
Human Primary Breast Cancer Stem Cells Are Characterized by Epithelial-Mesenchymal Plasticity

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
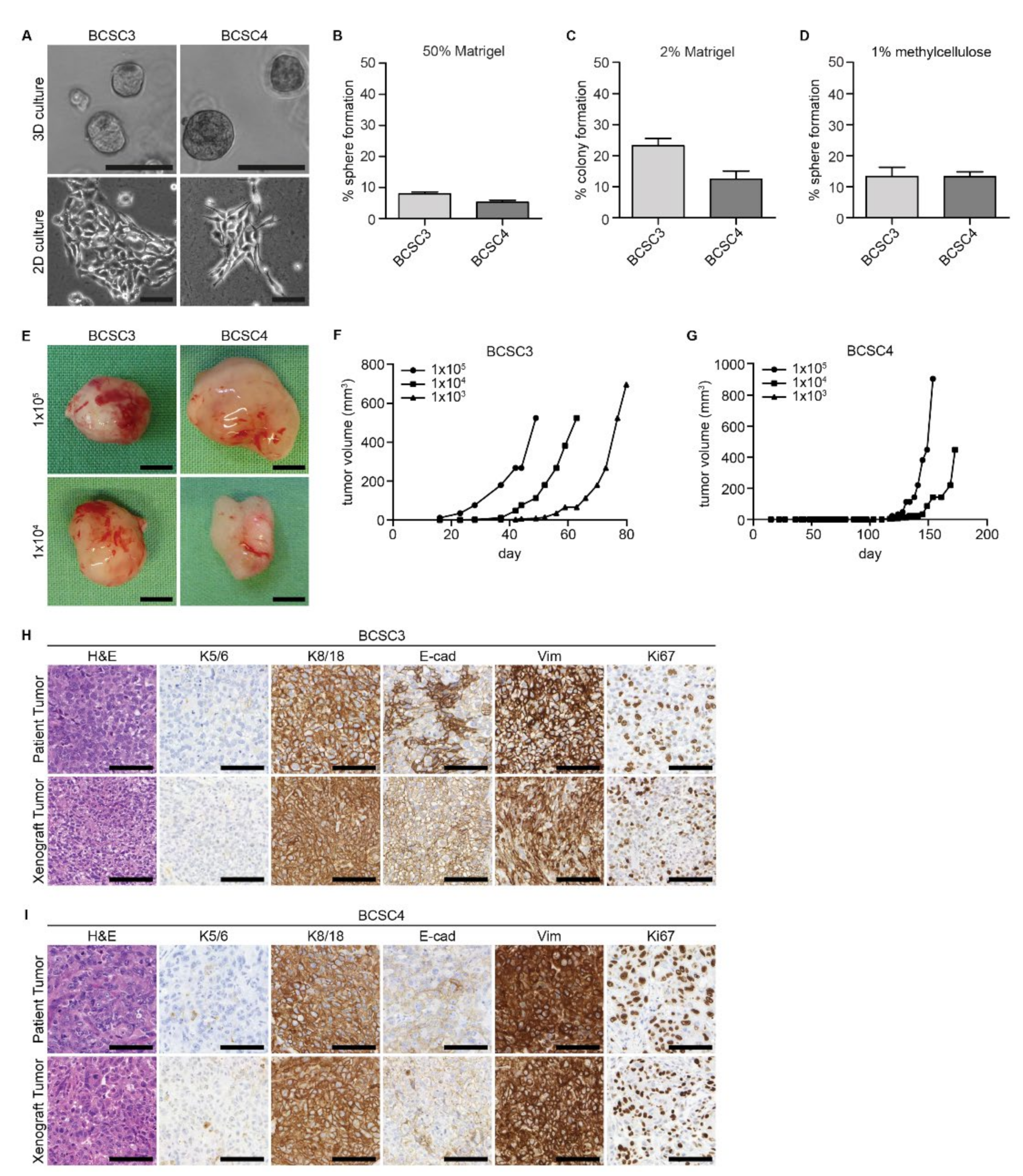
2.1. BCSCs Can Be Isolated from Patient Tumor Samples
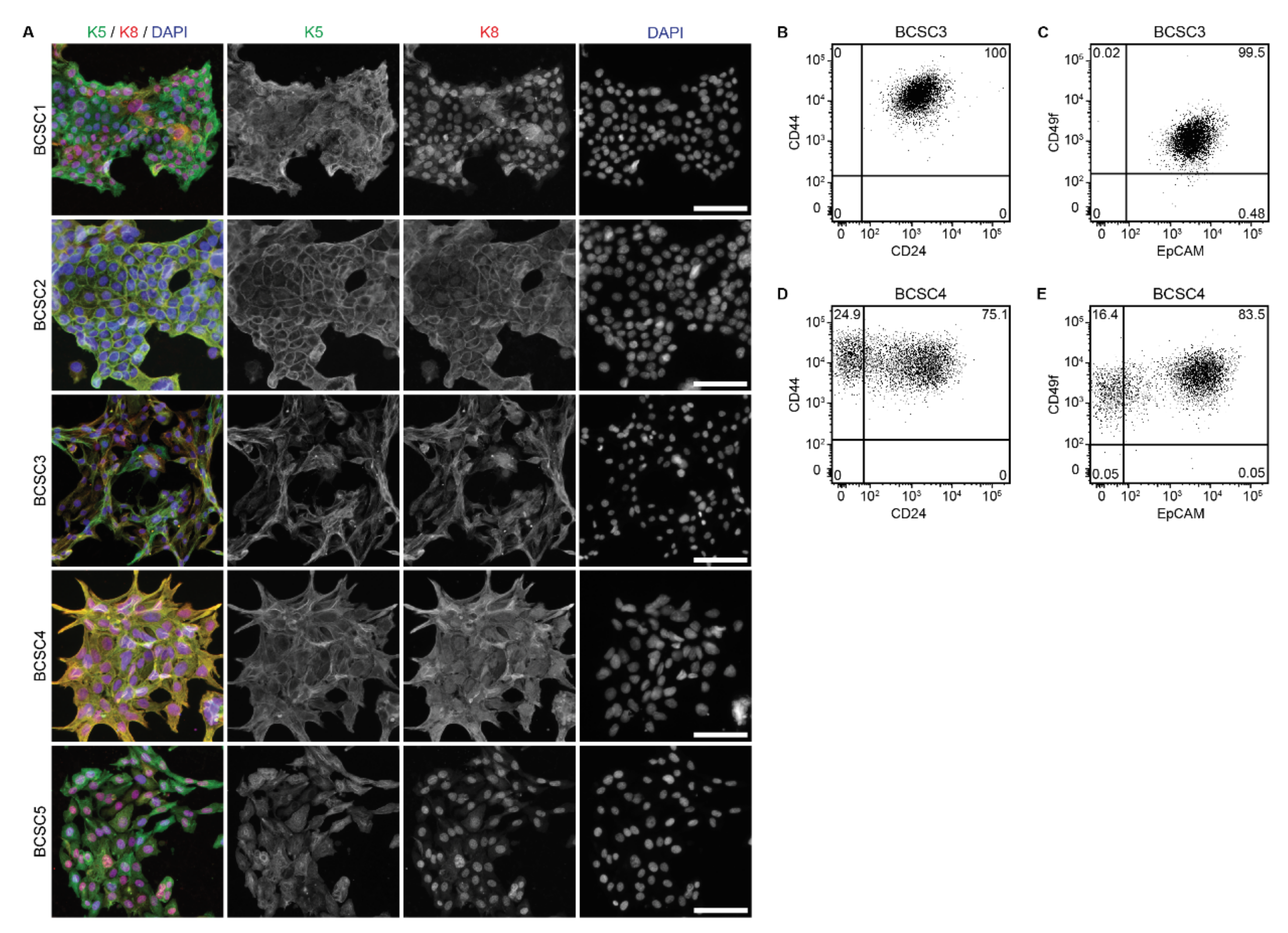
2.2. BCSCs Exhibit Cellular Heterogeneity In Vitro
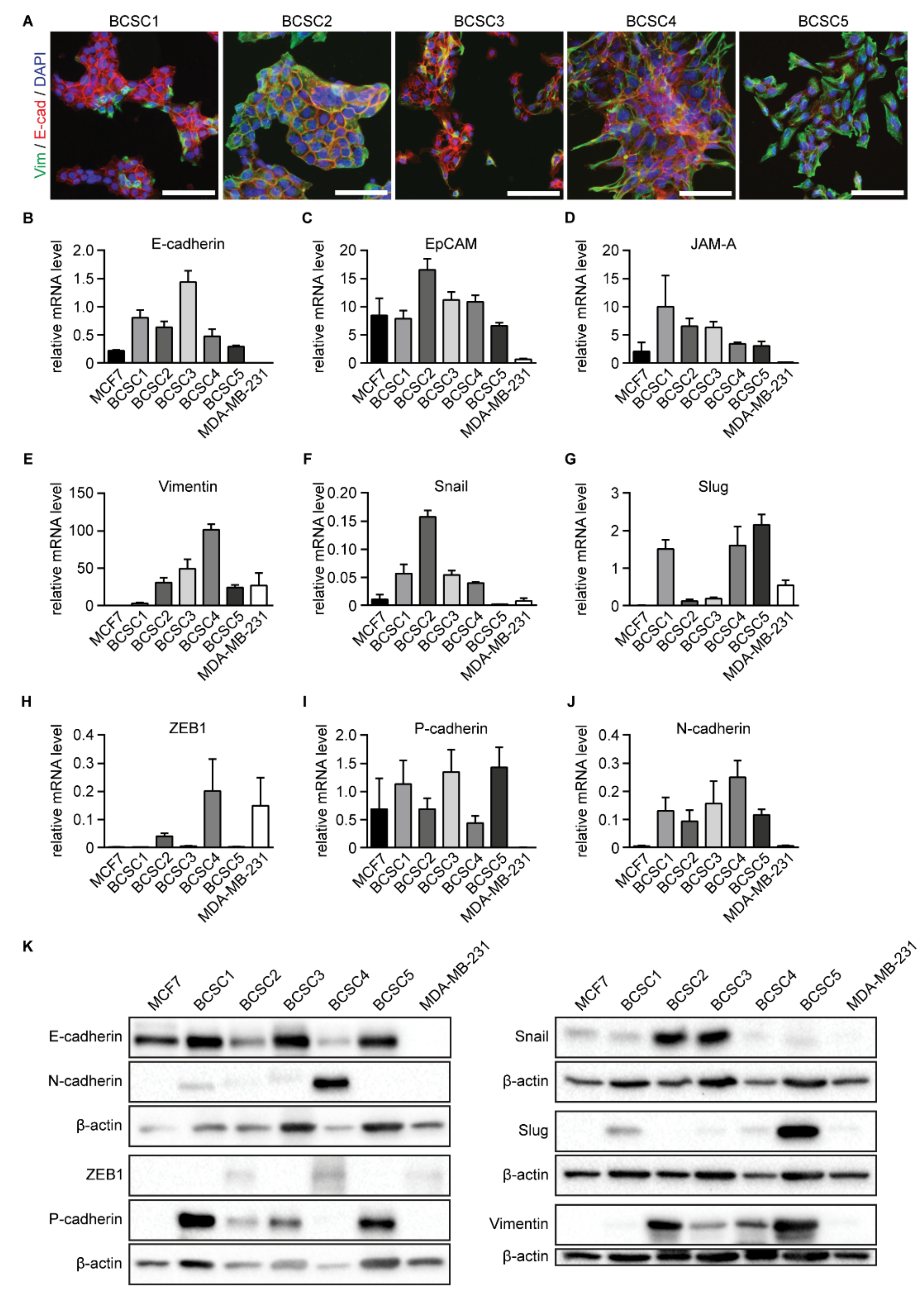
2.3. BCSCs Show an Intermediate EMT Phenotype
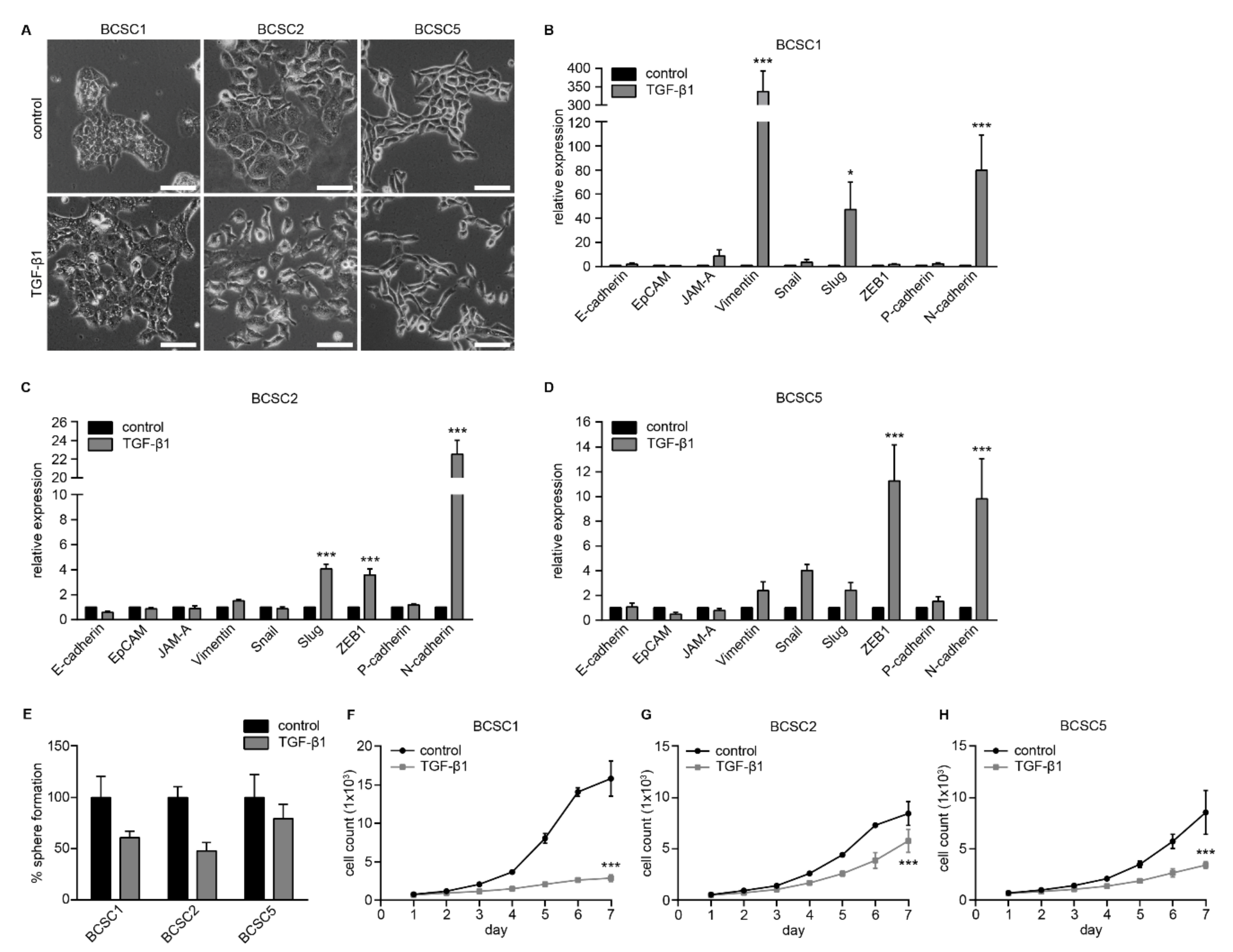
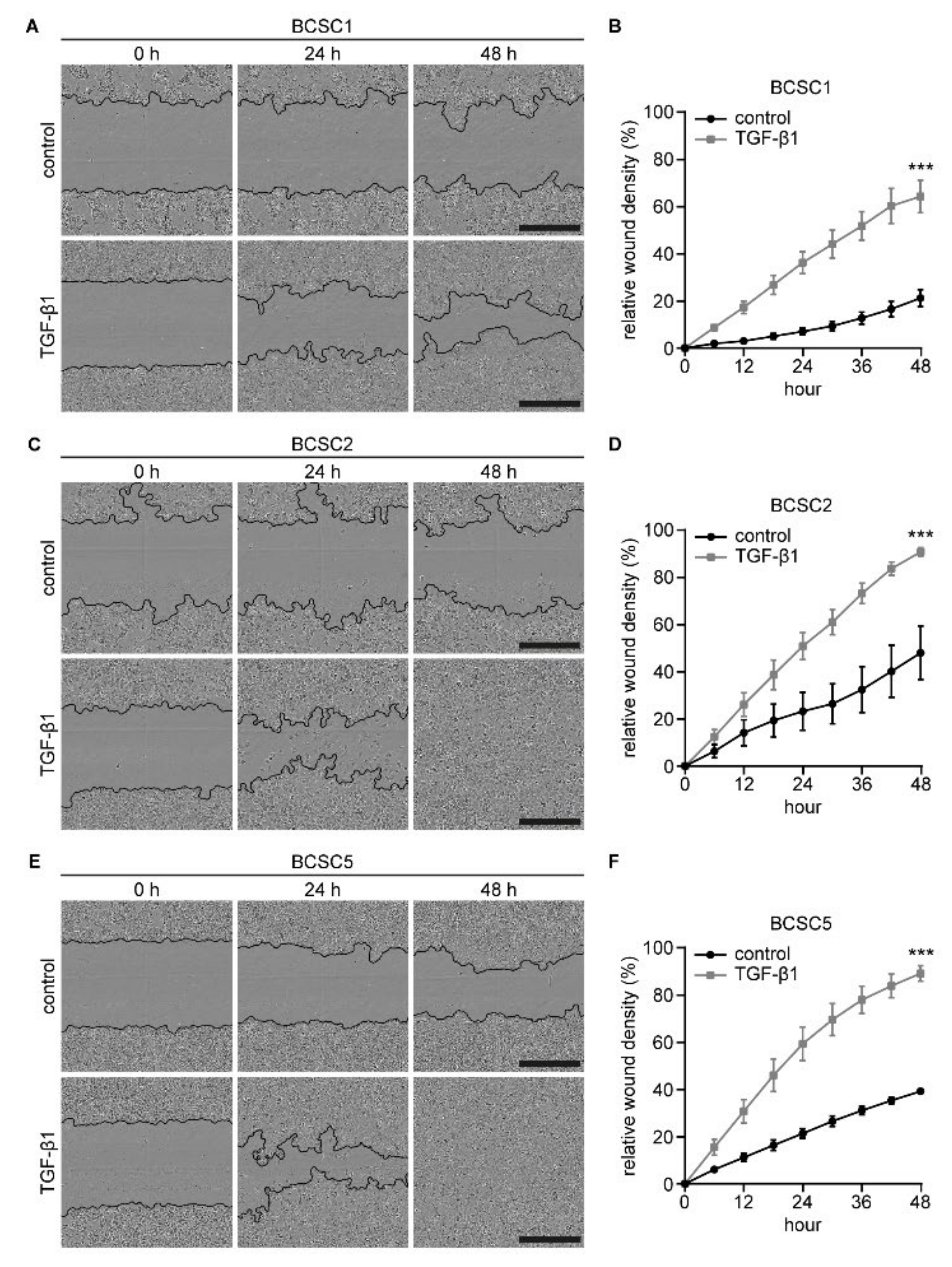
2.4. Primary BCSCs Exhibit Cellular Plasticity
3. Discussion
4. Materials and Methods
4.1. Tissue Specimens
4.2. BCSC Culture
4.3. Conventional Cell Culture
4.4. Cell Proliferation Assay
4.5. Colony Formation Assay
4.6. Sphere Assay in Matrigel
4.7. Sphere Assay in Methylcellulose
4.8. Immunofluorescence Staining
4.9. Flow Cytometry
4.10. Orthotopic Breast Cancer Xenografts
4.11. Immunohistochemistry
4.12. Quantitative Real-Time PCR Analysis
4.13. Western Blot Analysis
4.14. Scratch Wound Assay
4.15. Stimulation with TGF-β1
4.16. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wild, C.; Weiderpass, E.; Stewart, B. World Cancer Report: Cancer Research for Cancer Prevention; Wild, C., Weiderpass, E., Stewart, B., Eds.; International Agency for Research on Cancer: Lyon, France, 2020; ISBN 978-92-832-0447-3. [Google Scholar]
- Goldhirsch, A.; Winer, E.P.; Coates, A.S.; Gelber, R.D.; Piccart-Gebhart, M.; Thürlimann, B.; Senn, H.-J. Personalizing the treatment of women with early breast cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2013. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24, 2206–2223. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Pineda, E.; Adamo, B.; Galván, P.; Fernández, A.; Gaba, L.; Díez, M.; Viladot, M.; Arance, A.; Muñoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24 (Suppl. 2), S26–S35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geyer, F.C.; Pareja, F.; Weigelt, B.; Rakha, E.; Ellis, I.O.; Schnitt, S.J.; Reis-Filho, J.S. The Spectrum of Triple-Negative Breast Disease: High- and Low-Grade Lesions. Am. J. Pathol. 2017, 187, 2139–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.M.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [Green Version]
- Pardal, R.; Clarke, M.F.; Morrison, S.J. Applying the principles of stem-cell biology to cancer. Nat. Rev. Cancer 2003, 3, 895–902. [Google Scholar] [CrossRef]
- Jaggupilli, A.; Elkord, E. Significance of CD44 and CD24 as cancer stem cell markers: An enduring ambiguity. Clin. Dev. Immunol. 2012, 2012, 708036. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Nenutil, R.; Appleyard, M.V.; Murray, K.; Boylan, M.; Thompson, A.M.; Coates, P.J. Lack of correlation of stem cell markers in breast cancer stem cells. Br. J. Cancer 2014, 110, 2063–2071. [Google Scholar] [CrossRef] [Green Version]
- Da Cruz Paula, A.; Lopes, C. Implications of Different Cancer Stem Cell Phenotypes in Breast Cancer. Anticancer Res. 2017, 37, 2173–2183. [Google Scholar] [CrossRef] [Green Version]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cariati, M.; Naderi, A.; Brown, J.P.; Smalley, M.J.; Pinder, S.E.; Caldas, C.; Purushotham, A.D. Alpha-6 integrin is necessary for the tumourigenicity of a stem cell-like subpopulation within the MCF7 breast cancer cell line. Int. J. Cancer 2008, 122, 298–304. [Google Scholar] [CrossRef]
- Meyer, M.J.; Fleming, J.M.; Lin, A.F.; Hussnain, S.A.; Ginsburg, E.; Vonderhaar, B.K. CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor-negative breast cancer. Cancer Res. 2010, 70, 4624–4633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Morel, A.-P.; Lièvre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast Cancer Stem Cells Transition between Epithelial and Mesenchymal States Reflective of their Normal Counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Brooks, M.; Wicha, M.S. Epithelial-mesenchymal plasticity of breast cancer stem cells: Implications for metastasis and therapeutic resistance. Curr. Pharm. Des. 2015, 21, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosse-Wilde, A.; Fouquier d’Hérouël, A.; McIntosh, E.; Ertaylan, G.; Skupin, A.; Kuestner, R.E.; del Sol, A.; Walters, K.-A.; Huang, S. Stemness of the hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLoS ONE 2015, 10, e0126522. [Google Scholar] [CrossRef]
- Lacroix, M.; Leclercq, G. Relevance of breast cancer cell lines as models for breast tumours: An update. Breast Cancer Res. Treat. 2004, 83, 249–289. [Google Scholar] [CrossRef]
- Price, J.E.; Polyzos, A.; Zhang, R.D.; Daniels, L.M. Tumorigenicity and metastasis of human breast carcinoma cell lines in nude mice. Cancer Res. 1990, 50, 717–721. [Google Scholar]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.A.; Daidone, M.G. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillmore, C.M.; Kuperwasser, C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef] [Green Version]
- Metzger, E.; Stepputtis, S.S.; Strietz, J.; Preca, B.-T.; Urban, S.; Willmann, D.; Allen, A.; Zenk, F.; Iovino, N.; Bronsert, P.; et al. KDM4 Inhibition Targets Breast Cancer Stem-like Cells. Cancer Res. 2017, 77, 5900–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Strietz, J.; Bleilevens, A.; Stickeler, E.; Maurer, J. Chemotherapeutic Stress Influences Epithelial-Mesenchymal Transition and Stemness in Cancer Stem Cells of Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2020, 21, 404. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef]
- Wang, D.; Lu, P.; Zhang, H.; Luo, M.; Zhang, X.; Wei, X.; Gao, J.; Zhao, Z.; Liu, C. Oct-4 and Nanog promote the epithelial-mesenchymal transition of breast cancer stem cells and are associated with poor prognosis in breast cancer patients. Oncotarget 2014, 5, 10803–10815. [Google Scholar] [CrossRef] [Green Version]
- Domenici, G.; Aurrekoetxea-Rodríguez, I.; Simões, B.M.; Rábano, M.; Lee, S.Y.; Millán, J.S.; Comaills, V.; Oliemuller, E.; López-Ruiz, J.A.; Zabalza, I.; et al. A Sox2–Sox9 signalling axis maintains human breast luminal progenitor and breast cancer stem cells. Oncogene 2019, 38, 3151–3169. [Google Scholar] [CrossRef]
- Almozyan, S.; Colak, D.; Mansour, F.; Alaiya, A.; Al-Harazi, O.; Qattan, A.; Al-Mohanna, F.; Al-Alwan, M.; Ghebeh, H. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Int. J. Cancer 2017, 141, 1402–1412. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrallo-Gimeno, A.; Nieto, M.A. The Snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stemmler, M.P. Cadherins in development and cancer. Mol. Biosyst. 2008, 4, 835–850. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Zarzynska, J.M. Two faces of TGF-beta1 in breast cancer. Mediat. Inflamm. 2014, 2014, 141747. [Google Scholar] [CrossRef]
- Shipitsin, M.; Polyak, K. The cancer stem cell hypothesis: In search of definitions, markers, and relevance. Lab. Investig. 2008, 88, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003, 17, 1253–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farnie, G.; Clarke, R.B.; Spence, K.; Pinnock, N.; Brennan, K.; Anderson, N.G.; Bundred, N.J. Novel cell culture technique for primary ductal carcinoma in situ: Role of Notch and epidermal growth factor receptor signaling pathways. J. Natl. Cancer Inst. 2007, 99, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Harrison, H.; Farnie, G.; Howell, S.J.; Rock, R.E.; Stylianou, S.; Brennan, K.R.; Bundred, N.J.; Clarke, R.B. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010, 70, 709–718. [Google Scholar] [CrossRef] [Green Version]
- Drews-Elger, K.; Brinkman, J.A.; Miller, P.; Shah, S.H.; Harrell, J.C.; da Silva, T.G.; Ao, Z.; Schlater, A.; Azzam, D.J.; Diehl, K.; et al. Primary breast tumor-derived cellular models: Characterization of tumorigenic, metastatic, and cancer-associated fibroblasts in dissociated tumor (DT) cultures. Breast Cancer Res. Treat. 2014, 144, 503–517. [Google Scholar] [CrossRef]
- Wang, R.; Lv, Q.; Meng, W.; Tan, Q.; Zhang, S.; Mo, X.; Yang, X. Comparison of mammosphere formation from breast cancer cell lines and primary breast tumors. J. Thorac. Dis. 2014, 6, 829–837. [Google Scholar] [CrossRef]
- Kasai, F.; Hirayama, N.; Ozawa, M.; Iemura, M.; Kohara, A. Changes of heterogeneous cell populations in the Ishikawa cell line during long-term culture: Proposal for an in vitro clonal evolution model of tumor cells. Genomics 2016, 107, 259–266. [Google Scholar] [CrossRef]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef] [Green Version]
- Grimshaw, M.J.; Cooper, L.; Papazisis, K.; Coleman, J.A.; Bohnenkamp, H.R.; Chiapero-Stanke, L.; Taylor-Papadimitriou, J.; Burchell, J.M. Mammosphere culture of metastatic breast cancer cells enriches for tumorigenic breast cancer cells. Breast Cancer Res. 2008, 10, R52. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, C.; Jobs, G.; Thomas, M.; Burtscher, H.; Kubbies, M. Established breast cancer stem cell markers do not correlate with in vivo tumorigenicity of tumor-initiating cells. Int. J. Oncol. 2012, 41, 1932–1942. [Google Scholar] [CrossRef] [PubMed]
- Ricardo, S.; Vieira, A.F.; Gerhard, R.; Leitao, D.; Pinto, R.; Cameselle-Teijeiro, J.F.; Milanezi, F.; Schmitt, F.; Paredes, J. Breast cancer stem cell markers CD44, CD24 and ALDH1: Expression distribution within intrinsic molecular subtype. J. Clin. Pathol. 2011, 64, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.-L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef]
- Stingl, J.; Eaves, C.J.; Zandieh, I.; Emerman, J.T. Characterization of bipotent mammary epithelial progenitor cells in normal adult human breast tissue. Breast Cancer Res. Treat. 2001, 67, 93–109. [Google Scholar] [CrossRef]
- Abd El-Rehim, D.M.; Pinder, S.E.; Paish, C.E.; Bell, J.; Blamey, R.W.; Robertson, J.F.R.; Nicholson, R.I.; Ellis, I.O. Expression of luminal and basal cytokeratins in human breast carcinoma. J. Pathol. 2004, 203, 661–671. [Google Scholar] [CrossRef]
- Livasy, C.A.; Karaca, G.; Nanda, R.; Tretiakova, M.S.; Olopade, O.I.; Moore, D.T.; Perou, C.M. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod. Pathol. 2006, 19, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Rycaj, K.; Tang, D.G. Cell-of-Origin of Cancer versus Cancer Stem Cells: Assays and Interpretations. Cancer Res. 2015, 75, 4003–4011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadjimichael, C.; Chanoumidou, K.; Papadopoulou, N.; Arampatzi, P.; Papamatheakis, J.; Kretsovali, A. Common stemness regulators of embryonic and cancer stem cells. World J. Stem Cells 2015, 7, 1150–1184. [Google Scholar] [CrossRef] [PubMed]
- Leis, O.; Eguiara, A.; Lopez-Arribillaga, E.; Alberdi, M.J.; Hernandez-Garcia, S.; Elorriaga, K.; Pandiella, A.; Rezola, R.; Martin, A.G. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene 2012, 31, 1354–1365. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Pinilla, S.M.; Sarrio, D.; Moreno-Bueno, G.; Rodriguez-Gil, Y.; Martinez, M.A.; Hernandez, L.; Hardisson, D.; Reis-Filho, J.S.; Palacios, J. Sox2: A possible driver of the basal-like phenotype in sporadic breast cancer. Mod. Pathol. 2007, 20, 474–481. [Google Scholar] [CrossRef]
- Liu, C.; Lu, Y.; Wang, B.; Zhang, Y.; Zhang, R.; Lu, Y.; Chen, B.; Xu, H.; Jin, F.; Lu, P. Clinical implications of stem cell gene Oct-4 expression in breast cancer. Ann. Surg. 2011, 253, 1165–1171. [Google Scholar] [CrossRef]
- Nagata, T.; Shimada, Y.; Sekine, S.; Hori, R.; Matsui, K.; Okumura, T.; Sawada, S.; Fukuoka, J.; Tsukada, K. Prognostic significance of NANOG and KLF4 for breast cancer. Breast Cancer 2014, 21, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Assi, M.; Dauguet, N.; Jacquemin, P. DIE-RNA: A Reproducible Strategy for the Digestion of Normal and Injured Pancreas, Isolation of Pancreatic Cells from Genetically Engineered Mouse Models and Extraction of High Quality RNA. Front. Physiol. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Liu, Y.; Deng, L.; Wang, D.; He, X.; Zhou, L.; Wicha, M.S.; Bai, F.; Liu, S. Transcriptional profiles of different states of cancer stem cells in triple-negative breast cancer. Mol. Cancer 2018, 17, 65. [Google Scholar] [CrossRef]
- Brown, K.A.; Aakre, M.E.; Gorska, A.E.; Price, J.O.; Eltom, S.E.; Pietenpol, J.A.; Moses, H.L. Induction by transforming growth factor-β1 of epithelial to mesenchymal transition is a rare event in vitro. Breast Cancer Res. 2004, 6, R215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldhirsch, A.; Ingle, J.N.; Gelber, R.D.; Coates, A.S.; Thürlimann, B.; Senn, H.J. Thresholds for therapies: Highlights of the St Gallen international expert consensus on the primary therapy of early breast cancer 2009. Ann. Oncol. 2009, 20, 1319–1329. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strietz, J.; Stepputtis, S.S.; Follo, M.; Bronsert, P.; Stickeler, E.; Maurer, J. Human Primary Breast Cancer Stem Cells Are Characterized by Epithelial-Mesenchymal Plasticity. Int. J. Mol. Sci. 2021, 22, 1808. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041808
Strietz J, Stepputtis SS, Follo M, Bronsert P, Stickeler E, Maurer J. Human Primary Breast Cancer Stem Cells Are Characterized by Epithelial-Mesenchymal Plasticity. International Journal of Molecular Sciences. 2021; 22(4):1808. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041808
Chicago/Turabian StyleStrietz, Juliane, Stella S. Stepputtis, Marie Follo, Peter Bronsert, Elmar Stickeler, and Jochen Maurer. 2021. "Human Primary Breast Cancer Stem Cells Are Characterized by Epithelial-Mesenchymal Plasticity" International Journal of Molecular Sciences 22, no. 4: 1808. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041808