mol. Biol. 2017, 5, 73–82. [Google Scholar] [PubMed]
- Thiel, A.; Reumann, M.K.; Boskey, A.; Wischmann, J.; von Eisenhart-Rothe, R.; Mayer-Kuckuk, P. Osteoblast migration in vertebrate bone. Biol. Rev. Camb. Philos. Soc. 2018, 93, 350–363. [Google Scholar] [CrossRef] [PubMed]
- Langdahl, B.; Ferrari, S.; Dempster, D.W. Bone modeling and remodeling: Potential as therapeutic targets for the treatment of osteoporosis. Ther. Adv. Musculoskelet. Dis. 2016, 8, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Rucci, N.; Teti, A. The “love-hate” relationship between osteoclasts and bone matrix. Matrix Biol. 2016, 52–54, 176–190. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 2007, 9 (Suppl. S1), S1. [Google Scholar] [CrossRef] [Green Version]
- Hori, K.; Sen, A.; Artavanis-Tsakonas, S. Notch signaling at a glance. J. Cell. Sci. 2013, 126, 2135–2140. [Google Scholar] [CrossRef] [Green Version]
- Vlachakis, D.; Papageorgiou, L.; Papadaki, A.; Georga, M.; Kossida, S.; Eliopoulos, E. An updated evolutionary study of the Notch family reveals a new ancient origin and novel invariable motifs as potential pharmacological targets. PeerJ 2020, 8, e10334. [Google Scholar] [CrossRef]
- Chillakuri, C.R.; Sheppard, D.; Lea, S.M.; Handford, P.A. Notch receptor-ligand binding and activation: Insights from molecular studies. Semin. Cell. Dev. Biol. 2012, 23, 421–428. [Google Scholar] [CrossRef] [Green Version]
- Raffetseder, U.; Rauen, T.; Boor, P.; Ostendorf, T.; Hanssen, L.; Floege, J.; En-Nia, A.; Djudjaj, S.; Frye, B.C.; Mertens, P.R. Extracellular YB-1 blockade in experimental nephritis upregulates Notch-3 receptor expression and signaling. Nephron 2011, 118, e100–e108. [Google Scholar] [CrossRef] [PubMed]
- Borggrefe, T.; Liefke, R. Fine-tuning of the intracellular canonical Notch signaling pathway. Cell Cycle 2012, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Swiatek, P.J.; Lindsell, C.E.; del Amo, F.F.; Weinmaster, G.; Gridley, T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994, 8, 707–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, S.; Crabtree, J.S.; Golde, T.E.; Minter, L.M.; Osborne, B.A.; Miele, L. Targeting Notch in oncology: The path forward. Nat. Rev. Drug Discov. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ladi, E.; Nichols, J.T.; Ge, W.; Miyamoto, A.; Yao, C.; Yang, L.T.; Boulter, J.; Sun, Y.E.; Kintner, C.; Weinmaster, G. The divergent DSL ligand Dll3 does not activate Notch signaling but cell autonomously attenuates signaling induced by other DSL ligands. J. Cell. Biol. 2005, 170, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Rauen, T.; Raffetseder, U.; Frye, B.C.; Djudjaj, S.; Muhlenberg, P.J.; Eitner, F.; Lendahl, U.; Bernhagen, J.; Dooley, S.; Mertens, P.R. YB-1 acts as a ligand for Notch-3 receptors and modulates receptor activation. J. Biol. Chem. 2009, 284, 26928–26940. [Google Scholar] [CrossRef] [Green Version]
- Andersson, E.R.; Sandberg, R.; Lendahl, U. Notch signaling: Simplicity in design, versatility in function. Development 2011, 138, 3593–3612. [Google Scholar] [CrossRef] [Green Version]
- Vooijs, M.; Liu, Z.; Kopan, R. Notch: Architect, landscaper, and guardian of the intestine. Gastroenterology 2011, 141, 448–459. [Google Scholar] [CrossRef] [Green Version]
- Tiyanont, K.; Wales, T.E.; Siebel, C.W.; Engen, J.R.; Blacklow, S.C. Insights into Notch3 activation and inhibition mediated by antibodies directed against its negative regulatory region. J. Mol. Biol. 2013, 425, 3192–3204. [Google Scholar] [CrossRef] [Green Version]
- Leimeister, C.; Externbrink, A.; Klamt, B.; Gessler, M. Hey genes: A novel subfamily of hairy- and Enhancer of split related genes specifically expressed during mouse embryogenesis. Mech. Dev. 1999, 85, 173–177. [Google Scholar] [CrossRef]
- Zhou, M.; Yan, J.; Ma, Z.; Zhou, Y.; Abbood, N.N.; Liu, J.; Su, L.; Jia, H.; Guo, A.Y. Comparative and evolutionary analysis of the HES/HEY gene family reveal exon/intron loss and teleost specific duplication events. PLoS ONE 2012, 7, e40649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canalis, E.; Parker, K.; Feng, J.Q.; Zanotti, S. Osteoblast lineage-specific effects of notch activation in the skeleton. Endocrinology 2013, 154, 623–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciaudone, M.; Gazzerro, E.; Priest, L.; Delany, A.M.; Canalis, E. Notch 1 impairs osteoblastic cell differentiation. Endocrinology 2003, 144, 5631–5639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deregowski, V.; Gazzerro, E.; Priest, L.; Rydziel, S.; Canalis, E. Notch 1 overexpression inhibits osteoblastogenesis by suppressing Wnt/beta-catenin but not bone morphogenetic protein signaling. J. Biol. Chem. 2006, 281, 6203–6210. [Google Scholar] [CrossRef] [Green Version]
- Nobta, M.; Tsukazaki, T.; Shibata, Y.; Xin, C.; Moriishi, T.; Sakano, S.; Shindo, H.; Yamaguchi, A. Critical regulation of bone morphogenetic protein-induced osteoblastic differentiation by Delta1/Jagged1-activated Notch1 signaling. J. Biol. Chem. 2005, 280, 15842–15848. [Google Scholar] [CrossRef] [Green Version]
- Engin, F.; Yao, Z.; Yang, T.; Zhou, G.; Bertin, T.; Jiang, M.M.; Chen, Y.; Wang, L.; Zheng, H.; Sutton, R.E.; et al. Dimorphic effects of Notch signaling in bone homeostasis. Nat. Med. 2008, 14, 299–305. [Google Scholar] [CrossRef]
- Zanotti, S.; Smerdel-Ramoya, A.; Stadmeyer, L.; Durant, D.; Radtke, F.; Canalis, E. Notch inhibits osteoblast differentiation and causes osteopenia. Endocrinology 2008, 149, 3890–3899. [Google Scholar] [CrossRef] [Green Version]
- Bai, S.; Kopan, R.; Zou, W.; Hilton, M.J.; Ong, C.T.; Long, F.; Ross, F.P.; Teitelbaum, S.L. NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J. Biol. Chem. 2008, 283, 6509–6518. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, H.; Nakao, A.; Okamoto, F.; Shin, M.; Kajiya, H.; Sakano, S.; Bigas, A.; Jimi, E.; Okabe, K. The association of Notch2 and NF-kappaB accelerates RANKL-induced osteoclastogenesis. Mol. Cell. Biol. 2008, 28, 6402–6412. [Google Scholar] [CrossRef] [Green Version]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.Y.; Kang, N.; Yang, Y.M.; Hong, J.H.; Shin, D.M. The Role of Ca(2+)-NFATc1 Signaling and Its Modulation on Osteoclastogenesis. Int. J. Mol. Sci. 2020, 21, 3646. [Google Scholar] [CrossRef]
- Canalis, E.; Schilling, L.; Yee, S.P.; Lee, S.K.; Zanotti, S. Hajdu Cheney Mouse Mutants Exhibit Osteopenia, Increased Osteoclastogenesis, and Bone Resorption. J. Biol. Chem. 2016, 291, 1538–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vollersen, N.; Hermans-Borgmeyer, I.; Cornils, K.; Fehse, B.; Rolvien, T.; Triviai, I.; Jeschke, A.; Oheim, R.; Amling, M.; Schinke, T.; et al. High Bone Turnover in Mice Carrying a Pathogenic Notch2 Mutation Causing Hajdu-Cheney Syndrome. J. Bone Miner. Res. 2018, 33, 70–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yorgan, T.; Vollersen, N.; Riedel, C.; Jeschke, A.; Peters, S.; Busse, B.; Amling, M.; Schinke, T. Osteoblast-specific Notch2 inactivation causes increased trabecular bone mass at specific sites of the appendicular skeleton. Bone 2016, 87, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E.; Sanjay, A.; Yu, J.; Zanotti, S. An Antibody to Notch2 Reverses the Osteopenic Phenotype of Hajdu-Cheney Mutant Male Mice. Endocrinology 2017, 158, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jiang, Z.; Zhang, J.; Xie, Z.; Wang, Y.; Yang, G. Enhanced osteogenic differentiation of rat bone marrow mesenchymal stem cells on titanium substrates by inhibiting Notch3. Arch. Oral Biol. 2017, 80, 34–40. [Google Scholar] [CrossRef]
- Dou, X.W.; Park, W.; Lee, S.; Zhang, Q.Z.; Carrasco, L.R.; Le, A.D. Loss of Notch3 Signaling Enhances Osteogenesis of Mesenchymal Stem Cells from Mandibular Torus. J. Dent. Res. 2017, 96, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Long, F. Building strong bones: Molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell. Biol. 2011, 13, 27–38. [Google Scholar] [CrossRef]
- Matsuguchi, T.; Chiba, N.; Bandow, K.; Kakimoto, K.; Masuda, A.; Ohnishi, T. JNK activity is essential for Atf4 expression and late-stage osteoblast differentiation. J. Bone Miner. Res. 2009, 24, 398–410. [Google Scholar] [CrossRef]
- Bagheri, L.; Pellati, A.; Rizzo, P.; Aquila, G.; Massari, L.; De Mattei, M.; Ongaro, A. Notch pathway is active during osteogenic differentiation of human bone marrow mesenchymal stem cells induced by pulsed electromagnetic fields. J. Tissue Eng. Regen. Med. 2018, 12, 304–315. [Google Scholar] [CrossRef]
- Chakravorty, N.; Hamlet, S.; Jaiprakash, A.; Crawford, R.; Oloyede, A.; Alfarsi, M.; Xiao, Y.; Ivanovski, S. Pro-osteogenic topographical cues promote early activation of osteoprogenitor differentiation via enhanced TGFbeta, Wnt, and Notch signaling. Clin. Oral Implants Res. 2014, 25, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Ping, Y.; Ma, M.; Zhang, D.; Liu, C.; Zaidi, S.; Gao, S.; Ji, Y.; Lou, F.; Yu, F.; et al. Anabolic actions of Notch on mature bone. Proc. Natl. Acad. Sci. USA 2016, 113, E2152–E2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Ke, Y.; Gao, S. Intermittent activation of notch signaling promotes bone formation. Am. J. Transl. Res. 2017, 9, 2933–2944. [Google Scholar] [PubMed]
- Brandt, S.; Raffetseder, U.; Djudjaj, S.; Schreiter, A.; Kadereit, B.; Michele, M.; Pabst, M.; Zhu, C.; Mertens, P.R. Cold shock Y-box protein-1 participates in signaling circuits with auto-regulatory activities. Eur. J. Cell. Biol. 2012, 91, 464–471. [Google Scholar] [CrossRef]
- Claes, L.; Recknagel, S.; Ignatius, A. Fracture healing under healthy and inflammatory conditions. Nat. Rev. Rheumatol. 2012, 8, 133–143. [Google Scholar] [CrossRef]
- Einhorn, T.A. The cell and molecular biology of fracture healing. Clin. Orthop. Relat. Res. 1998, 355, S7–S21. [Google Scholar] [CrossRef]
- Garcia-Gareta, E.; Coathup, M.J.; Blunn, G.W. Osteoinduction of bone grafting materials for bone repair and regeneration. Bone 2015, 81, 112–121. [Google Scholar] [CrossRef]
- Dishowitz, M.I.; Terkhorn, S.P.; Bostic, S.A.; Hankenson, K.D. Notch signaling components are upregulated during both endochondral and intramembranous bone regeneration. J. Orthop. Res. 2012, 30, 296–303. [Google Scholar] [CrossRef] [Green Version]
- Zanotti, S.; Smerdel-Ramoya, A.; Canalis, E. HES1 (hairy and enhancer of split 1) is a determinant of bone mass. J. Biol. Chem. 2011, 286, 2648–2657. [Google Scholar] [CrossRef] [Green Version]
- Matthews, B.G.; Grcevic, D.; Wang, L.; Hagiwara, Y.; Roguljic, H.; Joshi, P.; Shin, D.G.; Adams, D.J.; Kalajzic, I. Analysis of alphaSMA-labeled progenitor cell commitment identifies notch signaling as an important pathway in fracture healing. J. Bone Miner. Res. 2014, 29, 1283–1294. [Google Scholar] [CrossRef]
- Wang, C.; Inzana, J.A.; Mirando, A.J.; Ren, Y.; Liu, Z.; Shen, J.; O’Keefe, R.J.; Awad, H.A.; Hilton, M.J. NOTCH signaling in skeletal progenitors is critical for fracture repair. J. Clin. Investig. 2016, 126, 1471–1481. [Google Scholar] [CrossRef] [PubMed]
- Dishowitz, M.I.; Mutyaba, P.L.; Takacs, J.D.; Barr, A.M.; Engiles, J.B.; Ahn, J.; Hankenson, K.D. Systemic inhibition of canonical Notch signaling results in sustained callus inflammation and alters multiple phases of fracture healing. PLoS ONE 2013, 8, e68726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Shen, J.; Yukata, K.; Inzana, J.A.; O’Keefe, R.J.; Awad, H.A.; Hilton, M.J. Transient gamma-secretase inhibition accelerates and enhances fracture repair likely via Notch signaling modulation. Bone 2015, 73, 77–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, T. Molecular Mechanism of Runx2-Dependent Bone Development. Mol. Cells 2020, 43, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.; Delgado-Calle, J.; Condon, K.W.; Maycas, M.; Zhang, H.; Carlesso, N.; Taketo, M.M.; Burr, D.B.; Plotkin, L.I.; Bellido, T. Osteocytes mediate the anabolic actions of canonical Wnt/beta-catenin signaling in bone. Proc. Natl. Acad. Sci. USA 2015, 112, E478–E486. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Zhou, Y.; Lin, J.; Nguyen, T.D.; Huang, R.; Gu, Y.; Friis, T.; Crawford, R.; Xiao, Y. Notch expressed by osteocytes plays a critical role in mineralisation. J. Mol. Med. 2018, 96, 333–347. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Zhou, Y.; Xiao, Y. The regulatory roles of Notch in osteocyte differentiation via the crosstalk with canonical Wnt pathways during the transition of osteoblasts to osteocytes. Bone 2018, 108, 165–178. [Google Scholar] [CrossRef]
- Zhang, K.; Barragan-Adjemian, C.; Ye, L.; Kotha, S.; Dallas, M.; Lu, Y.; Zhao, S.; Harris, M.; Harris, S.E.; Feng, J.Q.; et al. E11/gp38 selective expression in osteocytes: Regulation by mechanical strain and role in dendrite elongation. Mol. Cell. Biol. 2006, 26, 4539–4552. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Wang, Y.; Wang, W.; Chen, J.; Lai, P.; Liu, Z.; Yan, B.; Xu, S.; Zhang, Z.; Zeng, C.; et al. mTORC1 Prevents Preosteoblast Differentiation through the Notch Signaling Pathway. PLoS Genet. 2015, 11, e1005426. [Google Scholar] [CrossRef]
- Wang, R.N.; Green, J.; Wang, Z.; Deng, Y.; Qiao, M.; Peabody, M.; Zhang, Q.; Ye, J.; Yan, Z.; Denduluri, S.; et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis. 2014, 1, 87–105. [Google Scholar] [CrossRef] [Green Version]
- Borggrefe, T.; Lauth, M.; Zwijsen, A.; Huylebroeck, D.; Oswald, F.; Giaimo, B.D. The Notch intracellular domain integrates signals from Wnt, Hedgehog, TGFbeta/BMP and hypoxia pathways. Biochim. Biophys. Acta 2016, 1863, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lowther, W.; Kato, K.; Bianco, C.; Kenney, N.; Strizzi, L.; Raafat, D.; Hirota, M.; Khan, N.I.; Bargo, S.; et al. Notch4 intracellular domain binding to Smad3 and inhibition of the TGF-beta signaling. Oncogene 2005, 24, 5365–5374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Deng, C.; Li, Y.P. TGF-beta and BMP signaling in osteoblast differentiation and bone formation. Int. J. Biol. Sci. 2012, 8, 272–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Wei, Y.; Lian, J.; Yang, L.; Zhang, X.; Xie, J.; Liu, Q.; Luo, J.; He, B.; Tang, M. Notch signaling pathway promotes osteogenic differentiation of mesenchymal stem cells by enhancing BMP9/Smad signaling. Int. J. Mol. Med. 2017, 40, 378–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seong, C.H.; Chiba, N.; Kusuyama, J.; Amir, M.S.; Eiraku, N.; Yamashita, S.; Ohnishi, T.; Nakamura, N.; Matsuguchi, T. Bone morphogenetic protein 9 (BMP9) directly induces Notch effector molecule Hes1 through the SMAD signaling pathway in osteoblasts. FEBS Lett. 2020. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, J.; Deng, F.; Huang, E.; Yan, Z.; Wang, Z.; Deng, Y.; Zhang, Q.; Zhang, Z.; Ye, J.; et al. Canonical Wnt signaling acts synergistically on BMP9-induced osteo/odontoblastic differentiation of stem cells of dental apical papilla (SCAPs). Biomaterials 2015, 39, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Rodilla, V.; Villanueva, A.; Obrador-Hevia, A.; Robert-Moreno, A.; Fernandez-Majada, V.; Grilli, A.; Lopez-Bigas, N.; Bellora, N.; Alba, M.M.; Torres, F.; et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 6315–6320. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [Green Version]
- Sieiro, D.; Rios, A.C.; Hirst, C.E.; Marcelle, C. Cytoplasmic NOTCH and membrane-derived beta-catenin link cell fate choice to epithelial-mesenchymal transition during myogenesis. Elife 2016, 5, e14847. [Google Scholar] [CrossRef] [Green Version]
- Riddle, R.C.; Khatri, R.; Schipani, E.; Clemens, T.L. Role of hypoxia-inducible factor-1alpha in angiogenic-osteogenic coupling. J. Mol. Med. 2009, 87, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Tian, Q.; Xue, Y.; Zheng, W.; Sun, R.; Ji, W.; Wang, X.; An, R. Overexpression of hypoxia-inducible factor 1alpha induces migration and invasion through Notch signaling. Int. J. Oncol. 2015, 47, 728–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, R.; Sun, W.; Xia, L.; Zhao, A.; Yu, Y.; Zhao, L.; Wang, H.; Huang, C.; Sun, S. Hypoxia-induced down-regulation of microRNA-34a promotes EMT by targeting the Notch signaling pathway in tubular epithelial cells. PLoS ONE 2012, 7, e30771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domenga, V.; Fardoux, P.; Lacombe, P.; Monet, M.; Maciazek, J.; Krebs, L.T.; Klonjkowski, B.; Berrou, E.; Mericskay, M.; Li, Z.; et al. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 2004, 18, 2730–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Pan, L.; Moens, C.B.; Appel, B. Notch3 establishes brain vascular integrity by regulating pericyte number. Development 2014, 141, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stittrich, A.B.; Lehman, A.; Bodian, D.L.; Ashworth, J.; Zong, Z.; Li, H.; Lam, P.; Khromykh, A.; Iyer, R.K.; Vockley, J.G.; et al. Mutations in NOTCH1 cause Adams-Oliver syndrome. Am. J. Hum. Genet 2014, 95, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Lehman, A.; Wuyts, W.; Patel, M.S. Adams-Oliver Syndrome. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Kehrer, A.; Luther, C.; Exner, K. Das Adams-Oliver-Syndrom—Chirurgische Herausforderung eines seltenen Symptomenkomplexes. In Proceedings of the 130 Kongress der Deutschen Geseelschaft für Chirurgie, Munich, German Medical Science GMS Publishing House, Munich, Germany, 26 April 2013. [Google Scholar] [CrossRef]
- Sukalo, M.; Tilsen, F.; Kayserili, H.; Muller, D.; Tuysuz, B.; Ruddy, D.M.; Wakeling, E.; Orstavik, K.H.; Snape, K.M.; Trembath, R.; et al. DOCK6 mutations are responsible for a distinct autosomal-recessive variant of Adams-Oliver syndrome associated with brain and eye anomalies. Hum. Mutat. 2015, 36, 593–598. [Google Scholar] [CrossRef] [Green Version]
- Snape, K.M.; Ruddy, D.; Zenker, M.; Wuyts, W.; Whiteford, M.; Johnson, D.; Lam, W.; Trembath, R.C. The spectra of clinical phenotypes in aplasia cutis congenita and terminal transverse limb defects. Am. J. Med. Genet. A 2009, 149A, 1860–1881. [Google Scholar] [CrossRef]
- Madan, A.; Sardana, K.; Garg, V.K. Adams Oliver Syndrome. Indian Pediatr. 2015, 52, 633–634. [Google Scholar]
- Fraisse, A.; Losay, J.; Bourlon, F.; Agnoletti, G.; Lusson, J.R.; Godart, F.; De Geeter, B.; Petit, J.; Piechaud, J.F. Efficiency of transcatheter closure of atrial septal defects in small and symptomatic children. Cardiol. Young 2008, 18, 343–347. [Google Scholar] [CrossRef]
- Khashab, M.E.; Rhee, S.T.; Pierce, S.D.; Khashab, Y.E.; Nejat, F.; Fried, A. Management of large scalp and skull defects in a severe case of Adams-Oliver syndrome. J. Neurosurg. Pediatr. 2009, 4, 523–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heritage, M.L.; MacMillan, J.C.; Colliton, R.P.; Genin, A.; Spinner, N.B.; Anderson, G.J. Jagged1 (JAG1) mutation detection in an Australian Alagille syndrome population. Hum. Mutat. 2000, 16, 408–416. [Google Scholar] [CrossRef]
- Kamath, B.M.; Baker, A.; Houwen, R.; Todorova, L.; Kerkar, N. Systematic Review: The Epidemiology, Natural History, and Burden of Alagille Syndrome. J. Pediatr. Gastroenterol. Nutr. 2018, 67, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Turnpenny, P.D.; Ellard, S. Alagille syndrome: Pathogenesis, diagnosis and management. Eur. J. Hum. Genet. 2012, 20, 251–257. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.; Sweetwyne, M.T.; Hankenson, K.D. PKCdelta is required for Jagged-1 induction of human mesenchymal stem cell osteogenic differentiation. Stem Cells 2013, 31, 1181–1192. [Google Scholar] [CrossRef]
- Ayoub, M.D.; Kamath, B.M. Alagille Syndrome: Diagnostic Challenges and Advances in Management. Diagnostics 2020, 10, 907. [Google Scholar] [CrossRef]
- Descatoire, M.; Weller, S.; Irtan, S.; Sarnacki, S.; Feuillard, J.; Storck, S.; Guiochon-Mantel, A.; Bouligand, J.; Morali, A.; Cohen, J.; et al. Identification of a human splenic marginal zone B cell precursor with NOTCH2-dependent differentiation properties. J. Exp. Med. 2014, 211, 987–1000. [Google Scholar] [CrossRef]
- Spinner, N.B.; Gilbert, M.A.; Loomes, K.M.; Krantz, I.D. Alagille Syndrome; 19 May 2000 [updated 12 Dec 2019]; GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2021. [Google Scholar] [PubMed] [Green Version]
- Canalis, E. Clinical and experimental aspects of notch receptor signaling: Hajdu-Cheney syndrome and related disorders. Metabolism 2018, 80, 48–56. [Google Scholar] [CrossRef]
- Lee, S.Y.; Kumano, K.; Nakazaki, K.; Sanada, M.; Matsumoto, A.; Yamamoto, G.; Nannya, Y.; Suzuki, R.; Ota, S.; Ota, Y.; et al. Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma. Cancer Sci. 2009, 100, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Palav, S.; Vernekar, J.; Pereira, S.; Desai, A. Hajdu-Cheney syndrome: A case report with review of literature. J. Radiol. Case Rep. 2014, 8, 1–8. [Google Scholar] [CrossRef]
- Canalis, E.; Zanotti, S. Hajdu-Cheney syndrome: A review. Orphanet J. Rare Dis. 2014, 9, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanotti, S.; Yu, J.; Sanjay, A.; Schilling, L.; Schoenherr, C.; Economides, A.N.; Canalis, E. Sustained Notch2 signaling in osteoblasts, but not in osteoclasts, is linked to osteopenia in a mouse model of Hajdu-Cheney syndrome. J. Biol. Chem. 2017, 292, 12232–12244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, H.; Shimizu, K.; Watahiki, A.; Hoshikawa, S.; Kosho, T.; Oba, D.; Sakano, S.; Arakaki, M.; Yamada, A.; Nagashima, K.; et al. NOTCH2 Hajdu-Cheney Mutations Escape SCF(FBW7)-Dependent Proteolysis to Promote Osteoporosis. Mol. Cell. 2017, 68, 645–658.e5. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.; Zhang, G.; Wu, Y.; Stawicki, S.; Liang, W.C.; Chanthery, Y.; Kowalski, J.; Watts, R.J.; Callahan, C.; Kasman, I.; et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 2006, 444, 1083–1087. [Google Scholar] [CrossRef]
- Albright, C.F.; Dockens, R.C.; Meredith, J.E., Jr.; Olson, R.E.; Slemmon, R.; Lentz, K.A.; Wang, J.S.; Denton, R.R.; Pilcher, G.; Rhyne, P.W.; et al. Pharmacodynamics of selective inhibition of gamma-secretase by avagacestat. J. Pharmacol. Exp. Ther. 2013, 344, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Callahan, C.A.; Beyer, J.C.; Allamneni, K.P.; Zhang, G.; Ridgway, J.B.; Niessen, K.; Plowman, G.D. Chronic DLL4 blockade induces vascular neoplasms. Nature 2010, 463, E6–E7. [Google Scholar] [CrossRef]
- Adami, G.; Rossini, M.; Gatti, D.; Orsolini, G.; Idolazzi, L.; Viapiana, O.; Scarpa, A.; Canalis, E. Hajdu Cheney Syndrome; report of a novel NOTCH2 mutation and treatment with denosumab. Bone 2016, 92, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Adami, G.; Saag, K.G.; Chapurlat, R.D.; Guanabens, N.; Haugeberg, G.; Lems, W.F.; Matijevic, R.; Peel, N.; Poddubnyy, D.; Geusens, P. Balancing benefits and risks in the era of biologics. Ther. Adv. Musculoskelet. Dis. 2019, 11, 1759720X19883973. [Google Scholar] [CrossRef]
- Cappuccio, G.; Apuzzo, D.; Alagia, M.; Torella, A.; Pinelli, M.; Franco, B.; Corrado, B.; Del Giudice, E.; D’Amico, A.; Nigro, V.; et al. Expansion of the phenotype of lateral meningocele syndrome. Am. J. Med Genet A 2020, 182, 1259–1262. [Google Scholar] [CrossRef]
- Ejaz, R.; Qin, W.; Huang, L.; Blaser, S.; Tetreault, M.; Hartley, T.; Boycott, K.M.; Carter, M.T.; Care4Rare Canada, C. Lateral meningocele (Lehman) syndrome: A child with a novel NOTCH3 mutation. Am. J. Med. Genet A 2016, 170A, 1070–1075. [Google Scholar] [CrossRef]
- Brown, E.C.; Gupta, K.; Sayama, C. Neurosurgical management in lateral meningocele syndrome: Case report. J. Neurosurg. Pediatr. 2017, 19, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Gripp, K.W. Lateral meningocele syndrome and Hajdu-Cheney syndrome: Different disorders with overlapping phenotypes. Am. J. Med. Genet A 2011, 155A, 1773–1774. [Google Scholar] [CrossRef] [PubMed]
- Gripp, K.W.; Robbins, K.M.; Sobreira, N.L.; Witmer, P.D.; Bird, L.M.; Avela, K.; Makitie, O.; Alves, D.; Hogue, J.S.; Zackai, E.H.; et al. Truncating mutations in the last exon of NOTCH3 cause lateral meningocele syndrome. Am. J. Med. Genet A 2015, 167A, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehman, R.A.; Stears, J.C.; Wesenberg, R.L.; Nusbaum, E.D. Familial osteosclerosis with abnormalities of the nervous system and meninges. J. Pediatr. 1977, 90, 49–54. [Google Scholar] [CrossRef]
- Yu, J.; Siebel, C.W.; Schilling, L.; Canalis, E. An antibody to Notch3 reverses the skeletal phenotype of lateral meningocele syndrome in male mice. J. Cell. Physiol. 2020, 235, 210–220. [Google Scholar] [CrossRef]
- Chabriat, H.; Joutel, A.; Dichgans, M.; Tournier-Lasserve, E.; Bousser, M.G. Cadasil. Lancet 2009, 8, 643–653. [Google Scholar] [CrossRef]
- Di Donato, I.; Bianchi, S.; De Stefano, N.; Dichgans, M.; Dotti, M.T.; Duering, M.; Jouvent, E.; Korczyn, A.D.; Lesnik-Oberstein, S.A.; Malandrini, A.; et al. Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL) as a model of small vessel disease: Update on clinical, diagnostic, and management aspects. BMC Med. 2017, 15, 41. [Google Scholar] [CrossRef] [Green Version]
- Joutel, A.; Dodick, D.D.; Parisi, J.E.; Cecillon, M.; Tournier-Lasserve, E.; Bousser, M.G. De novo mutation in the Notch3 gene causing CADASIL. Ann. Neurol. 2000, 47, 388–391. [Google Scholar] [CrossRef]
- Brennan-Krohn, T.; Salloway, S.; Correia, S.; Dong, M.; de la Monte, S.M. Glial vascular degeneration in CADASIL. J. Alzheimers Dis. 2010, 21, 1393–1402. [Google Scholar] [CrossRef] [Green Version]
- Toni-Uebari, T.K. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL): A rare cause of dementia. BMJ Case Rep. 2013, 2013. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ballhause, T.M.; Jiang, S.; Baranowsky, A.; Brandt, S.; Mertens, P.R.; Frosch, K.-H.; Yorgan, T.; Keller, J. Relevance of Notch Signaling for Bone Metabolism and Regeneration. Int. J. Mol. Sci. 2021, 22, 1325. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031325
Ballhause TM, Jiang S, Baranowsky A, Brandt S, Mertens PR, Frosch K-H, Yorgan T, Keller J. Relevance of Notch Signaling for Bone Metabolism and Regeneration. International Journal of Molecular Sciences. 2021; 22(3):1325. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031325
Chicago/Turabian StyleBallhause, Tobias M., Shan Jiang, Anke Baranowsky, Sabine Brandt, Peter R. Mertens, Karl-Heinz Frosch, Timur Yorgan, and Johannes Keller. 2021. "Relevance of Notch Signaling for Bone Metabolism and Regeneration" International Journal of Molecular Sciences 22, no. 3: 1325. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22031325