
The Alter Retina: Alternative Splicing of Retinal Genes in Health and Disease


Abstract
:1. Introduction
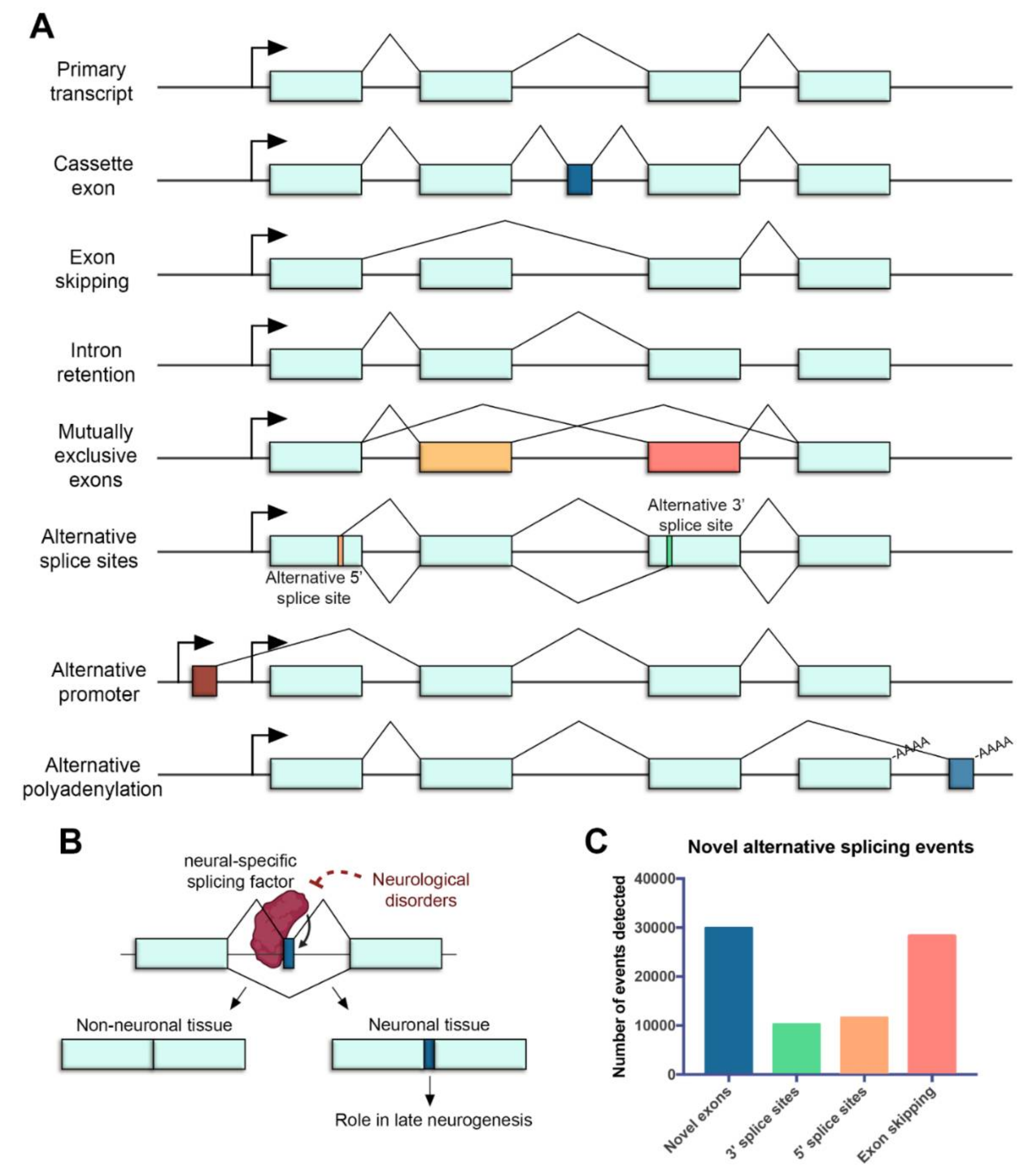
2. Alternative Splicing of Retinal Genes
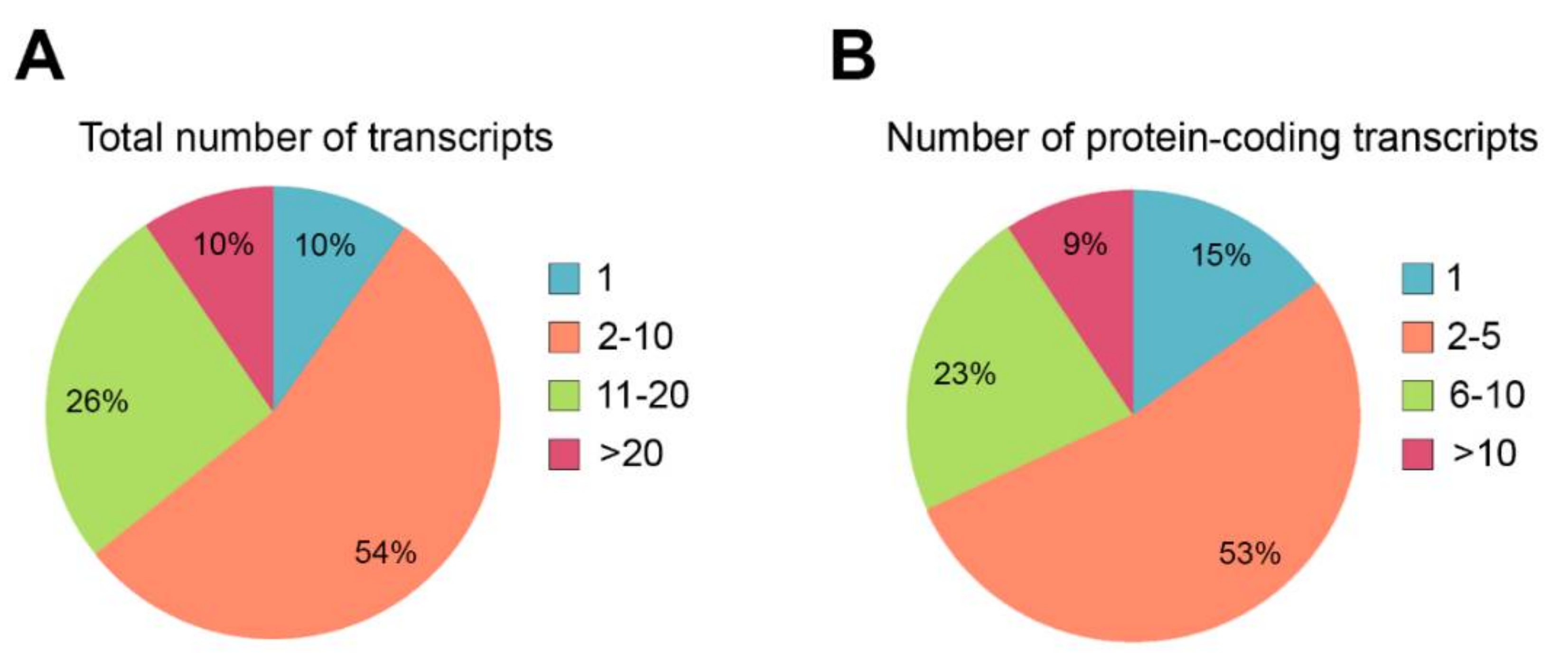
2.1. Alternative Splicing in the Retina
2.2. Novel Approaches to Detect Alternative Splicing Events
2.3. Implications of Retinal Alternative Splicing in Functional Analyses of IRD Genes
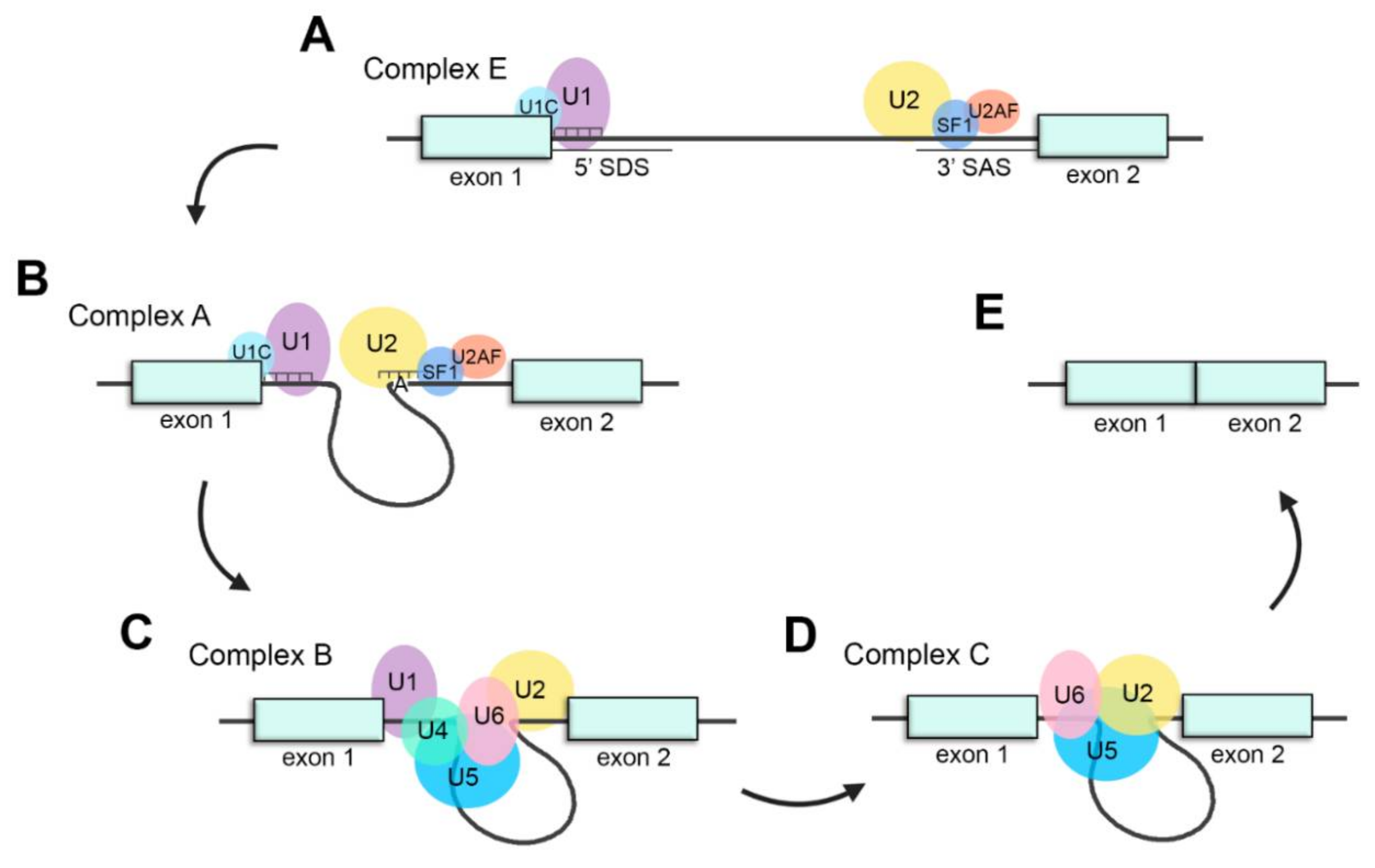
2.4. Splicing Factors Involved in the Processing of Retinal Transcripts
2.5. Regulation of the Splicing
2.5.1. Trans-Regulatory Elements: RNA-Binding Proteins
2.5.2. Cis-Regulatory Elements: Enhancers and Silencers
3. The Role of Alternative Splicing in Retinal Disease
3.1. Trans-Acting Mutations in Splicing Factors: PRPF31
3.2. Cis-Acting Mutations Altering the Splicing
3.2.1. Non-Canonical Splice Site Variants (NCSS): ABCA4
3.2.2. Deep Intronic Variants: ABCA4, CEP290 and USH2A
3.2.3. Deep Exonic Variants: RHO
3.3. Mutations in Retina-Specific Exons and Microexons: BBS8, RPGR and DYNC2H1
4. Therapeutic Strategies to Modulate Aberrant Splicing
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patrushev, L.I.; Kovalenko, T.F. Functions of noncoding sequences in mammalian genomes. Biochemistry 2014, 79, 1442–1469. [Google Scholar] [CrossRef]
- Lee, H.; Zhang, Z.; Krause, H.M. Long Noncoding RNAs and Repetitive Elements: Junk or Intimate Evolutionary Partners? Trends Genet. 2019, 35, 892–902. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Gonzàlez-Porta, M.; Santos, S.; Brazma, A.; Marioni, J.C.; Aebersold, R.; Venkitaraman, A.R.; Wickramasinghe, V.O. Impact of Alternative Splicing on the Human Proteome. Cell Rep. 2017, 20, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Vaquero-Garcia, J.; Barrera, A.; Gazzara, M.R.; Gonzalez-Vallinas, J.; Lahens, N.F.; Hogenesch, J.B.; Lynch, K.W.; Barash, Y. A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife 2016, 5. [Google Scholar] [CrossRef]
- Lee, Y.; Rio, D.C. Mechanisms and regulation of alternative Pre-mRNA splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S. SR proteins: Binders, regulators, and connectors of RNA. Mol. Cells 2017, 40, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Tellier, M.; Maudlin, I.; Murphy, S. Transcription and splicing: A two-way street. Wiley Interdiscip. Rev. RNA 2020, 11. [Google Scholar] [CrossRef]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Merkin, J.; Russell, C.; Chen, P.; Burge, C.B. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science 2012, 338, 1593–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Kim, H.K.; Pham, M.H.C.; Ko, K.S.; Rhee, B.D.; Han, J. Alternative splicing isoforms in health and disease. Pflugers Arch. Eur. J. Physiol. 2018, 470, 995–1016. [Google Scholar] [CrossRef]
- Furlanis, E.; Scheiffele, P. Regulation of Neuronal Differentiation, Function, and Plasticity by Alternative Splicing. Annu. Rev. Cell Dev. Biol. 2018, 34, 451–469. [Google Scholar] [CrossRef]
- Hermey, G.; Blüthgen, N.; Kuhl, D. Neuronal activity-regulated alternative mRNA splicing. Int. J. Biochem. Cell Biol. 2017, 91, 184–193. [Google Scholar] [CrossRef]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Bergsma, A.J.; van der Wal, E.; Broeders, M.; van der Ploeg, A.T.; Pim Pijnappel, W.W.M. Alternative Splicing in Genetic Diseases: Improved Diagnosis and Novel Treatment Options. Int. Rev. Cell Mol. Biol. 2018, 335, 85–141. [Google Scholar] [PubMed]
- Sterne-Weiler, T.; Howard, J.; Mort, M.; Cooper, D.N.; Sanford, J.R. Loss of exon identity is a common mechanism of human inherited disease. Genome Res. 2011, 21, 1563–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Wan, J.; Masuda, T.; Hackler, L.; Torres, K.M.; Merbs, S.L.; Zack, D.J.; Qian, J. Dynamic usage of alternative splicing exons during mouse retina development. Nucleic Acids Res. 2011, 39, 7920–7930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, H.K.W.; Lin, M.Y.; Pandey, S. Exon-skipping variant of RGR opsin in human retina and pigment epithelium. Exp. Eye Res. 2006, 83, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Aísa-Marín, I.; López-Iniesta, M.J.; Milla, S.; Lillo, J.; Navarro, G.; de la Villa, P.; Marfany, G. Nr2e3 functional domain ablation by CRISPR-Cas9D10Aidentifies a new isoform and generates retinitis pigmentosa and enhanced S-cone syndrome models. Neurobiol. Dis. 2020, 146. [Google Scholar] [CrossRef]
- Mellough, C.B.; Bauer, R.; Collin, J.; Dorgau, B.; Zerti, D.; Dolan, D.W.P.; Jones, C.M.; Izuogu, O.G.; Yu, M.; Hallam, D.; et al. An integrated transcriptional analysis of the developing human retina. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stojic, J.; Stöhr, H.; Weber, B.H.F. Three novel ABCC5 splice variants in human retina and their role as regulators of ABCC5 gene expression. BMC Mol. Biol. 2007, 8. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Swaminathan, A.; Zhang, X.; Hughes, B.A. Expression of Kir7.1 and a novel Kir7.1 splice variant in native human retinal pigment epithelium. Exp. Eye Res. 2008, 86, 81–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campla, C.K.; Mast, H.; Dong, L.; Lei, J.; Halford, S.; Sekaran, S.; Swaroop, A. Targeted deletion of an NRL- and CRX-regulated alternative promoter specifically silences FERM and PDZ domain containing 1 (Frmpd1) in rod photoreceptors. Hum. Mol. Genet. 2019, 28, 804–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, H.; Tummala, P.; Guzman, E.; Mali, R.S.; Gregorski, J.; Swaroop, A.; Mitton, K.P. The transcription factor Neural Retina Leucine Zipper (NRL) controls photoreceptor-specific expression of myocyte enhancer factor Mef2c from an alternative promoter. J. Biol. Chem. 2011, 286, 34893–34902. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Li, S.; Park, J.Y.; Boppana, S.; Ni, T.; Li, M.; Zhu, J.; Xie, Z.; Xiang, M. Dynamic landscape of alternative polyadenylation during retinal development HHS Public Access. Cell Mol. Life Sci. 2017, 74, 1721–1739. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Chen, L.L. Microexons go big. Cell 2014, 159, 1488–1489. [Google Scholar] [CrossRef] [Green Version]
- Irimia, M.; Weatheritt, R.J.; Ellis, J.D.; Parikshak, N.N.; Gonatopoulos-Pournatzis, T.; Babor, M.; Quesnel-Vallières, M.; Tapial, J.; Raj, B.; O’Hanlon, D.; et al. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 2014, 159, 1511–1523. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.I.; Sanchez-Pulido, L.; Haerty, W.; Ponting, C.P. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res. 2015, 25, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Capponi, S.; Stöffler, N.; Irimia, M.; Van Schaik, F.M.A.; Ondik, M.M.; Biniossek, M.L.; Lehmann, L.; Mitschke, J.; Vermunt, M.W.; Creyghton, M.P.; et al. Neuronal-specific microexon splicing of TAF1 mRNA is directly regulated by SRRM4/nSR100. RNA Biol. 2020, 17, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Gonatopoulos-Pournatzis, T.; Blencowe, B.J. Microexons: At the nexus of nervous system development, behaviour and autism spectrum disorder. Curr. Opin. Genet. Dev. 2020, 65, 22–33. [Google Scholar] [CrossRef]
- Porter, R.S.; Jaamour, F.; Iwase, S. Neuron-specific alternative splicing of transcriptional machineries: Implications for neurodevelopmental disorders. Mol. Cell. Neurosci. 2018, 87, 35–45. [Google Scholar] [CrossRef]
- da Costa, P.J.; Menezes, J.; Romão, L. The role of alternative splicing coupled to nonsense-mediated mRNA decay in human disease. Int. J. Biochem. Cell Biol. 2017, 91, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Farkas, M.H.; Grant, G.R.; White, J.A.; Sousa, M.E.; Consugar, M.B.; Pierce, E.A. Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 Mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [Green Version]
- Aragón, J.; González-Reyes, M.; Romo-Yáñez, J.; Vacca, O.; Aguilar-González, G.; Rendón, A.; Vaillend, C.; Montañez, C. Dystrophin Dp71 Isoforms Are Differentially Expressed in the Mouse Brain and Retina: Report of New Alternative Splicing and a Novel Nomenclature for Dp71 Isoforms. Mol. Neurobiol. 2018, 55, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Whitney, I.E.; Kautzman, A.G.; Reese, B.E. Alternative splicing of the LIM-homeodomain transcription factor Isl1 in the mouse retina. Mol. Cell. Neurosci. 2015, 65, 102–113. [Google Scholar] [CrossRef] [Green Version]
- Kole, C.; Berdugo, N.; Silva, C.D.; Aït-Ali, N.; Millet-Puel, G.; Pagan, D.; Blond, F.; Poidevin, L.; Ripp, R.; Fontaine, V.; et al. Identification of an alternative splicing product of the OtX2 gene expressed in the neural retina and retinal pigmented epithelial cells. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayega, A.; Wang, Y.C.; Oikonomopoulos, S.; Djambazian, H.; Fahiminiya, S.; Ragoussis, J. Transcript Profiling Using Long-Read Sequencing Technologies. Methods Mol. Biol. 2018, 1783, 121–147. [Google Scholar]
- Midha, M.K.; Wu, M.; Chiu, K.P. Long-read sequencing in deciphering human genetics to a greater depth. Hum. Genet. 2019, 138, 1201–1215. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the potential of full-length transcriptome sequencing. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, T.A.; Cochran, K.; Kozlowski, C.; Wang, J.; Alexander, G.; Cady, M.A.; Spencer, W.J.; Ruzycki, P.A.; Clark, B.S.; Laeremans, A.; et al. Comprehensive identification of mRNA isoforms reveals the diversity of neural cell-surface molecules with roles in retinal development and disease. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Quinn, P.M.; Pellissier, L.P.; Wijnholds, J. The CRB1 complex: Following the trail of crumbs to a feasible gene therapy strategy. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef]
- van de Pavert, S.A.; Kantardzhieva, A.; Malysheva, A.; Meuleman, J.; Versteeg, I.; Levelt, C.; Klooster, J.; Geiger, S.; Seeliger, M.W.; Rashbass, P.; et al. Crumbs homologue 1 is required for maintenance of photoreceptor cell polarization and adhesion during light exposure. J. Cell Sci. 2004, 117, 4169–4177. [Google Scholar] [CrossRef] [Green Version]
- Garanto, A.; Riera, M.; Pomares, E.; Permanyer, J.; de Castro-Miró, M.; Sava, F.; Abril, J.F.; Marfany, G.; Gonzàlez-Duarte, R. High transcriptional complexity of the retinitis pigmentosa CERKL gene in human and mouse. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5202–5214. [Google Scholar] [CrossRef] [Green Version]
- Garanto, A.; Vicente-Tejedor, J.; Riera, M.; De la Villa, P.; Gonzàlez-Duarte, R.; Blanco, R.; Marfany, G. Targeted knockdown of Cerkl, a retinal dystrophy gene, causes mild affectation of the retinal ganglion cell layer. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1258–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domènech, E.B.; Andres, R.; López-Iniesta, M.J.; Mirra, S.; Arroyo, R.G.; Milla, S.; Sava, F.; Andilla, J.; Alvarez, P.L.; De La Villa, P.; et al. A New cerkl mouse model generated by crispr-cas9 shows progressive retinal degeneration and altered morphological and electrophysiological phenotype. Investig. Ophthalmol. Vis. Sci. 2020, 61, 14. [Google Scholar] [CrossRef]
- Wilkinson, M.E.; Charenton, C.; Nagai, K. RNA Splicing by the Spliceosome. Annu. Rev. Biochem. 2020, 89, 359–388. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Oubridge, C.; van Roon, A.M.M.; Nagai, K. Crystal structure of human U1 snRNP, a small nuclear ribonucleoprotein particle, reveals the mechanism of 5′ splice site recognition. Elife 2015, 4. [Google Scholar] [CrossRef]
- Sickmier, E.A.; Frato, K.E.; Shen, H.; Paranawithana, S.R.; Green, M.R.; Kielkopf, C.L. Structural Basis for Polypyrimidine Tract Recognition by the Essential Pre-mRNA Splicing Factor U2AF65. Mol. Cell 2006, 23, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Manley, J.L. Mammalian pre-mRNA branch site selection by U2 snRNP involves base pairing. Genes Dev. 1989, 3, 1553–1561. [Google Scholar] [CrossRef] [Green Version]
- Sterner, D.A.; Carlo, T.; Berget, S.M. Architectural limits on split genes. Proc. Natl. Acad. Sci. USA 1996, 93, 15081–15085. [Google Scholar] [CrossRef] [Green Version]
- Wan, R.; Yan, C.; Bai, R.; Wang, L.; Huang, M.; Wong, C.C.L.; Shi, Y. The 3.8 Å structure of the U4/U6.U5 tri-snRNP: Insights into spliceosome assembly and catalysis. Science 2016, 351, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Raghunathan, P.L.; Guthrie, C. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr. Biol. 1998, 8, 847–855. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.S.; Manley, J.L. A novel U2-U6 snRNA structure is necessary for mammalian mRNA splicing. Genes Dev. 1995, 9, 843–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourmann, J.B.; Schmitzová, J.; Christian, H.; Urlaub, H.; Ficner, R.; Boon, K.L.; Fabrizio, P.; Lührmann, R. Dissection of the factor requirements for spliceosome disassembly and the elucidation of its dissociation products using a purified splicing system. Genes Dev. 2013, 27, 413–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Růžičková, Š.; Staněk, D. Mutations in spliceosomal proteins and retina degeneration. RNA Biol. 2017, 14, 544–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Wan, R.; Bai, R.; Huang, G.; Shi, Y. Structure of a yeast activated spliceosome at 3.5 Å resolution. Science 2016, 353, 904–912. [Google Scholar] [CrossRef]
- Xu, M.; Xie, Y.; Abouzeid, H.; Gordon, C.T.; Fiorentino, A.; Sun, Z.; Lehman, A.; Osman, I.S.; Dharmat, R.; Riveiro-Alvarez, R.; et al. Mutations in the Spliceosome Component CWC27 Cause Retinal Degeneration with or without Additional Developmental Anomalies. Am. J. Hum. Genet. 2017, 100, 592–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef] [Green Version]
- Shenasa, H.; Hertel, K.J. Combinatorial regulation of alternative splicing. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862. [Google Scholar] [CrossRef]
- Murphy, D.; Cieply, B.; Carstens, R.; Ramamurthy, V.; Stoilov, P. The Musashi 1 Controls the Splicing of Photoreceptor-Specific Exons in the Vertebrate Retina. PLoS Genet. 2016, 12. [Google Scholar] [CrossRef] [PubMed]
- Qi, X. The role of miR-9 during neuron differentiation of mouse retinal stem cells. Artif. Cells Nanomed. Biotechnol. 2016, 44, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Bok, D.; Yu, F.; Caprioli, J.; Piri, N. Downregulation of splicing regulator RBFOX1 compromises visual depth perception. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Lin, Y.S.; Kuo, K.T.; Chen, S.K.; Huang, H.S. RBFOX3/NeuN is dispensable for visual function. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Gu, L.; Kawaguchi, R.; Caprioli, J.; Piri, N. The effect of Rbfox2 modulation on retinal transcriptome and visual function. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Sundar, J.; Matalkah, F.; Jeong, B.; Stoilov, P.; Ramamurthy, V. The Musashi proteins MSI1 and MSI2 are required for photoreceptor morphogenesis and vision in mice. J. Biol. Chem. 2020, 100048. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.P.; Wilks, C.; Charles, R.; Leavey, P.J.; Ghosh, D.; Jiang, L.; Santiago, C.P.; Pang, B.; Venkataraman, A.; Clark, B.S.; et al. ASCOT identifies key regulators of neuronal subtype-specific splicing. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.M.; Zack, D.J. Alternative splicing and retinal degeneration. Clin. Genet. 2013, 84, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Buvoli, M.; Buvoli, A.; Leinwand, L.A. Interplay between exonic splicing enhancers, mRNA processing, and mRNA surveillance in the dystrophic Mdx mouse. PLoS ONE 2007, 2. [Google Scholar] [CrossRef] [Green Version]
- Lam, B.J.; Hertel, K.J. A general role for splicing enhancers in exon definition. RNA 2002, 8, 1233–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheway, G.; Lord, J.; Baralle, D. Splicing in the pathogenesis, diagnosis and treatment of ciliopathies. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862. [Google Scholar] [CrossRef]
- Van Cauwenbergh, C.; Coppieters, F.; Roels, D.; De Jaegere, S.; Flipts, H.; De Zaeytijd, J.; Walraedt, S.; Claes, C.; Fransen, E.; Van Camp, G.; et al. Mutations in Splicing Factor Genes Are a Major Cause of Autosomal Dominant Retinitis Pigmentosa in Belgian Families. PLoS ONE 2017, 12, e0170038. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Wu, J.; Lam, S.; Duan, R.; Newnham, C.; Molday, R.S.; Graziotto, J.J.; Pierce, E.A.; Hu, J. Temporal and Tissue Specific Regulation of RP-Associated Splicing Factor Genes PRPF3, PRPF31 and PRPC8—Implications in the Pathogenesis of RP. PLoS ONE 2011, 6, e15860. [Google Scholar] [CrossRef] [Green Version]
- Tanackovic, G.; Ransijn, A.; Thibault, P.; Elela, S.A.; Klinck, R.; Berson, E.L.; Chabot, B.; Rivolta, C. PRPF mutations are associated with generalized defects in spliceosome formation and pre-mRNA splicing in patients with retinitis pigmentosa. Hum. Mol. Genet. 2011, 20, 2116–2130. [Google Scholar] [CrossRef] [Green Version]
- Comitato, A.; Spampanato, C.; Chakarova, C.; Sanges, D.; Bhattacharya, S.S.; Marigo, V. Mutations in splicing factor PRPF3, causing retinal degeneration, form detrimental aggregates in photoreceptor cells. Hum. Mol. Genet. 2007, 16, 1699–1707. [Google Scholar] [CrossRef]
- Shinde, V.; Kotla, P.; Strang, C.; Gorbatyuk, M. Unfolded protein response-induced dysregulation of calcium homeostasis promotes retinal degeneration in rat models of autosomal dominant retinitis pigmentosa. Cell Death Dis. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Vithana, E.N.; Abu-Safieh, L.; Allen, M.J.; Carey, A.; Papaioannou, M.; Chakarova, C.; Al-Maghtheh, M.; Ebenezer, N.D.; Willis, C.; Moore, A.T.; et al. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11). Mol. Cell 2001, 8, 375–381. [Google Scholar] [CrossRef]
- Sato, H.; Wada, Y.; Itabashi, T.; Nakamura, M.; Kawamura, M.; Tamai, M. Mutations in the pre-mRNA splicing gene, PRPF31, in Japanese families with autosomal dominant retinitis pigmentosa. Am. J. Ophthalmol. 2005, 140, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ribaudo, M.; Zhao, K.; Yu, N.; Chen, Q.; Sun, Q.; Wang, L.; Wang, Q. Novel deletion in the pre-mRNA splicing gene PRPF31 causes autosomal dominant retinitis pigmentosa in a large Chinese family. Am. J. Med. Genet. 2003, 121 A, 235–239. [Google Scholar] [CrossRef]
- Sullivan, L.S.; Bowne, S.J.; Seaman, C.R.; Blanton, S.H.; Lewis, R.A.; Heckenlively, J.R.; Birch, D.G.; Hughbanks-Wheaton, D.; Daiger, S.P. Genomic rearrangements of the PRPF31 gene account for 2.5% of autosomal dominant retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4579–4588. [Google Scholar] [CrossRef] [Green Version]
- Vithana, E.N.; Abu-Safieh, L.; Pelosini, L.; Winchester, E.; Hornan, D.; Bird, A.C.; Hunt, D.M.; Bustin, S.A.; Bhattacharya, S.S. Expression of PRPF31 mRNA in patients with autosomal dominant retinitis pigmentosa: A molecular clue for incomplete penetrance? Investig. Ophthalmol. Vis. Sci. 2003, 44, 4204–4209. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.M.; Shah, A.Z.; Venturini, G.; Krishna, A.; Chakravarti, A.; Rivolta, C.; Bhattacharya, S.S. Transcriptional regulation of PRPF31 gene expression by MSR1 repeat elements causes incomplete penetrance in retinitis pigmentosa. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Wilkie, S.E.; Vaclavik, V.; Wu, H.; Bujakowska, K.; Chakarova, C.F.; Bhattacharya, S.S.; Warren, M.J.; Hunt, D.M. Disease mechanism for retinitis pigmentosa (RP11) caused by missense mutations in the splicing factor gene PRPF31. Mol. Vis. 2008, 14, 683–690. [Google Scholar] [PubMed]
- Huranová, M.; Hnilicová, J.; Fleischer, B.; Cvačková, Z.; Staněk, D. A mutation linked to retinitis pigmentosa in HPRP31 causes protein instability and impairs its interactions with spliceosomal snRNPs. Hum. Mol. Genet. 2009, 18, 2014–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azizzadeh Pormehr, L.; Ahmadian, S.; Daftarian, N.; Mousavi, S.A.; Shafiezadeh, M. PRPF31 reduction causes mis-splicing of the phototransduction genes in human organotypic retinal culture. Eur. J. Hum. Genet. 2020, 28, 491–498. [Google Scholar] [CrossRef]
- Buskin, A.; Zhu, L.; Chichagova, V.; Basu, B.; Mozaffari-Jovin, S.; Dolan, D.; Droop, A.; Collin, J.; Bronstein, R.; Mehrotra, S.; et al. Disrupted alternative splicing for genes implicated in splicing and ciliogenesis causes PRPF31 retinitis pigmentosa. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Toulis, V.; Cortés-González, V.; de Castro-Miró, M.; Sallum, J.F.; Català-Mora, J.; Villanueva-Mendoza, C.; Ciccioli, M.; Gonzàlez-Duarte, R.; Valero, R.; Marfany, G. Increasing the genetic diagnosis yield in inherited retinal dystrophies: Assigning pathogenicity to novel non-canonical splice site variants. Genes 2020, 11, 378. [Google Scholar] [CrossRef] [Green Version]
- Sangermano, R.; Bax, N.M.; Bauwens, M.; Van Den Born, L.I.; De Baere, E.; Garanto, A.; Collin, R.W.J.; Goercharn-Ramlal, A.S.A.; Den Engelsman-Van Dijk, A.H.A.; Rohrschneider, K.; et al. Photoreceptor Progenitor mRNA Analysis Reveals Exon Skipping Resulting from the ABCA4 c.5461-10T→C Mutation in Stargardt Disease. Ophthalmology 2016, 123, 1375–1385. [Google Scholar] [CrossRef]
- Sangermano, R.; Khan, M.; Cornelis, S.S.; Richelle, V.; Albert, S.; Garanto, A.; Elmelik, D.; Qamar, R.; Lugtenberg, D.; Ingeborgh van den Born, L.; et al. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res. 2018, 28, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Albert, S.; Garanto, A.; Sangermano, R.; Khan, M.; Bax, N.M.; Hoyng, C.B.; Zernant, J.; Lee, W.; Allikmets, R.; Collin, R.W.J.; et al. Identification and Rescue of Splice Defects Caused by Two Neighboring Deep-Intronic ABCA4 Mutations Underlying Stargardt Disease. Am. J. Hum. Genet. 2018, 102, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Zernant, J.; Lee, W.; Nagasaki, T.; Collison, F.T.; Fishman, G.A.; Bertelsen, M.; Rosenberg, T.; Gouras, P.; Tsang, S.H.; Allikmets, R. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Cold Spring Harb. Mol. Case Stud. 2018, 4. [Google Scholar] [CrossRef] [Green Version]
- Zernant, J.; Lee, W.; Collison, F.T.; Fishman, G.A.; Sergeev, Y.V.; Schuerch, K.; Sparrow, J.R.; Tsang, S.H.; Allikmets, R. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J. Med. Genet. 2017, 54, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Cremers, F.P.M.; Lee, W.; Collin, R.W.J.; Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Strauss, R.W.; Chiang, J.; Audo, I.S.; Bernstein, P.S.; Birch, D.G.; Bomotti, S.M.; Cideciyan, A.V.; Ervin, A.M.; Marino, M.J.; et al. Detailed genetic characteristics of an international large cohort of patients with Stargardt disease: ProgStar study report 8. Br. J. Ophthalmol. 2019, 103, 390–397. [Google Scholar] [CrossRef]
- Fadaie, Z.; Khan, M.; Del Pozo-Valero, M.; Cornelis, S.S.; Ayuso, C.; Cremers, F.P.M.; Roosing, S.; The ABCA4 Study Group. Identification of splice defects due to noncanonical splice site or deep-intronic variants in ABCA4. Hum. Mutat. 2019, 40, 2365–2376. [Google Scholar] [CrossRef] [Green Version]
- Chiang, J.P.W.; Trzupek, K. The current status of molecular diagnosis of inherited retinal dystrophies. Curr. Opin. Ophthalmol. 2015, 26, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Xie, Y.; Zernant, J.; Yuan, B.; Bearelly, S.; Tsang, S.H.; Lupski, J.R.; Allikmets, R. Complex inheritance of ABCA4 disease: Four mutations in a family with multiple macular phenotypes. Hum. Genet. 2016, 135, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, H.L.; Grassmann, F.; Kellner, U.; Spital, G.; Rüther, K.; Jägle, H.; Hufendiek, K.; Rating, P.; Huchzermeyer, C.; Baier, M.J.; et al. Mutation spectrum of the ABCA4 gene in 335 stargardt disease patients from a multicenter German cohort—impact of selected deep intronic variants and common SNPs. Investig. Ophthalmol. Vis. Sci. 2017, 58, 394–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaz-Drago, R.; Custódio, N.; Carmo-Fonseca, M. Deep intronic mutations and human disease. Hum. Genet. 2017, 136, 1093–1111. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, F.; Lefever, S.; Leroy, B.P.; De Baere, E. CEP290, a gene with many faces: Mutation overview and presentation of CEP290base. Hum. Mutat. 2010, 31, 1097–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drivas, T.G.; Wojno, A.P.; Tucker, B.A.; Stone, E.M.; Bennett, J. Basal exon skipping and genetic pleiotropy: A predictive model of disease pathogenesis. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Hollander, A.I.; Koenekoop, R.K.; Yzer, S.; Lopez, I.; Arends, M.L.; Voesenek, K.E.J.; Zonneveld, M.N.; Strom, T.M.; Meitinger, T.; Brunner, H.G.; et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of leber congenital amaurosis. Am. J. Hum. Genet. 2006, 79, 556–561. [Google Scholar] [CrossRef] [Green Version]
- Vaché, C.; Besnard, T.; le Berre, P.; García-García, G.; Baux, D.; Larrieu, L.; Abadie, C.; Blanchet, C.; Bolz, H.J.; Millan, J.; et al. Usher syndrome type 2 caused by activation of an USH2A pseudoexon: Implications for diagnosis and therapy. Hum. Mutat. 2012, 33, 104–108. [Google Scholar] [CrossRef]
- Slijkerman, R.W.; Vaché, C.; Dona, M.; García-García, G.; Claustres, M.; Hetterschijt, L.; Peters, T.A.; Hartel, B.P.; Pennings, R.J.; Millan, J.M.; et al. Antisense Oligonucleotide-based Splice Correction for USH2A-associated Retinal Degeneration Caused by a Frequent Deep-intronic Mutation. Mol. Ther. Nucleic Acids 2016, 5, e381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin. Eye Res. 2018, 62, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrar, G.J.; Findlay, J.B.C.; Kumar-singh, R.; Kenna, P.; Humphries, M.M.; Sharpe, E.; Humphries, P. Autosomal dominant retinitis pigmentosa: A novel mutation in the rhodopsin gene in the original 3q linked family. Hum. Mol. Genet. 1992, 1, 769–771. [Google Scholar] [CrossRef] [PubMed]
- McWilliam, P.; Farrar, G.J.; Kenna, P.; Bradley, D.G.; Humphries, M.M.; Sharp, E.M.; McConnell, D.J.; Lawler, M.; Sheils, D.; Ryan, C.; et al. Autosomal dominant retinitis pigmentosa (ADRP): Localization of an ADRP gene to the long arm of chromosome 3. Genomics 1989, 5, 619–622. [Google Scholar] [CrossRef]
- Audo, I.; Friedrich, A.; Mohand-Saïd, S.; Lancelot, M.E.; Antonio, A.; Moskova-Doumanova, V.; Poch, O.; Bhattacharya, S.; Sahel, J.A.; Zeitz, C. An unusual retinal phenotype associated with a novel mutation in RHO. Arch. Ophthalmol. 2010, 128, 1036–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedmayr, L.M.; Böhm, S.; Biel, M.; Becirovic, E. Enigmatic rhodopsin mutation creates an exceptionally strong splice acceptor site. Hum. Mol. Genet. 2020, 29, 295–304. [Google Scholar] [CrossRef]
- Schmid, F.; Glaus, E.; Cremers, F.P.M.; Kloeckener-Gruissem, B.; Berger, W.; Neidhardt, J. Mutation- and tissue-specific alterations of RPGR transcripts. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1628–1635. [Google Scholar] [CrossRef] [Green Version]
- Neidhardt, J.; Glaus, E.; Barthelmes, D.; Zeitz, C.; Fleischhauer, J.; Berger, W. Identification and characterization of a novel RPGR isoform in human retina. Hum. Mutat. 2007, 28, 797–807. [Google Scholar] [CrossRef]
- Vervoort, R.; Lennon, A.; Bird, A.C.; Tulloch, B.; Axton, R.; Miano, M.G.; Meindl, A.; Meitinger, T.; Ciccodicola, A.; Wright, A.F. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 2000, 25, 462–466. [Google Scholar] [CrossRef]
- Wright, R.N.; Hong, D.H.; Perkins, B. Misexpression of the Constitutive Rpgr ex1-19 Variant Leads to Severe Photoreceptor Degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5189–5201. [Google Scholar] [CrossRef] [Green Version]
- Riazuddin, S.A.; Iqbal, M.; Wang, Y.; Masuda, T.; Chen, Y.; Bowne, S.; Sullivan, L.S.; Waseem, N.H.; Bhattacharya, S.; Daiger, S.P.; et al. A Splice-Site Mutation in a Retina-Specific Exon of BBS8 Causes Nonsyndromic Retinitis Pigmentosa. Am. J. Hum. Genet. 2010, 86, 805–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, D.; Singh, R.; Kolandaivelu, S.; Ramamurthy, V.; Stoilov, P. Alternative Splicing Shapes the Phenotype of a Mutation in BBS8 To Cause Nonsyndromic Retinitis Pigmentosa. Mol. Cell. Biol. 2015, 35, 1860–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vig, A.; Poulter, J.A.; Ottaviani, D.; Tavares, E.; Toropova, K.; Tracewska, A.M.; Mollica, A.; Kang, J.; Kehelwathugoda, O.; Paton, T.; et al. DYNC2H1 hypomorphic or retina-predominant variants cause nonsyndromic retinal degeneration. Genet. Med. 2020, 22. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, G.; Glaus, E.; Barthelmes, D.; Ader, M.; Fleischhauer, J.; Pagani, F.; Berger, W.; Neidhardt, J. Therapeutic strategy to rescue mutation-induced exon skipping in rhodopsin by adaptation of U1 snRNA. Hum. Mutat. 2009, 30, 255–263. [Google Scholar] [CrossRef]
- Glaus, E.; Schmid, F.; Da Costa, R.; Berger, W.; Neidhardt, J. Gene therapeutic approach using mutation-adapted U1 snRNA to correct a RPGR Splice defect in patient-derived cells. Mol. Ther. 2011, 19, 936–941. [Google Scholar] [CrossRef]
- Puttaraju, M.; Jamison, S.F.; Mansfield, S.G.; Garcia-Blanco, M.A.; Mitchell, L.G. Spliceosome-mediated RNA trans-splicing as a tool for gene therapy. Nat. Biotechnol. 1999, 17, 246–252. [Google Scholar] [CrossRef]
- Finta, C.; Zaphiropoulos, P.G. Intergenic mRNA molecules resulting from trans-splicing. J. Biol. Chem. 2002, 277, 5882–5890. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.S.; Yu, C.Y.; Chuang, C.Y.; Hsiao, M.; Kao, C.F.; Kuo, H.C.; Chuang, T.J. Integrative transcriptome sequencing identifies trans-splicing events with important roles in human embryonic stem cell pluripotency. Genome Res. 2014, 24, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, A.; Lorain, S.; Joséphine, C.; Desrosiers, M.; Peccate, C.; Voit, T.; Garcia, L.; Sahel, J.A.; Bemelmans, A.P. Repair of rhodopsin mRNA by spliceosome-mediated RNA trans-splicing: A new approach for autosomal dominant retinitis pigmentosa. Mol. Ther. 2015, 23, 918–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooley, S.J.; McDougald, D.S.; Fisher, K.J.; Bennicelli, J.L.; Mitchell, L.G.; Bennett, J. Spliceosome-Mediated Pre-mRNA trans-Splicing Can Repair CEP290 mRNA. Mol. Ther. Nucleic Acids 2018, 12, 294–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gemayel, M.C.; Bhatwadekar, A.D.; Ciulla, T. RNA therapeutics for retinal diseases. Expert Opin. Biol. Ther. 2020. [Google Scholar] [CrossRef]
- Jiang, J.; Zhang, X.; Tang, Y.; Li, S.; Chen, J. Progress on ocular siRNA gene-silencing therapy and drug delivery systems. Fundam. Clin. Pharmacol. 2020. fcp.12561. [Google Scholar] [CrossRef]
- Kleinman, M.E.; Kaneko, H.; Cho, W.G.; Dridi, S.; Fowler, B.J.; Blandford, A.D.; Albuquerque, R.J.C.; Hirano, Y.; Terasaki, H.; Kondo, M.; et al. Short-interfering RNAs induce retinal degeneration via TLR3 and IRF3. Mol. Ther. 2012, 20, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsay, E.; Raviña, M.; Sarkhel, S.; Hehir, S.; Cameron, N.R.; Ilmarinen, T.; Skottman, H.; Kjems, J.; Urtti, A.; Ruponen, M.; et al. Avoiding the pitfalls of siRNA delivery to the retinal pigment epithelium with physiologically relevant cell models. Pharmaceutics 2020, 12, 667. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Sudharsan, R.; Dufour, V.L.; Massengill, M.T.; Iwabe, S.; Swider, M.; Lisi, B.; Sumaroka, A.; Marinho, L.F.; Appelbaum, T.; et al. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc. Natl. Acad. Sci. USA 2018, 115, E8547–E8556. [Google Scholar] [CrossRef] [Green Version]
- Askou, A.L.; Pournaras, J.A.C.; Pihlmann, M.; Svalgaard, J.D.; Arsenijevic, Y.; Kostic, C.; Bek, T.; Dagnæs-Hansen, F.; Mikkelsen, J.G.; Jensen, T.G.; et al. Reduction of choroidal neovascularization in mice by adeno-associated virus-delivered anti-vascular endothelial growth factor short hairpin RNA. J. Gene Med. 2012, 14, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Gerard, X.; Garanto, A.; Rozet, J.M.; Collin, R.W.J. Antisense oligonucleotide therapy for inherited retinal dystrophies. Adv. Exp. Med. Biol. 2016, 854, 517–524. [Google Scholar]
- Garanto, A. RNA-Based Therapeutic Strategies for Inherited Retinal Dystrophies. Adv. Exp. Med. Biol. 2019, 1185, 71–77. [Google Scholar] [PubMed]
- Vázquez-Domínguez, I.; Garanto, A.; Collin, R.W.J. Molecular Therapies for Inherited Retinal Diseases—Current Standing, Opportunities and Challenges. Genes 2019, 10, 654. [Google Scholar] [CrossRef] [Green Version]
- Collin, R.W.; Den Hollander, A.I.; Der Velde-Visser, S.D.V.; Bennicelli, J.; Bennett, J.; Cremers, F.P. Antisense oligonucleotide (AON)-based therapy for leber congenital amaurosis caused by a frequent mutation in CEP290. Mol. Ther. Nucleic Acids 2012, 1, e14. [Google Scholar] [CrossRef] [PubMed]
- Gerard, X.; Perrault, I.; Hanein, S.; Silva, E.; Bigot, K.; Defoort-Delhemmes, S.; Rio, M.; Munnich, A.; Scherman, D.; Kaplan, J.; et al. AON-mediated exon skipping restores ciliation in fibroblasts harboring the common leber congenital amaurosis CEP290 mutation. Mol. Ther. Nucleic Acids 2012, 1, e29. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Jacobson, S.G.; Drack, A.V.; Ho, A.C.; Charng, J.; Garafalo, A.V.; Roman, A.J.; Sumaroka, A.; Han, I.C.; Hochstedler, M.D.; et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat. Med. 2019, 25, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Bonifert, T.; Gonzalez Menendez, I.; Battke, F.; Theurer, Y.; Synofzik, M.; Schöls, L.; Wissinger, B. Antisense Oligonucleotide Mediated Splice Correction of a Deep Intronic Mutation in OPA1. Mol. Ther. Nucleic Acids 2016, 5, e390. [Google Scholar] [CrossRef] [Green Version]
- Garanto, A.; van der Velde-Visser, S.D.; Cremers, F.P.M.; Collin, R.W.J. Antisense oligonucleotide-based splice correction of a deep-intronic mutation in CHM underlying choroideremia. Adv. Exp. Med. Biol. 2018, 1074, 83–89. [Google Scholar] [PubMed]
- Sangermano, R.; Garanto, A.; Khan, M.; Runhart, E.H.; Bauwens, M.; Bax, N.M.; van den Born, L.I.; Khan, M.I.; Cornelis, S.S.; Verheij, J.B.G.M.; et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med. 2019, 21, 1751–1760. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Location | Associated Disease | Splice Variants | Coding Transcripts |
|---|---|---|---|---|
| ABCA4 | 1p22.1 | Recessive Stargardt disease, juvenile and late onset; recessive macular dystrophy; recessive retinitis pigmentosa; recessive fundus flavimaculatus; recessive cone-rod dystrophy | 8 | 3 |
| ABCC6 | 16p13.11 | Recessive pseudoxanthoma elasticum; dominant pseudoxanthoma elasticum | 9 | 4 |
| ABHD12 | 20p11.21 | Recessive syndromic PHARC; recessive Usher syndrome, type 3-like | 24 | 17 |
| ACBD5 | 10p12.1 | Recessive cone-rod dystrophy with psychomotor delay | 8 | 7 |
| ACO2 | 22q13.2 | Recessive optic atrophy; recessive cerebellar degeneration with optic atrophy | 7 | 2 |
| ADAM9 | 8p11.23 | Recessive cone-rod dystrophy | 9 | 3 |
| ADAMTS18 | 16q23.1 | Recessive Knobloch syndrome; recessive retinal dystrophy, early onset | 9 | 3 |
| ADGRA3 | 4p15.2 | Recessive retinitis pigmentosa | 13 | 4 |
| ADGRV1 | 5q14.3 | Recessive Usher syndrome, type 2; dominant/recessive febrile convulsions | 37 | 10 |
| ADIPOR1 | 1q32.1 | Recessive retinitis pigmentosa, syndromic, Bardet-Biedl like; dominant retinitis pigmentosa | 5 | 4 |
| AFG3L2 | 18p11.21 | Dominant optic atrophy, non-syndromic; dominant spinocerebellar ataxia; recessive spastic ataxia | 4 | 2 |
| AGBL5 | 2p23.3 | Recessive retinitis pigmentosa | 10 | 7 |
| AHI1 | 6q23.3 | Recessive Joubert syndrome | 17 | 10 |
| AHR | 7p21.1 | Recessive retinitis pigmentosa | 7 | 2 |
| AIPL1 | 17p13.2 | Recessive Leber congenital amaurosis; dominant cone-rod dystrophy | 11 | 10 |
| ALMS1 | 2p13.1 | Recessive Alström syndrome | 12 | 4 |
| ARHGEF18 | 19p13.2 | Recessive retinitis pigmentosa | 6 | 5 |
| ARL2BP | 16q13.3 | Recessive retinitis pigmentosa | 4 | 3 |
| ARL3 | 10q24.32 | Dominant retinitis pigmentosa | 1 | 1 |
| ARL6 | 3q11.2 | Recessive Bardet-Biedl syndrome; recessive retinitis pigmentosa | 9 | 6 |
| ARMS2 | 10q26.13 | Age-related macular degeneration, complex etiology | 1 | 1 |
| ARSG | 17q24.2 | Recessive Usher syndrome, atypical | 9 | 4 |
| ASRGL1 | 11q12.3 | Recessive retinal degeneration | 11 | 6 |
| ATF6 | 1q23.3 | Recessive achromatopsia | 2 | 1 |
| ATOH7 | 10q21 | Recessive nonsyndromal congenital retinal nonattachment | 1 | 1 |
| ATXN7 | 3p14.1 | Dominant spinocerebellar ataxia w/ macular dystrophy or retinal degeneration | 20 | 7 |
| BBIP1 | 10q25.2 | Recessive Bardet-Biedl syndrome | 15 | 10 |
| BBS1 | 11q13 | Recessive Bardet-Biedl syndrome; recessive retinitis pigmentosa | 25 | 7 |
| BBS10 | 12q21.2 | Recessive Bardet-Biedl syndrome | 1 | 1 |
| BBS12 | 4q27 | Recessive Bardet-Biedl syndrome | 3 | 3 |
| BBS2 | 16q13 | Recessive Bardet-Biedl syndrome; recessive retinitis pigmentosa | 21 | 5 |
| BBS4 | 15q24.1 | Recessive Bardet-Biedl syndrome | 18 | 5 |
| BBS5 | 2q31.1 | Recessive Bardet-Biedl syndrome | 6 | 2 |
| BBS7 | 4q27 | Recessive Bardet Biedl syndrome | 6 | 3 |
| BBS8 | 14q31.3 | Recessive Bardet Biedl syndrome | 13 | 7 |
| BBS9 | 7p14.3 | Recessive Bardet Biedl syndrome | 29 | 14 |
| BEST1 | 11q12.3 | Dominant macular dystrophy, Best type; dominant vitreoretinochoroidopathy; recessive bestrophinopathy; recessive retinitis pigmentosa; dominant retinitis pigmentosa | 8 | 4 |
| C12orf65 | 12q24.31 | Recessive spastic paraplegia, neuropathy and optic atrophy | 8 | 6 |
| C1QTNF5 | 11q23.3 | Dominant macular dystrophy, late onset; dominant macular dystrophy with lens zonules | 4 | 3 |
| C2 | 6p21.32 | Age-related macular degeneration, complex etiology | 17 | 11 |
| C3 | 19p13.3 | Age-related macular degeneration, complex etiology | 18 | 5 |
| C8orf37 | 8q22.1 | Recessive cone-rod dystrophy; recessive retinitis pigmentosa with early macular involvement; recessive Bardet-Biedl syndrome | 1 | 1 |
| CA4 | 17q23.2 | Dominant retinitis pigmentosa | 6 | 4 |
| CABP4 | 11q13.1 | Recessive congenital stationary night blindness; recessive congenital cone-rod synaptic disease; recessive Leber congenital amaurosis | 7 | 2 |
| CACNA1F | Xp11.23 | X-linked congenital stationary night blindness, incomplete; AIED-like disease; severe congenital stationary night blindness; X-linked progressive cone-rod dystrophy | 6 | 4 |
| CACNA2D4 | 12p13.33 | Recessive cone dystrophy | 26 | 9 |
| CAPN5 | 11q13.5 | Dominant neovascular inflammatory vitreoretinopathy | 8 | 5 |
| CC2D2A | 4p15.33 | Recessive retinitis pigmentosa and mental retardation; recessive Joubert syndrome | 23 | 13 |
| CCT2 | 12q15 | Recessive Leber congenital amaurosis | 14 | 3 |
| CDH23 | 10q22.1 | Recessive Usher syndrome, type 1d; recessive deafness without retinitis pigmentosa; digenic Usher syndrome with PCDH15 | 19 | 14 |
| CDH3 | 16q22.1 | Recessive macular dystrophy, juvenile with hypotrichosis | 10 | 4 |
| CDHR1 | 10q23.1 | Recessive cone-rod dystrophy | 7 | 4 |
| CEP164 | 11q23.3 | Recessive nephronophthisis with retinal degeneration | 14 | 7 |
| CEP19 | 3q29 | Recessive Bardet-Biedl syndrome | 2 | 2 |
| CEP250 | 20q11.22 | Recessive Usher syndrome, atypical | 14 | 8 |
| CEP290 | 12q21.32 | Recessive Senior-Loken syndrome; recessive Joubert syndrome; recessive Leber congenital amaurosis; recessive Meckel syndrome | 34 | 13 |
| CEP78 | 9q21.2 | Recessive cone-rod dystrophy with hearing loss; recessive Usher syndrome, atypical | 24 | 14 |
| CERKL | 2q31.3 | Recessive retinitis pigmentosa; recessive cone-rod dystrophy with inner retinopathy | 14 | 5 |
| CFAP410 | 21q22.3 | Recessive cone-rod dystrophy | 7 | 3 |
| CFB | 6p21.32 | Age-related macular degeneration, complex etiology | 14 | 4 |
| CFH | 1q31.3 | Age-related macular degeneration, complex etiology; recessive drusen, early-onset | 6 | 3 |
| CHM | Xq21.2 | Choroideremia | 5 | 2 |
| CIB2 | 15q25.1 | Recessive Usher syndrome, type 1J | 10 | 7 |
| CISD2 | 4q22-q24 | Recessive Wolfram syndrome | 5 | 2 |
| CLCC1 | 1p13.3 | Recessive retinitis pigmentosa, severe | 30 | 21 |
| CLN3 | 16p11.2 | Recessive Batten disease (ceroid-lipofuscinosis, neuronal 3), juvenile | 62 | 20 |
| CLRN1 | 3q25.1 | Recessive Usher syndrome, type 3; recessive retinitis pigmentosa | 8 | 4 |
| CLUAP1 | 16p13.3 | Recessive Leber congenital amaurosis | 12 | 7 |
| CNGA1 | 4p12 | Recessive retinitis pigmentosa | 7 | 6 |
| CNGA3 | 2q11.2 | Recessive achromatopsia; recessive cone-rod dystrophy; protein: cone photoreceptor cgmp-gated cation channel alpha subunit [Gene] | 4 | 2 |
| CNGB1 | 16q21 | Recessive retinitis pigmentosa | 9 | 6 |
| CNGB3 | 8q21.3 | Recessive achromatopsia Pingelapese; recessive progressive cone dystrophy | 3 | 2 |
| CNNM4 | 2q11.2 | Recessive cone-rod dystrophy and amelogenesis imperfecta syndrome | 4 | 1 |
| COL11A1 | 1p21.1 | Dominant Stickler syndrome, type II; dominant Marshall syndrome | 14 | 10 |
| COL2A1 | 12q13.11 | Dominant Stickler syndrome, type I; dominant bone dysplasias, developmental disorders, osteoarthritic diseases, and syndromic disorders | 9 | 2 |
| COL9A1 | 6q13 | Recessive Stickler syndrome; dominant multiple epiphyseal dysplasia (MED) | 11 | 3 |
| CRB1 | 1q31.3 | Recessive retinitis pigmentosa with para-arteriolar preservation of the RPE (PPRPE); recessive retinitis pigmentosa; recessive Leber congenital amaurosis; dominant pigmented paravenous chorioretinal atrophy | 11 | 7 |
| CRX | 19q13.32 | Dominant cone-rod dystrophy; recessive, dominant and de novo Leber congenital amaurosis; dominant retinitis pigmentosa | 7 | 4 |
| CSPP1 | 8q13.1-q13.2 | Recessive Jobert syndrome | 16 | 8 |
| CTNNA1 | 5q31.2 | Dominant macular dystrophy, butterfly-shaped | 44 | 27 |
| CWC27 | 5q12.3 | Retinitis pigmentosa with or without skeletal anomalies | 5 | 2 |
| CYP4V2 | 4q35.2 | Recessive Bietti crystalline corneoretinal dystrophy; recessive retinitis pigmentosa | 4 | 1 |
| DHDDS | 1p36.11 | Recessive retinitis pigmentosa | 25 | 16 |
| DHX38 | 16q22.2 | Recessive retinitis pigmentosa, early onset with macular coloboma | 14 | 6 |
| DMD | Xp21.2-p21.1 | Oregon eye disease (probably) | 32 | 20 |
| DNM1L | 22q12.1-q13.1 | Dominant optic atrophy | 30 | 11 |
| DRAM2 | 1p13.3 | Recessive macular dystrophy, early adult onset | 11 | 2 |
| DTHD1 | 4p14 | Recessive Leber congenital amaurosis with myopathy | 6 | 4 |
| DYNC2H1 | 11q22.3 | Syndromic and non syndromic retinal degeneration | 11 | 5 |
| EFEMP1 | 2p16.1 | Dominant radial, macular drusen; dominant Doyne honeycomb retinal degeneration (Malattia Leventinese) | 14 | 10 |
| ELOVL1 | 1p34.2 | Dominant optic atrophy, deafness, ichthyosis and neuronal disorders | 16 | 3 |
| ELOVL4 | 6q14.1 | Dominant macular dystrophy, Stargardt-like; recessive spinocerebellar ataxia; recessive ichthyosis, quadriplegia and retardation | 1 | 1 |
| EMC1 | 1p36.13 | Recessive retinitis pigmentosa | 13 | 5 |
| ERCC6 | 10q11.23 | Age-related macular degeneration, complex etiology; Cockayne syndrome, recessive | 12 | 4 |
| ESPN | 1p36.31 | Recessive Usher syndrome | 17 | 14 |
| EXOSC2 | 9q34.12 | Recessive retinitis pigmentosa with hearing loss and additional disabilities | 11 | 6 |
| EYS | 6q12 | Recessive retinitis pigmentosa | 11 | 5 |
| FAM161A | 2p15 | Recessive retinitis pigmentosa | 7 | 2 |
| FBLN5 | 14q32.12 | Familial macular dystrophy, age-related | 9 | 4 |
| FLVCR1 | 1q32.3 | Recessive retinitis pigmentosa with posterior column ataxia (PCARP) | 5 | 2 |
| FSCN2 | 17q25.3 | Dominant retinitis pigmentosa; dominant macular dystrophy | 3 | 2 |
| FZD4 | 11p13-p12 | Dominant familial exudative vitreoretinopathy | 1 | 1 |
| FZD4 | 11q14.2 | Dominant familial exudative vitreoretinopathy | 1 | 1 |
| GDF6 | 8q22.1 | Recessive Leber congenital amaurosis; dominant Klippel-Feil syndrome; dominant microphthalmia | 3 | 3 |
| GNAT1 | 3p21.31 | Dominant congenital stationary night blindness, Nougaret type; recessive congenital stationary night blindness | 5 | 3 |
| GNAT2 | 1p13.3 | Recessive achromatopsia | 2 | 2 |
| GNB3 | 12p13.31 | Recessive congenital stationary night blindness | 10 | 6 |
| GNPTG | 16p13.3 | Recessive retinitis pigmentosa and skeletal abnormalities; recessive mucolipidosis III gamma | 9 | 3 |
| GPR179 | 17q12 | Recessive complete congenital stationary night blindness | 1 | 1 |
| GRK1 | 13q34 | Recessive congenital stationary night blindness, Oguchi type | 3 | 1 |
| GRM6 | 5q35.3 | Recessive congenital stationary night blindness | 6 | 3 |
| GUCA1A | 6p21.1 | Dominant cone dystrophy; dominant cone-rod dystrophy | 2 | 2 |
| GUCA1B | 6p21.1 | Dominant retinitis pigmentosa; dominant macular dystrophy | 1 | 1 |
| GUCY2D | 17p13 | Dominant central areolar choroidal dystrophy | 2 | 1 |
| GUCY2D | 17p13.1 | Recessive Leber congenital amaurosis; dominant cone-rod dystrophy | 2 | 1 |
| HARS1 | 5q31.3 | Recessive Usher syndrome | 30 | 13 |
| HGSNAT | 8p11.21-p11.1 | Recessive retinitis pigmentosa, non-syndromic; recessive mucopolysaccharidosis | 10 | 4 |
| HK1 | 10q22.1 | Dominant retinitis pigmentosa; recessive nonspherocytic hemolytic anemia; recessive hereditary neuropathy (Russe type) | 18 | 10 |
| HMCN1 | 1q25.3-q31.1 | Dominant macular dystrophy, age-related | 5 | 2 |
| HMX1 | 4p16.1 | Recessive oculoauricular syndrome | 2 | 2 |
| HTRA1 | 10q26.13 | Age-related macular degeneration, complex etiology | 3 | 3 |
| IDH3B | 20p13 | Recessive retinitis pigmentosa | 12 | 4 |
| IFT140 | 16p13.3 | Recessive Mainzer-Saldino syndrome; recessive retinitis pigmentosa; recessive Leber congenital amaurosis | 11 | 5 |
| IFT172 | 2p33.3 | Recessive Bardet-Biedl syndrome; recessive retinitis pigmentosa | 34 | 6 |
| IFT27 | 22q12.3 | Recessive Bardet-Biedl syndrome | 12 | 5 |
| IFT81 | 12q24.11 | Recessive cone-rod dystrophy; recessive spectrum of ciliopathies including retinal dystrophy | 9 | 4 |
| IMPDH1 | 7q32.1 | Dominant retinitis pigmentosa; dominant Leber congenital amaurosis | 18 | 11 |
| IMPG1 | 6q14.1 | Dominant macular dystrophy, vitelliform; recessive macular dystrophy, vitelliform; dominant retinitis pigmentosa | 4 | 4 |
| IMPG2 | 3q12.3 | Recessive retinitis pigmentosa | 1 | 1 |
| INPP5E | 9q34.3 | Recessive Joubert syndrome; recessive MORM syndrome | 6 | 3 |
| INVS | 9q31.1 | Recessive Senior-Loken syndrome; recessive nephronophthisis | 7 | 3 |
| IQCB1 | 3q13.33 | Recessive Senior-Loken syndrome; recessive Leber congenital amaurosis | 7 | 5 |
| ITM2B | 13q14.2 | Dominant retinal dystrophy; dominant dementia, familial | 11 | 4 |
| JAG1 | 20p12.2 | Dominant Alagille syndrome | 9 | 2 |
| KCNJ13 | 2q37.1 | Dominant vitreoretinal degeneration, snowflake; recessive Leber congenital amaurosis | 5 | 5 |
| KCNV2 | 9p24.2 | Recessive cone dystrophy with supernormal rod electroretinogram | 1 | 1 |
| KIAA1549 | 7q34 | Recessive retinitis pigmentosa; protein: KIAA1549 protein | 2 | 2 |
| KIF11 | 10q23.33 | Dominant microcephaly, lymphedema and chorioretinopathy | 1 | 1 |
| KIZ | 20p11.23 | Recessive retinitis pigmentosa | 15 | 8 |
| KLHL7 | 7p15.3 | Dominant retinitis pigmentosa | 13 | 5 |
| LAMA1 | 18p11.31-p11.23 | Recessive retinal dystrophy and cerebellar dysplasia | 9 | 2 |
| LCA5 | 6q14.1 | Recessive Leber congenital amaurosis | 3 | 3 |
| LRAT | 4q32.1 | Recessive retinitis pigmentosa, severe early-onset; recessive Leber congenital amaurosis | 8 | 3 |
| LRIT3 | 4q25 | Recessive congenital stationary night blindness | 2 | 2 |
| LRP5 | 11q13.2 | Dominant familial exudative vitreoretinopathy; dominant high bone mass trait; recessive osteoporosis-pseudoglioma syndrome; recessive familial exudative vitreoretinopathy | 7 | 2 |
| LZTFL1 | 3p21.31 | Recessive Bardet-Biedl syndrome with developmental anomalies | 16 | 5 |
| MAK | 6p24.2 | Recessive retinits pigmentosa | 7 | 5 |
| MAPKAPK3 | 3p21.2 | Dominant Martinique retinal dystrophy and retinitis pigmentosa | 8 | 6 |
| MERTK | 2q13 | Recessive retinitis pigmentosa; recessive rod-cone dystrophy, early onset | 7 | 5 |
| MFN2 | 1p36.22 | Dominant optic atrophy with neuropathy and myopathy; dominant Charcot-Marie-Tooth disease | 33 | 17 |
| MFRP | 11q23.3 | Recessive microphthalmos and retinal disease syndrome; recessive nanophthalmos | 6 | 3 |
| MFSD8 | 4q28.2 | Recessive macular dystrophy | 62 | 24 |
| MKKS | 20p12.2 | Recessive Bardet-Biedl syndrome | 4 | 3 |
| MKS1 | 17q22 | Recessive Bardet-Biedl syndrome; recessive Meckel syndrome | 13 | 7 |
| MMP19 | 12q13.13-q14.3 | Dominant cavitary optic disc anomalies | 9 | 3 |
| MT-ATP6 | mitochondrion | Retinitis pigmentosa with developmental and neurological abnormalities; Leigh syndrome; Leber hereditary optic neuropathy | 1 | 1 |
| MT-TH | mitochondrion | Pigmentary retinopathy and sensorineural hearing loss | 1 | - |
| MT-TL1 | mitochondrion | Macular pattern dystrophy with type II diabetes and deafness | 1 | - |
| MT-TP | mitochondrion | Retinitis pigmentosa with deafness and neurological abnormalities | 1 | - |
| MT-TS2 | mitochondrion | Retinitis pigmentosa with progressive sensorineural hearing loss | 1 | - |
| MTTP | 4q23 | Recessive abetalipoproteinemia | 11 | 5 |
| MVK | 12q24.11 | Recessive retinitis pigmentosa; recessive mevalonic aciduria; recessive hyper-igd syndrome | 17 | 10 |
| MYO7A | 11q13.5 | Recessive Usher syndrome, type 1b; recessive congenital deafness without retinitis pigmentosa; recessive atypical Usher syndrome (USH3-like) | 14 | 8 |
| NBAS | 2p24.3 | Recessive optic atrophy and retinal dystrophy, syndromic; | 9 | 7 |
| NDP | Xp11.3 | Norrie disease; familial exudative vitreoretinopathy; Coats disease | 3 | 2 |
| NEK2 | 1q32.3 | Recessive retinitis pigmentosa; protein: NIMA (never in mitosis gene A)-related kinase 2 [Gene] | 5 | 3 |
| NEUROD1 | 2q31.3 | Recessive retinitis pigmentosa | 2 | 1 |
| NMNAT1 | 1p36.22 | Recessive Leber congenital amaurosis | 5 | 3 |
| NPHP1 | 2q13 | Recessive Senior-Loken syndrome; recessive nephronophthisis, juvenile; recessive Joubert syndrome; recessive Bardet-Biedl syndrome | 22 | 12 |
| NPHP3 | 3q22.1 | Recessive Senior-Loken syndrome; recessive nephronophthisis, adolescent | 11 | 3 |
| NPHP4 | 1p36.31 | Recessive Senior-Loken syndrome, recessive nephronophthisis | 11 | 2 |
| NR2E3 | 15q23 | Recessive enhanced S-cone syndrome (ESCS); recessive retinitis pigmentosa in Portuguese Crypto Jews; recessive Goldmann-Favre syndrome; dominant retinitis pigmentosa; combined dominant and recessive retinopathy | 4 | 3 |
| NR2F1 | 5q15 | Dominant optic atrophy with intellectual disability and developmental delay | 6 | 3 |
| NRL | 14q11.2 | Dominant retinitis pigmentosa; recessive retinitis pigmentosa | 6 | 6 |
| NYX | Xp11.4 | X-linked congenital stationary night blindness | 3 | 2 |
| OAT | 10q26.13 | Recessive gyrate atrophy | 8 | 2 |
| OFD1 | Xp22.2 | Jobert syndrome; orofaciodigital syndrome 1, Simpson-Golabi-Behmel syndrome 2; X-linked retinitis pigmentosa, severe | 9 | 4 |
| OPA1 | 3q29 | Dominant optic atrophy, Kjer type; dominant optic atrophy with sensorineural hearing loss | 32 | 14 |
| OPA3 | 19q13.32 | Recessive optic atrophy with ataxia and 3-methylglutaconic aciduria; dominant optic atrophy with cataract, ataxia and areflexia | 3 | 3 |
| OPN1LW | Xq28 | Deuteranopia and rare macular dystrophy in blue cone monochromacy with loss of locus control element | 3 | 2 |
| OPN1MW | Xq28 | Protanopia and rare macular dystrophy in blue cone monochromacy with loss of locus control element | 3 | 2 |
| OPN1SW | 7q32.1 | Dominant tritanopia | 1 | 1 |
| OTX2 | 14q22.3 | Dominant Leber congenital amaurosis and pituitary dysfunction; recessive microphthalmia; dominant pattern dystrophy | 11 | 11 |
| PANK2 | 20p13 | Recessive HARP (hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and palladial degeneration); recessive Hallervorden-Spatz syndrome | 12 | 6 |
| PAX2 | 10q24.31 | Dominant renal-coloboma syndrome | 9 | 6 |
| PCARE | 2p23.2 | Recessive retinitis pigmentosa | 2 | 1 |
| PCDH15 | 10q21.1 | Recessive Usher syndrome, type 1f; recessive deafness without retinitis pigmentosa; digenic Usher syndrome with CDH23 | 36 | 31 |
| PCYT1A | 3q29 | Recessive cone-rod dystrophy with skeletal disease | 16 | 9 |
| PDE6A | 5q33.1 | Recessive retinitis pigmentosa | 5 | 3 |
| PDE6B | 4p16.3 | Recessive retinitis pigmentosa; dominant congenital stationary night blindness | 12 | 8 |
| PDE6C | 10q23.33 | Recessive cone dystrophy, early onset; recessive complete and incomplete achromatopsia | 2 | 1 |
| PDE6G | 17q25.3 | Recessive retinitis pigmentosa | 5 | 3 |
| PDE6H | 12p12.3 | Recessive achromatopsia, incomplete | 1 | 1 |
| PDZD7 | 10q24.31 | Recessive non-syndromic deafness | 9 | 7 |
| PEX1 | 7q21.2 | Recessive Refsum disease, infantile form | 10 | 3 |
| PEX2 | 8q21.13 | Recessive Refsum disease, infantile form | 6 | 5 |
| PEX7 | 6q23.3 | Recessive Refsum disease, adult form | 3 | 3 |
| PGK1 | Xq21.1 | Retinitis pigmentosa with myopathy | 6 | 2 |
| PHYH | 10p13 | Recessive Refsum disease, adult form | 7 | 5 |
| PITPNM3 | 17p13.2 | Dominant cone-rod dystrophy | 5 | 3 |
| PLA2G5 | 1p36.13-p36.12 | Recessive benign fleck retina | 8 | 1 |
| PLK4 | 4q28.2 | Recessive microcephaly, growth failure and retinopathy | 11 | 6 |
| PNPLA6 | 19p13.2 | Recessive Boucher-Neuhauser syndrome with chorioretinal dystrophy | 25 | 14 |
| POC1B | 12q21.33 | Recessive cone-rod dystrophy; recessive Joubert syndrome | 14 | 4 |
| POC5 | 5q13.3 | Recessive syndromic disease with retinitis pigmentosa | 14 | 7 |
| POMGNT1 | 1p34.1 | Recessive retinitis pigmentosa | 10 | 3 |
| PRCD | 17q25.1 | Recessive retinitis pigmentosa | 13 | 2 |
| PRDM13 | 6q16.2 | Dominant macular dystrophy, North Carolina type; dominant progressive bifocal chorioretinal atrophy | 2 | 1 |
| PROM1 | 4p15.32 | Recessive retinitis pigmentosa with macular degeneration; dominant Stargardt-like macular dystrophy; dominant macular dystrophy, bull’s-eye; dominant cone-rod dystrophy | 22 | 11 |
| PRPF3 | 1q21.2 | Dominant retinitis pigmentosa | 7 | 1 |
| PRPF31 | 19q13.42 | Dominant retinitis pigmentosa | 9 | 6 |
| PRPF4 | 9q32 | Dominant retinitis pigmentosa | 3 | 2 |
| PRPF6 | 20q13.33 | Dominant retinits pigmentosa | 1 | 1 |
| PRPF8 | 17p13.3 | Dominant retinitis pigmentosa | 16 | 5 |
| PRPH2 | 6p21.1 | Dominant retinitis pigmentosa; dominant macular dystrophy; digenic RP with ROM1; dominant adult vitelliform macular dystrophy; dominant cone-rod dystrophy; dominant central areolar choroidal dystrophy; recessive LCA | 1 | 1 |
| PRPS1 | Xq22.3 | Neuropathy, optic atrophy, deafness and retinitis pigmentosa | 25 | 10 |
| RAB28 | 4p15.33 | Recessive cone-rod dystrophy | 8 | 7 |
| RAX2 | 19p13.3 | Cone-rod dystrophy, isolated; age-related macular degeneration, isolated | 2 | 2 |
| RB1 | 13q14.2 | Dominant germline or somatic retinoblastoma; benign retinoma; pinealoma; osteogenic sarcoma | 9 | 4 |
| RBP3 | 10q11.22 | Recessive retinitis pigmentosa | 1 | 1 |
| RBP4 | 10q23.33 | Recessive RPE degeneration | 4 | 4 |
| RCBTB1 | 13q14.2 | Recessive syndromic and non-syndromic retinal dystrophy; dominant familial exudative vitreoretinopathy and Coats disease | 4 | 2 |
| RD3 | 1q32.3 | Recessive Leber congenital amaurosis | 2 | 1 |
| RDH11 | 14q24.1 | Recessive retinitis pigmentosa, syndromicxd | 11 | 6 |
| RDH12 | 14q24.1 | Recessive Leber congenital amaurosis with severe childhood retinal dystrophy; dominant retinitis pigmentosaxd | 4 | 2 |
| RDH5 | 12q13.2 | Recessive fundus albipunctatus; recessive cone dystrophy, late onset | 11 | 4 |
| REEP6 | 19p13.3 | Recessive retinitis pigmentosa | 4 | 2 |
| RGR | 10q23.1 | Recessive retinitis pigmentosa; dominant choroidal sclerosis | 18 | 9 |
| RGS9 | 17q24.1 | Recessive delayed cone adaptation | 12 | 4 |
| RGS9BP | 19q13.12 | Recessive delayed cone adaptation | 1 | 1 |
| RHO | 3q22.1 | Dominant retinitis pigmentosa; dominant congenital stationary night blindness; recessive retinitis pigmentosa | 1 | 1 |
| RIMS1 | 6q13 | Dominant cone-rod dystrophy | 20 | 16 |
| RLBP1 | 15q26.1 | Recessive retinitis pigmentosa; recessive Bothnia dystrophy; recessive retinitis punctata albescens; recessive Newfoundland rod-cone dystrophy | 4 | 2 |
| ROM1 | 11q12.3 | Dominant retinitis pigmentosa; digenic retinitis pigmentosa with PRPH2 | 5 | 4 |
| RP1 | 8q12.1 | Dominant retinitis pigmentosa; recessive retinitis pigmentosa | 8 | 3 |
| RP1L1 | 8p23.1 | Dominant occult macular dystrophy; recessive retinitis pigmentosa | 2 | 1 |
| RP2 | Xp11.23 | X-linked retinitis pigmentosa; X-linked retinitis pigmentosa, dominant | 1 | 1 |
| RP9 | 7p14.3 | Dominant retinitis pigmentosa | 4 | 2 |
| RPE65 | 1p31.2 | Recessive Leber congenital amaurosis; recessive retinitis pigmentosa; dominant retinitis pigmentosa with choroidal involvement | 1 | 1 |
| RPGR | Xp11.4 | X-linked retinitis pigmentosa, recessive; X-linked retinitis pigmentosa, dominant; X-linked cone dystrophy 1; X-linked atrophic macular dystrophy, recessive | 10 | 9 |
| RPGRIP1 | 14q11.2 | Recessive Leber congenital amaurosis; recessive cone-rod dystrophy | 13 | 8 |
| RPGRIP1L | 16q12.2 | Recessive Joubert syndrome; recesssive Meckel syndrome | 12 | 10 |
| RS1 | Xp22.13 | Retinoschisis | 2 | 1 |
| RTN4IP1 | 6q21 | Recessive optic atrophy, non-syndromic and syndromic | 4 | 2 |
| SAG | 2q37.1 | Recessive Oguchi disease; recessive retinitis pigmentosa; dominant retinitis pigmentosa | 16 | 3 |
| SAMD11 | 1p36.33 | Recessive retinitis pigmentosa | 17 | 13 |
| SDCCAG8 | 1q43 | Recessive nephronophthisis, ciliopathy-related; recessive Bardet-Biedl syndrome | 11 | 3 |
| SEMA4A | 1q22 | Dominant retinitis pigmentosa; dominant cone-rod dystrophy | 16 | 9 |
| SLC24A1 | 15q22.31 | Recessive congenital stationary night blindness | 12 | 7 |
| SLC25A46 | 5q22.1 | Recessive syndromic optic atrophy; protein | 8 | 5 |
| SLC38A8 | 16q23.2-q24.2 | Recessive foveal hypoplasia and anterior segment dysgenesis | 4 | 3 |
| SLC7A14 | 3q26.2 | Recessive retinitis pigmentosa | 2 | 1 |
| SNRNP200 | 2q11.2 | Dominant retinitis pigmentosa | 9 | 2 |
| SPATA7 | 14q31.3 | Recessive Leber congenital amaurosis; recessive RP, juvenile | 22 | 8 |
| SPP2 | 2q37.1 | Dominant retinitis pigmentosa | 4 | 3 |
| TEAD1 | 11p15.3 | Dominant atrophia areata | 6 | 5 |
| TIMM8A | Xq22.1 | Optic atrophy with deafness-dystonia syndrome | 4 | 2 |
| TIMP3 | 22q12.3 | Dominant Sorsby’s fundus dystrophy | 1 | 1 |
| TLR3 | 4q35.1 | Age-related macular degeneration, complex etiology | 5 | 3 |
| TLR4 | 9q33.1 | Age-related macular degeneration, complex etiology | 4 | 3 |
| TMEM126A | 11q14.1 | Recessive non-syndromic optic atrophy | 6 | 3 |
| TMEM216 | 11q12.2 | Recessive Joubert syndrome; recessive Meckel syndrome | 5 | 2 |
| TMEM237 | 2q33.1 | Recessive Jobert syndrome | 13 | 3 |
| TOPORS | 9p21.1 | Dominant retinitis pigmentosa | 2 | 2 |
| TREX1 | 3p21.31 | Dominant retinal vasculopathy with cerebral leukodystrophy; dominant Aicardi-Goutiere syndrome 1, dominant chilblain lupus | 6 | 6 |
| TRIM32 | 9q33.1 | Recessive Bardet-Biedl syndrome; recessive limb-girdle muscular dystrophy | 3 | 3 |
| TRNT1 | 3p26.2 | Recessive retinitis pigmentosa with erythrocytic microcytosis; recessive retinitis pigmentosa, non-syndromic | 20 | 7 |
| TRPM1 | 15q13.3 | Recessive congenital stationary night blindness, complete | 12 | 7 |
| TSPAN12 | 7q31.31 | Dominant familial exudative vitreoretinopathy | 7 | 6 |
| TTC8 | 14q32.11 | Recessive Bardet-Biedl syndrome; recessive retinitis pigmentosa | 13 | 7 |
| TTLL5 | 14q24.3 | Recessive cone and cone-rod dystrophy | 22 | 7 |
| TTPA | 8q12.3 | Recessive retinitis pigmentosa and/or recessive or dominant ataxia | 2 | 1 |
| TUB | 11p15.4 | Recessive retinal dystrophy and obesity | 3 | 3 |
| TUBGCP4 | 15q15.3 | Recessive chorioretinopathy and microcephaly | 12 | 4 |
| TUBGCP6 | 22q13.33 | Recessive microcephaly with chorioretinopathy | 8 | 4 |
| TULP1 | 6p21.31 | Recessive retinitis pigmentosa; recessive Leber congenital amaurosis | 8 | 4 |
| UNC119 | 17q11.2 | Dominant cone-rod dystrophy | 8 | 6 |
| USH1C | 11p15.1 | Recessive Usher syndrome, Acadian; recessive deafness without retinitis pigmentosa | 11 | 5 |
| USH1G | 17q25.1 | Recessive Usher syndrome | 2 | 1 |
| USH2A | 1q41 | Recessive Usher syndrome, type 2a; recessive retinitis pigmentosa | 5 | 2 |
| VCAN | 5q14.3 | Dominant Wagner disease and erosive vitreoretinopathy | 12 | 7 |
| WDPCP | 2p15 | Recessive Bardet-Biedl syndrome | 17 | 7 |
| WDR19 | 4p14 | Recessive renal, skeletal and retinal anomalies; recessive Senior-Loken syndrome | 18 | 4 |
| WFS1 | 4p16.1 | Recessive Wolfram syndrome; dominant low frequency sensorineural hearing loss | 9 | 6 |
| WHRN | 9q32 | Recessive Usher syndrome, type 2; recessive deafness without retinitis pigmentosa | 8 | 5 |
| ZNF408 | 11p11.2 | Dominant familial exudative vitreoretinopathy; recessive retinitis pigmentosa with vitreal alterations | 5 | 1 |
| ZNF423 | 16q12.1 | Recessive Jobert syndrome; recessive nephronophthisis | 8 | 8 |
| ZNF513 | 2p23.3 | Recessive retinitis pigmentosa | 4 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aísa-Marín, I.; García-Arroyo, R.; Mirra, S.; Marfany, G. The Alter Retina: Alternative Splicing of Retinal Genes in Health and Disease. Int. J. Mol. Sci. 2021, 22, 1855. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041855
Aísa-Marín I, García-Arroyo R, Mirra S, Marfany G. The Alter Retina: Alternative Splicing of Retinal Genes in Health and Disease. International Journal of Molecular Sciences. 2021; 22(4):1855. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041855
Chicago/Turabian StyleAísa-Marín, Izarbe, Rocío García-Arroyo, Serena Mirra, and Gemma Marfany. 2021. "The Alter Retina: Alternative Splicing of Retinal Genes in Health and Disease" International Journal of Molecular Sciences 22, no. 4: 1855. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22041855