
In Vitro Suppression of T Cell Proliferation Is a Conserved Function of Primary and Immortalized Human Cancer-Associated Fibroblasts

 and
and 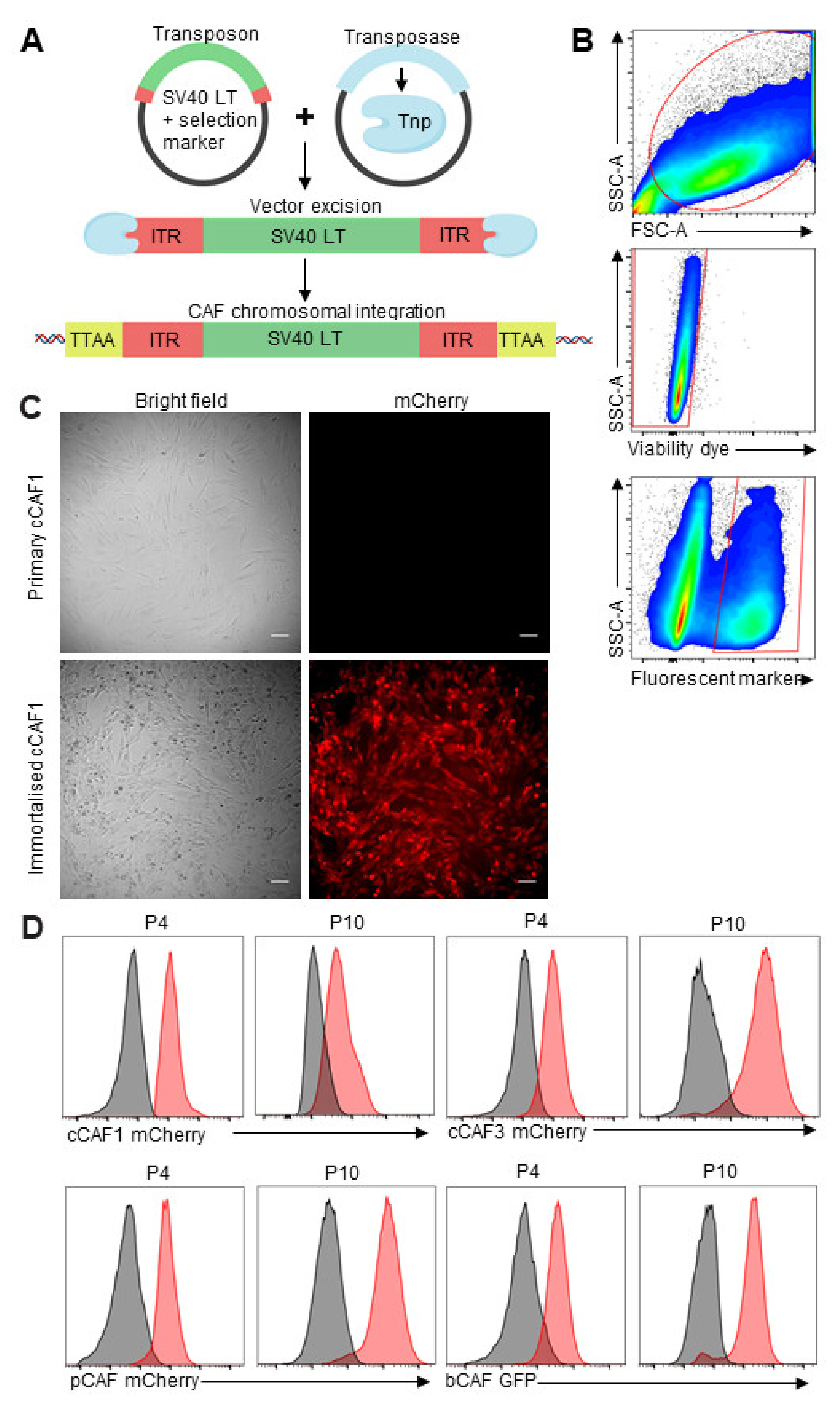{kind=link}
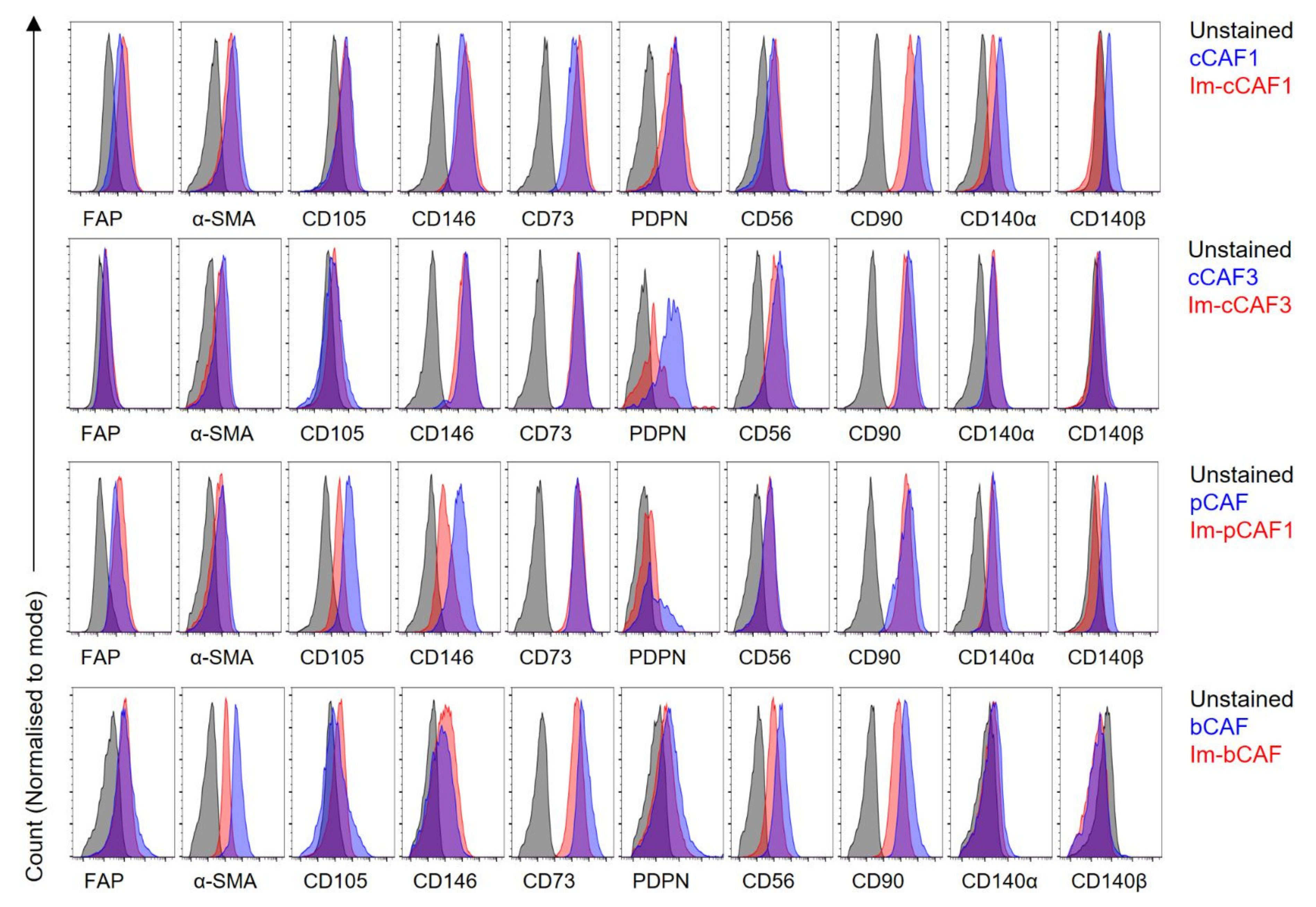{kind=link}
{kind=link}
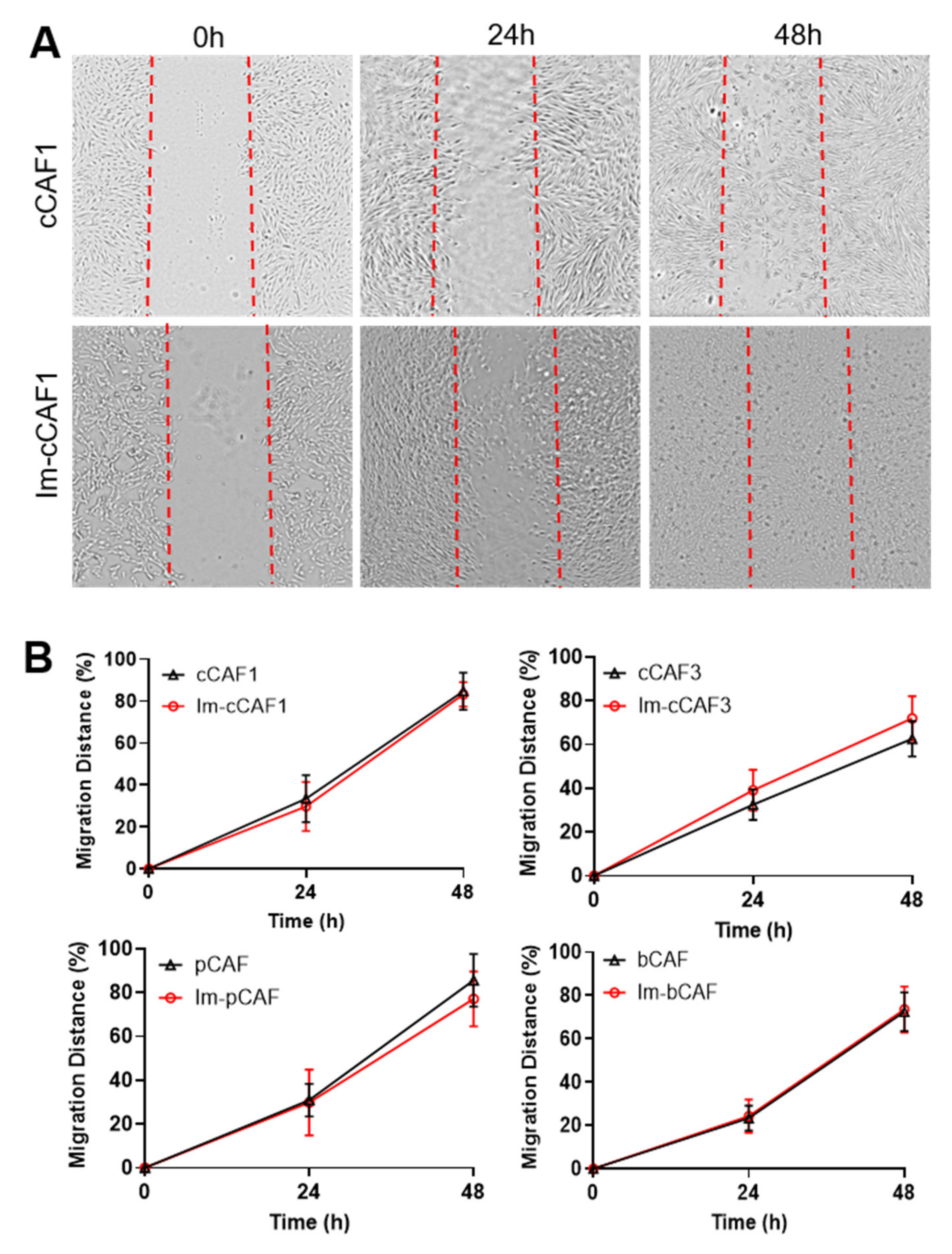{kind=link}
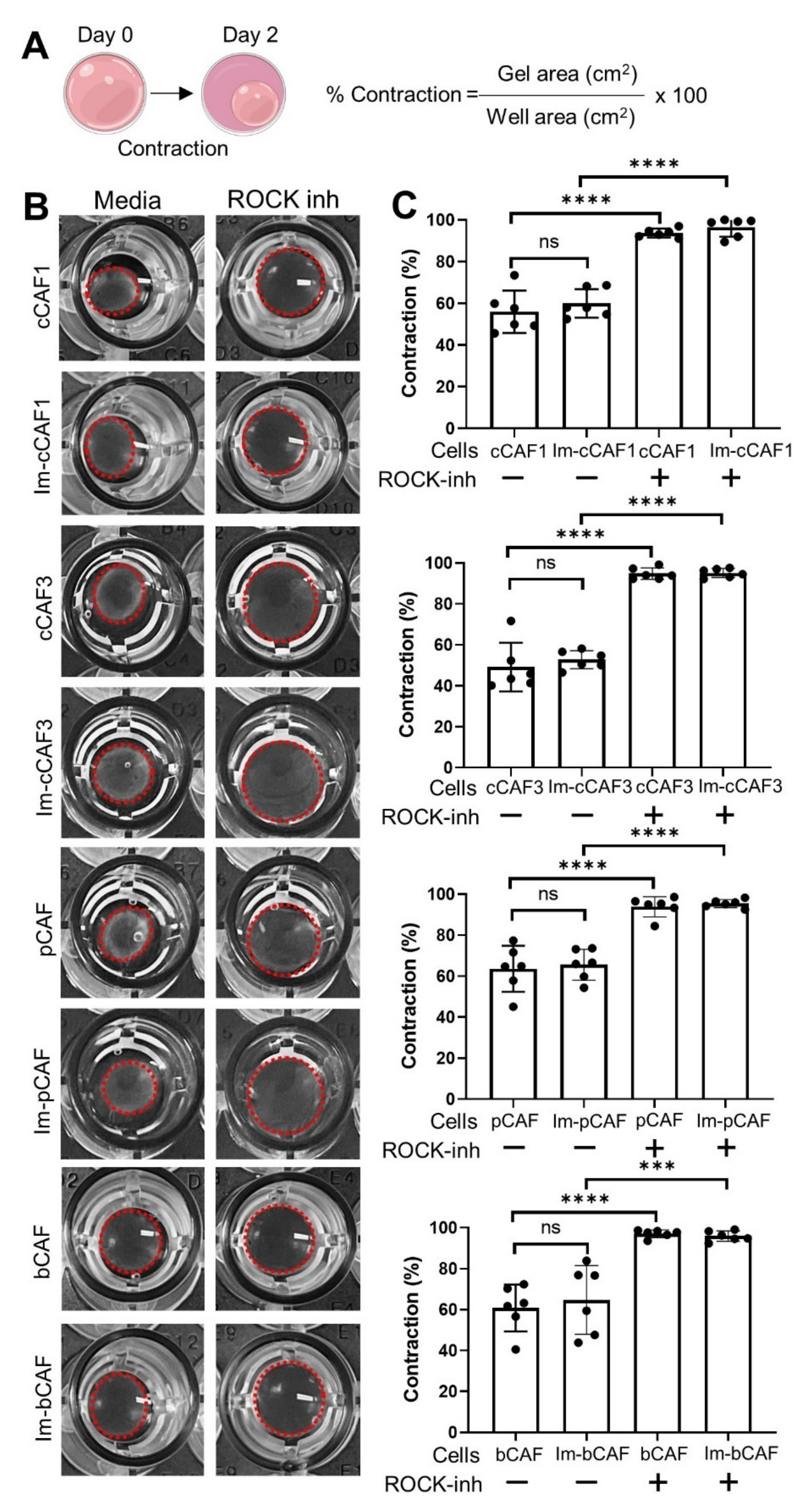{kind=link}
{kind=link}
Abstract
:1. Introduction
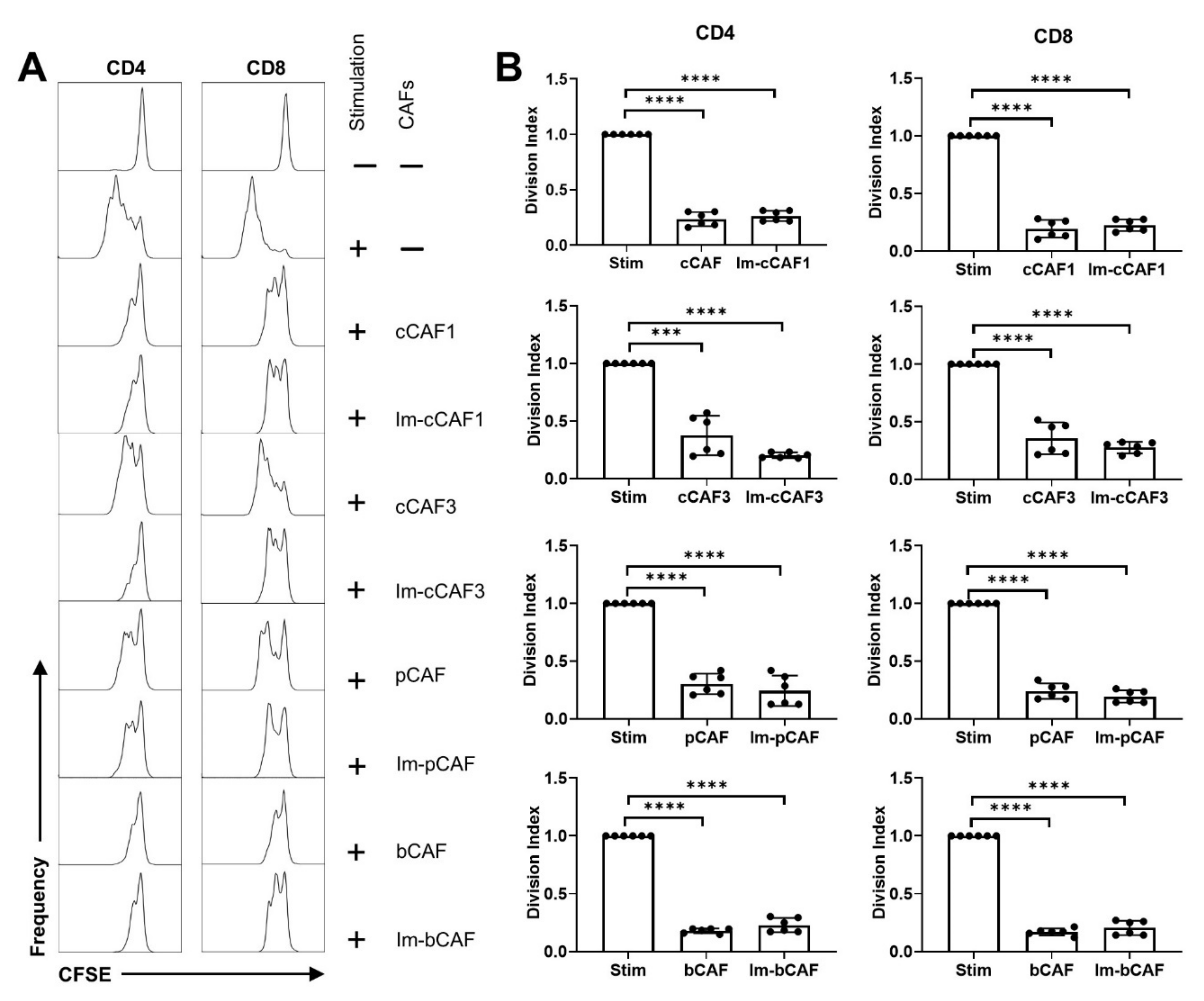
2. Results
3. Discussion
4. Materials and Methods
4.1. Primary Cells
4.2. Construction of PiggyBac Transposon Vectors
4.3. Establishment of Immortalized CAFs
4.4. Scratch Assay
4.5. T Cell Suppression Assay
4.6. Collagen Gel Contraction Assay
4.7. Cell Proliferation
4.8. Flow Cytometry
4.9. Statistical Analysi
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CAF | Cancer-associated fibroblast |
| Im-CAFs | Immortalized CAFs |
| CAR | Chimeric antigen receptor |
| bCAFs | Breast carcinoma CAFs |
| cCAFs | Colorectal carcinoma CAFs |
| pCAFs | Pancreatic adenocarcinoma CAFs |
| Im-bCAFs | Immortalized breast carcinoma CAFs |
| Im-cCAFs | Immortalized colorectal carcinoma CAFs |
| Im-pCAFs | Immortalized pancreatic adenocarcinoma CAFs |
| SV40LT | Simian virus 40 large T |
| IRES | Internal ribosome entry site |
| ITR | Inverted terminal repeat |
| FACS | Fluorescence Activated Cell Sorting |
| PDPN | Podoplanin |
| FAP | Fibroblast activation protein |
| αSMA | α-smooth muscle actin |
| CD146/MCAM | Melanoma cell adhesion molecule |
| CD105 | Endoglin |
| CD73 | Ecto-5′-nucleotidase |
| CD56/NCAM | Neural-cell adhesion molecule |
| CD140α | Platelet-derived growth factor receptor- α |
| CD140β | Platelet-derived growth factor receptor- β |
| CD90/Thy1 | Thymus cell antigen 1 |
| ROCK | Rho-associated, coiled-coil containing protein kinase |
| CFSE | Carboxyfluorescein succinimidyl ester |
| RB | retinoblastoma suppressor gene |
References
- Finn, O.J. A Believer’s Overview of Cancer Immunosurveillance and Immunotherapy. J. Immunol. 2018, 200, 385–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darvin, P.; Toor, S.M.; Nair, V.S.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.E.; Mackall, C.L. CAR T cell therapy: Inroads to response and resistance. Nat. Rev. Immunol. 2019, 19, 73–74. [Google Scholar] [CrossRef]
- Chen, L.; Qiu, X.; Wang, X.; He, J. FAP positive fibroblasts induce immune checkpoint blockade resistance in colorectal cancer via promoting immunosuppression. Biochem. Biophys. Res. Commun. 2017, 487, 8–14. [Google Scholar] [CrossRef]
- Su, S.; Chen, J.; Yao, H.; Liu, J.; Yu, S.; Lao, L.; Wang, M.; Luo, M.; Xing, Y.; Chen, F.; et al. CD10+GPR77+ Cancer-Associated Fibroblasts Promote Cancer Formation and Chemoresistance by Sustaining Cancer Stemness. Cell 2018, 172, 841–856.e16. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; DeNardo, D.G.; Affara, N.I.; Coussens, L.M. Lymphocytes in cancer development: Polarization towards pro-tumor immunity. Cytokine Growth Factor Rev. 2010, 21, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffell, B.; Coussens, L.M. Macrophages and Therapeutic Resistance in Cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Cha, Y.J.; Koo, J.S. Adipocyte biology in breast cancer: From silent bystander to active facilitator. Prog. Lipid Res. 2018, 69, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Schaaf, M.B.; Garg, A.D.; Agostinis, P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hida, K.; Maishi, N.; Annan, D.A.; Hida, Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 1272. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, A.L.; Okamoto, O.K. Combined Effects of Pericytes in the Tumor Microenvironment. Stem Cells Int. 2015, 2015, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Saci, A.; Szabo, P.M.; Chasalow, S.D.; Castillo-Martin, M.; Domingo-Domenech, J.; Siefker-Radtke, A.; Sharma, P.; Sfakianos, J.P.; Gong, Y.; et al. EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Aruga, N.; Kijima, H.; Masuda, R.; Onozawa, H.; Yoshizawa, T.; Tanaka, M.; Inokuchi, S.; Iwazaki, M. Epithelial-mesenchymal Transition (EMT) is Correlated with Patient’s Prognosis of Lung Squamous Cell Carcinoma. Tokai J. Exp. Clin. Med. 2018, 43, 5–13. [Google Scholar]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.F.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Surowiak, P.; Murawa, D.; Materna, V.; Maciejczyk, A.; Pudelko, M.; Ciesla, S.; Breborowicz, J.; Murawa, P.; Zabel, M.; Dietel, M.; et al. Occurence of stromal myofibroblasts in the invasive ductal breast cancer tissue is an unfavourable prognostic factor. Anticancer Res. 2007, 27, 2917–2924. [Google Scholar]
- Marsh, D.; Suchak, K.; Moutasim, K.A.; Vallath, S.; Hopper, C.; Jerjes, W.; Upile, T.; Kalavrezos, N.; Violette, S.M.; Weinreb, P.H.; et al. Stromal features are predictive of disease mortality in oral cancer patients. J. Pathol. 2011, 223, 470–481. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, J.; Cukierman, E. Stromal dynamic reciprocity in cancer: Intricacies of fibroblastic-ECM interactions. Curr. Opin. Cell Biol. 2016, 42, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel III, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Wu, S.Z.; Roden, D.L.; Wang, C.; Holliday, H.; Harvey, K.; Cazet, A.S.; Murphy, K.J.; Pereira, B.; Al-Eryani, G.; Bartonicek, N.; et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. EMBO J. 2020, 39, 104063. [Google Scholar] [CrossRef]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated Pancreatic Stellate Cells Sequester CD8+ T Cells to Reduce Their Infiltration of the Juxtatumoral Compartment of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef] [Green Version]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018, 33, 463–479. [Google Scholar] [CrossRef] [Green Version]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Hanley, C.J.; Noble, F.; Ward, M.; Bullock, M.; Drifka, C.; Mellone, M.; Manousopoulou, A.; Johnston, H.E.; Hayden, A.; Thirdborough, S.; et al. A subset of myofibroblastic cancer-associated fibroblasts regulate collagen fiber elongation, which is prognostic in multiple cancers. Oncotarget 2015, 7, 6159–6174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Yu, D.-H.; Chen, Y.; Zhao, C.-Y.; Zhang, J.; Liu, Q.-H.; Ni, C.-R.; Zhu, M.-H. Expression of fibroblast activation protein in human pancreatic adenocarcinoma and its clinicopathological significance. World J. Gastroenterol. 2012, 18, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-O.; Mullins, S.R.; Franco-Barraza, J.; Valianou, M.; Cukierman, E.; Cheng, J.D. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer 2011, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Tejada, M.L.; Yu, L.; Dong, J.; Jung, K.; Meng, G.; Peale, F.V.; Frantz, G.D.; Hall, L.; Liang, X.; Gerber, H.-P.; et al. Tumor-Driven Paracrine Platelet-Derived Growth Factor Receptor α Signaling Is a Key Determinant of Stromal Cell Recruitment in a Model of Human Lung Carcinoma. Clin. Cancer Res. 2006, 12, 2676–2688. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, K.; Valyi-Nagy, I.; Heldin, C.H.; Herlyn, M.; Westermark, B. Platelet-derived growth factor (PDGF) in oncogenesis: Development of a vascular connective tissue stroma in xenotransplanted human melanoma producing PDGF-BB. Proc. Natl. Acad. Sci. USA 1993, 90, 393–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Enomoto, A.; Woods, S.L.; Burt, A.D.; Takahashi, M.; Worthley, D.L. Cancer-associated fibroblasts in gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 282–295. [Google Scholar] [CrossRef]
- Bork, S.; Pfister, S.; Witt, H.; Horn, P.; Korn, B.; Ho, A.D.; Wagner, W. DNA methylation pattern changes upon long-term culture and aging of human mesenchymal stromal cells. Aging Cell 2010, 9, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Wagner, W. Implications of long-term culture for mesenchymal stem cells: Genetic defects or epigenetic regulation? Stem Cell Res. Ther. 2012, 3, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Davalos, A.R.; Coppe, J.-P.; Campisi, J.; Desprez, P.-Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010, 29, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Wu, X.; Li, G.; Han, M.; Zhuang, Y.; Xu, T. Efficient Transposition of the piggyBac (PB) Transposon in Mammalian Cells and Mice. Cell 2005, 122, 473–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Jiang, E.; Chen, S.; Gu, Y.; Shangguan, A.J.; Lv, T.; Luo, L.; Yu, Z. PiggyBac transposon vectors: The tools of the human gene encoding. Transl. Lung Cancer Res. 2016, 5, 120–125. [Google Scholar] [PubMed]
- Henke, A.; Franco, O.E.; Stewart, G.D.; Riddick, A.C.; Katz, E.; Hayward, S.W.; Thomson, A.A. Reduced Contractility and Motility of Prostatic Cancer-Associated Fibroblasts after Inhibition of Heat Shock Protein 90. Cancers 2016, 8, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Cremasco, V.; Astarita, J.L.; Grauel, A.L.; Keerthivasan, S.; MacIsaac, K.D.; Woodruff, M.C.; Wu, M.; Spel, L.; Santoro, S.; Amoozgar, Z.; et al. FAP Delineates Heterogeneous and Functionally Divergent Stromal Cells in Immune-Excluded Breast Tumors. Cancer Immunol. Res. 2018, 6, 1472–1485. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.-C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, T.M.N.W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Érsek, B.; Silló, P.; Cakir, U.; Molnár, V.; Bencsik, A.; Mayer, B.; Mezey, E.; Kárpáti, S.; Pós, Z.; Németh, K. Melanoma-associated fibroblasts impair CD8+ T cell function and modify expression of immune checkpoint regulators via increased arginase activity. Cell. Mol. Life Sci. 2020, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goehrig, D.; Nigri, J.; Samain, R.; Wu, Z.; Cappello, P.; Gabiane, G.; Zhang, X.; Zhao, Y.; Kim, I.-S.; Chanal, M.; et al. Stromal protein βig-h3 reprogrammes tumour microenvironment in pancreatic cancer. Gut 2019, 68, 693–707. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.H.; Coates, C.J.; George, A.L. PiggyBac Transposon-mediated Gene Transfer in Human Cells. Mol. Ther. 2007, 15, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Inada, E.; Saitoh, I.; Matsumoto, Y.; Ohtsuka, M.; Miura, H.; Nakamura, S.; Sakurai, T.; Watanabe, S. A combination of targeted toxin technology and the piggyBac-mediated gene transfer system enables efficient isolation of stable transfectants in nonhuman mammalian cells. Biotechnol. J. 2014, 10, 143–153. [Google Scholar] [CrossRef]
- Sullivan, C.S.; Grundhoff, A.T.; Tevethia, S.; Pipas, J.M.; Ganem, D. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nat. Cell Biol. 2005, 435, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Umehara, K.; Sun, Y.; Hiura, S.; Hamada, K.; Itoh, M.; Kitamura, K.; Oshima, M.; Iwama, A.; Saito, K.; Anzai, N.; et al. A New Conditionally Immortalized Human Fetal Brain Pericyte Cell Line: Establishment and Functional Characterization as a Promising Tool for Human Brain Pericyte Studies. Mol. Neurobiol. 2017, 55, 5993–6006. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Yoshida, N.; Takanashi, M.; Ito, Y.; Fukami, K.; Yanagihara, K.; Yashiro, M.; Sakai, R. Stromal Fibroblasts Mediate Extracellular Matrix Remodeling and Invasion of Scirrhous Gastric Carcinoma Cells. PLoS ONE 2014, 9, e85485. [Google Scholar] [CrossRef] [Green Version]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 1–19. [Google Scholar] [CrossRef]
- Patel, A.K.; Vipparthi, K.; Thatikonda, V.; Arun, I.; Bhattacharjee, S.; Sharan, R.; Arun, P.; Singh, S. A subtype of cancer-associated fibroblasts with lower expression of alpha-smooth muscle actin suppresses stemness through BMP4 in oral carcinoma. Oncogenesis 2018, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ridge, K.M.; Shumaker, D.; Robert, A.; Hookway, C.; Gelfand, V.I.; Janmey, P.A.; Lowery, J.; Guo, M.; Weitz, D.A.; Kuczmarski, E.; et al. Methods for Determining the Cellular Functions of Vimentin Intermediate Filaments. Methods Enzymol. 2016, 568, 389–426. [Google Scholar]
- O’Malley, G.; Treacy, O.; Lynch, K.; Naicker, S.D.; Leonard, N.A.; Lohan, P.; Dunne, P.D.; Ritter, T.; Egan, L.J.; Ryan, A.E. Stromal Cell PD-L1 Inhibits CD8(+) T-cell Antitumor Immune Responses and Promotes Colon Cancer. Cancer Immunol. Res. 2018, 6, 1426–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorchs, L.; Moro, C.F.; Bankhead, P.; Kern, K.P.; Sadeak, I.; Meng, Q.; Rangelova, E.; Kaipe, H. Human Pancreatic Carcinoma-Associated Fibroblasts Promote Expression of Co-inhibitory Markers on CD4+ and CD8+ T-Cells. Front. Immunol. 2019, 10, 847. [Google Scholar] [CrossRef]
- Fletcher, A.L.; Lukacs-Kornek, V.; Reynoso, E.D.; Pinner, S.E.; Bellemare-Pelletier, A.; Curry, M.S.; Collier, A.-R.; Boyd, R.L.; Turley, S.J. Lymph node fibroblastic reticular cells directly present peripheral tissue antigen under steady-state and inflammatory conditions. J. Exp. Med. 2010, 207, 689–697. [Google Scholar] [CrossRef]
- Dubrot, J.; Duraes, F.D.V.; Potin, L.; Capotosti, F.; Brighouse, D.; Suter, T.; LeibundGut-Landmann, S.; Garbi, N.; Reith, W.; Swartz, M.A.; et al. Lymph node stromal cells acquire peptide-MHCII complexes from dendritic cells and induce antigen-specific CD4+ T cell tolerance. J. Exp. Med. 2014, 211, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Knoblich, K.; Migoni, S.C.; Siew, S.M.; Jinks, E.; Kaul, B.; Jeffery, H.C.; Baker, A.T.; Suliman, M.; Vrzalikova, K.; Mehenna, H.; et al. The human lymph node microenvironment unilaterally regulates T-cell activation and differentiation. PLoS Biol. 2018, 16, e2005046. [Google Scholar] [CrossRef]
- Schaeuble, K.; Cannelle, H.; Favre, S.; Huang, H.-Y.; Oberle, S.G.; Speiser, D.E.; Zehn, D.; Luther, S.A. Attenuation of chronic antiviral T-cell responses through constitutive COX2-dependent prostanoid synthesis by lymph node fibroblasts. PLoS Biol. 2019, 17, e3000072. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, A.L.; Acton, S.E.; Knoblich, K. Lymph node fibroblastic reticular cells in health and disease. Nat. Rev. Immunol. 2015, 15, 350–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohr, J.; Ratliff, T.; Huppertz, A.; Ge, Y.; Dictus, C.; Ahmadi, R.; Grau, S.; Hiraoka, N.; Eckstein, V.; Ecker, R.C.; et al. Effector T-Cell Infiltration Positively Impacts Survival of Glioblastoma Patients and Is Impaired by Tumor-Derived TGF-β. Clin. Cancer Res. 2011, 17, 4296–4308. [Google Scholar] [CrossRef] [Green Version]
- Fukunaga, A.; Miyamoto, M.; Cho, Y.; Murakami, S.; Kawarada, Y.; Oshikiri, T.; Kato, K.; Kurokawa, T.; Suzuoki, M.; Nakakubo, Y.; et al. CD8+ Tumor-Infiltrating Lymphocytes Together with CD4+ Tumor-Infiltrating Lymphocytes and Dendritic Cells Improve the Prognosis of Patients with Pancreatic Adenocarcinoma. Pancreas 2004, 28, e26–e31. [Google Scholar] [CrossRef]
- Narayanan, S.; Kawaguchi, T.; Yan, L.; Peng, X.; Qi, Q.; Takabe, K. Cytolytic Activity Score to Assess Anticancer Immunity in Colorectal Cancer. Ann. Surg. Oncol. 2018, 25, 2323–2331. [Google Scholar] [CrossRef]
- Carstens, J.L.; De Sampaio, P.C.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Sheih, A.; Voillet, V.; Hanafi, L.-A.; DeBerg, H.A.; Yajima, M.; Hawkins, R.; Gersuk, V.; Riddell, S.R.; Maloney, D.G.; Wohlfahrt, M.E.; et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lakins, M.A.; Ghorani, E.; Munir, H.; Martins, C.P.; Shields, J.D. Cancer-associated fibroblasts induce antigen-specific deletion of CD8+ T Cells to protect tumour cells. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Fox, A.; Harland, K.L.; Kedzierska, K.; Kelso, A. Exposure of Human CD8+ T Cells to Type-2 Cytokines Impairs Division and Differentiation and Induces Limited Polarization. Front. Immunol. 2018, 9, 1141. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Luo, Y.; Markowitz, D.; Xiang, R.; Reisfeld, R.A. Cancer Associated Fibroblasts Promote Tumor Growth and Metastasis by Modulating the Tumor Immune Microenvironment in a 4T1 Murine Breast Cancer Model. PLoS ONE 2009, 4, e7965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, T.; Noma, K.; Ohara, T.; Kashima, H.; Katsura, Y.; Sato, H.; Komoto, S.; Katsube, R.; Ninomiya, T.; Tazawa, H.; et al. Cancer-Associated Fibroblasts Affect Intratumoral CD8+ and FoxP3+ T Cells Via IL6 in the Tumor Microenvironment. Clin. Cancer Res. 2018, 24, 4820–4833. [Google Scholar] [CrossRef] [Green Version]
- Zelenay, S.; Van Der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [Green Version]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGF drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.-C.; Park, A.Y.; Guan, J.-L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäcker, V. ImageJ Macro Toolsets for Biological Image Analysis. In Proceedings of the ImageJ User and Developer Conference 2012, Luxembourg, 24–26 October 2012. [Google Scholar]
- Quah, B.J.C.; Warren, H.S.; Parish, C.R. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat. Protoc. 2007, 2, 2049–2056. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abuwarwar, M.H.; Baker, A.T.; Harding, J.; Payne, N.L.; Nagy, A.; Knoblich, K.; Fletcher, A.L. In Vitro Suppression of T Cell Proliferation Is a Conserved Function of Primary and Immortalized Human Cancer-Associated Fibroblasts. Int. J. Mol. Sci. 2021, 22, 1827. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041827
Abuwarwar MH, Baker AT, Harding J, Payne NL, Nagy A, Knoblich K, Fletcher AL. In Vitro Suppression of T Cell Proliferation Is a Conserved Function of Primary and Immortalized Human Cancer-Associated Fibroblasts. International Journal of Molecular Sciences. 2021; 22(4):1827. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041827
Chicago/Turabian StyleAbuwarwar, Mohammed H., Alfie T. Baker, Jeffrey Harding, Natalie L. Payne, Andras Nagy, Konstantin Knoblich, and Anne L. Fletcher. 2021. "In Vitro Suppression of T Cell Proliferation Is a Conserved Function of Primary and Immortalized Human Cancer-Associated Fibroblasts" International Journal of Molecular Sciences 22, no. 4: 1827. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041827