
Molecular Alterations in Pancreatic Cancer: Transfer to the Clinic


 , , and
, , and 
Abstract
:
1. Introduction
2. PDA Genetic Alterations
2.1. KRAS
2.2. CDKN2A
2.3. TP53
2.4. SMAD4
2.5. c-MYC
2.6. GATA6
3. PDA Epigenetic Alterations
4. PDA Molecular Subtypes
5. Intratumoral Heterogeneity and Metastasis
6. Genetic Alterations Associated with Familial Pancreatic Cancer
7. Molecular Alterations as a Predictive Response Factor and New Targets
8. Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Malvezzi, M.; Bertuccio, P.; Levi, F.; Vecchia, C.L.; Negri, E. European Cancer Mortality Predictions for the Year 2014. Ann. Oncol. 2014, 25, 1650–1656. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellam, F.; Harir, N.; Khaled, M.B.; Mrabent, N.M.; Salah, R.; Diaf, M. Epidemiology and Risk Factors for Exocrine Pancreatic Cancer in a Northern African Population. J. Gastrointest. Cancer 2015, 46, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.-B.; Brown, A.M.K.; Kim, J.C.; et al. A Renewed Model of Pancreatic Cancer Evolution Based on Genomic Rearrangement Patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.-C.; Mansour, J.; Mollaee, M.; Wagner, K.-U.; Koduru, P.; Yopp, A.; et al. Whole-Exome Sequencing of Pancreatic Cancer Defines Genetic Diversity and Therapeutic Targets. Nature Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Riva, G.; Pea, A.; Pilati, C.; Fiadone, G.; Lawlor, R.T.; Scarpa, A.; Luchini, C. Histo-Molecular Oncogenesis of Pancreatic Cancer: From Precancerous Lesions to Invasive Ductal Adenocarcinoma. World J. Gastrointest. Oncol. 2018, 10, 317–327. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Kuboki, Y.; Hatori, T.; Yamamoto, M.; Shiratori, K.; Kawamura, S.; Kobayashi, M.; Shimizu, M.; Ban, S.; Koyama, I.; et al. Somatic Mutations in PIK3CA and Activation of AKT in Intraductal Tubulopapillary Neoplasms of the Pancreas. Am. J. Surg. Pathol. 2011, 35, 1812–1817. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; Wu, J.; et al. Pancreatic Cancer Genomes Reveal Aberrations in Axon Guidance Pathway Genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole Genomes Redefine the Mutational Landscape of Pancreatic Cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [Green Version]
- Iacobuzio-Donahue, C.A.; Ryu, B.; Hruban, R.H.; Kern, S.E. Exploring the Host Desmoplastic Response to Pancreatic Carcinoma: Gene Expression of Stromal and Neoplastic Cells at the Site of Primary Invasion. Am. J. Pathol. 2002, 160, 91–99. [Google Scholar] [CrossRef]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.-M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of Somatic Mutations in Most Early-Stage Pancreatic Intraepithelial Neoplasia. Gastroenterology 2012, 142, 730–733.e9. [Google Scholar] [CrossRef] [Green Version]
- Chan-Seng-Yue, M.; Kim, J.C.; Wilson, G.W.; Ng, K.; Figueroa, E.F.; O’Kane, G.M.; Connor, A.A.; Denroche, R.E.; Grant, R.C.; McLeod, J.; et al. Transcription Phenotypes of Pancreatic Cancer Are Driven by Genomic Events during Tumor Evolution. Nat. Genet. 2020, 52, 231–240. [Google Scholar] [CrossRef]
- Mueller, S.; Engleitner, T.; Maresch, R.; Zukowska, M.; Lange, S.; Kaltenbacher, T.; Konukiewitz, B.; Öllinger, R.; Zwiebel, M.; Strong, A.; et al. Evolutionary Routes and KRAS Dosage Define Pancreatic Cancer Phenotypes. Nature 2018, 554, 62–68. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Roeth, A.A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.M.; Wang, X.; Bronson, R.T.; Lyssiotis, C.A.; Ying, H.; et al. Oncogenic KRAS Supports Pancreatic Cancer through Regulation of Nucleotide Synthesis. Nature Commun. 2018, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.Y.L.; Dubois, C.L.; Sarai, K.; Zarei, S.; Schaeffer, D.F.; Sander, M.; Kopp, J.L. Cell of Origin Affects Tumour Development and Phenotype in Pancreatic Ductal Adenocarcinoma. Gut 2019, 68, 487. [Google Scholar] [CrossRef]
- Li, Y.; He, Y.; Peng, J.; Su, Z.; Li, Z.; Zhang, B.; Ma, J.; Zhuo, M.; Zou, D.; Liu, X.; et al. Mutant Kras Co-Opts a Proto-Oncogenic Enhancer Network in Inflammation-Induced Metaplastic Progenitor Cells to Initiate Pancreatic Cancer. Nat. Cancer 2020, 1–17. [Google Scholar] [CrossRef]
- McAllister, F.; Bailey, J.M.; Alsina, J.; Nirschl, C.J.; Sharma, R.; Fan, H.; Rattigan, Y.; Roeser, J.C.; Lankapalli, R.H.; Zhang, H.; et al. Oncogenic Kras Activates a Hematopoietic-to-Epithelial IL-17 Signaling Axis in Preinvasive Pancreatic Neoplasia. Cancer Cell 2014, 25, 621–637. [Google Scholar] [CrossRef] [Green Version]
- Dey, P.; Li, J.; Zhang, J.; Chaurasiya, S.; Strom, A.; Wang, H.; Liao, W.-T.; Cavallaro, F.; Denz, P.; Bernard, V.; et al. Oncogenic KRAS-Driven Metabolic Reprogramming in Pancreatic Cancer Cells Utilizes Cytokines from the Tumor Microenvironment. Cancer Discov. 2020, 10, 608–625. [Google Scholar] [CrossRef]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.-H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both P16(Ink4a) and the P19(Arf)-P53 Pathway Constrain Progression of Pancreatic Adenocarcinoma in the Mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hustinx, S.R.; Leoni, L.M.; Yeo, C.J.; Brown, P.N.; Goggins, M.; Kern, S.E.; Hruban, R.H.; Maitra, A. Concordant Loss of MTAP and P16/CDKN2A Expression in Pancreatic Intraepithelial Neoplasia: Evidence of Homozygous Deletion in a Noninvasive Precursor Lesion. Mod. Pathol. 2005, 18, 959–963. [Google Scholar] [CrossRef]
- Fukushima, N.; Sato, N.; Ueki, T.; Rosty, C.; Walter, K.M.; Wilentz, R.E.; Yeo, C.J.; Hruban, R.H.; Goggins, M. Aberrant Methylation of Preproenkephalin and P16 Genes in Pancreatic Intraepithelial Neoplasia and Pancreatic Ductal Adenocarcinoma. Am. J. Pathol. 2002, 160, 1573–1581. [Google Scholar] [CrossRef] [Green Version]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; DePinho, R.A. Genetics and Biology of Pancreatic Ductal Adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [Green Version]
- Scarlett, C.J.; Salisbury, E.L.; Biankin, A.V.; Kench, J. Precursor Lesions in Pancreatic Cancer: Morphological and Molecular Pathology. Pathology 2011, 43, 183–200. [Google Scholar] [CrossRef]
- Wörmann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Görgülü, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2–STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology 2016, 151, 180–193.e12. [Google Scholar] [CrossRef] [Green Version]
- Escobar-Hoyos, L.F.; Penson, A.; Kannan, R.; Cho, H.; Pan, C.-H.; Singh, R.K.; Apken, L.H.; Hobbs, G.A.; Luo, R.; Lecomte, N.; et al. Altered RNA Splicing by Mutant P53 Activates Oncogenic RAS Signaling in Pancreatic Cancer. Cancer Cell 2020, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Chen, H.; Sun, L.; Wang, M.; Wu, X.; Xiao, Z.-X.J. Hotspot Mutant P53-R273H Inhibits KLF6 Expression to Promote Cell Migration and Tumor Metastasis. Cell Death Dis. 2020, 11, 595. [Google Scholar] [CrossRef]
- Butera, G.; Pacchiana, R.; Mullappilly, N.; Margiotta, M.; Bruno, S.; Conti, P.; Riganti, C.; Donadelli, M. Mutant P53 Prevents GAPDH Nuclear Translocation in Pancreatic Cancer Cells Favoring Glycolysis and 2-Deoxyglucose Sensitivity. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1914–1923. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and Invasive Ductal Pancreatic Cancer and Its Early Detection in the Mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.; Wagenblast, E.; Davis, C.A.; Moon, S.-H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant P53 Drives Pancreatic Cancer Metastasis through Cell-Autonomous PDGF Receptor β Signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [Green Version]
- Wartenberg, M.; Cibin, S.; Zlobec, I.; Vassella, E.; Eppenberger-Castori, S.; Terracciano, L.; Eichmann, M.D.; Worni, M.; Gloor, B.; Perren, A.; et al. Integrated Genomic and Immunophenotypic Classification of Pancreatic Cancer Reveals Three Distinct Subtypes with Prognostic/Predictive Significance. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4444–4454. [Google Scholar] [CrossRef] [Green Version]
- Iacobuzio-Donahue, C.A.; Fu, B.; Yachida, S.; Luo, M.; Abe, H.; Henderson, C.M.; Vilardell, F.; Wang, Z.; Keller, J.W.; Banerjee, P.; et al. DPC4 Gene Status of the Primary Carcinoma Correlates with Patterns of Failure in Patients with Pancreatic Cancer. J. Clin. Oncol. 2009, 27, 1806–1813. [Google Scholar] [CrossRef] [Green Version]
- Schwarte-Waldhoff, I.; Volpert, O.V.; Bouck, N.P.; Sipos, B.; Hahn, S.A.; Klein-Scory, S.; Lüttges, J.; Klöppel, G.; Graeven, U.; Eilert-Micus, C.; et al. Smad4/DPC4-Mediated Tumor Suppression through Suppression of Angiogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 9624–9629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-W.; Hsiao, P.-J.; Weng, C.-C.; Kuo, K.-K.; Kuo, T.-L.; Wu, D.-C.; Hung, W.-C.; Cheng, K.-H. SMAD4 Loss Triggers the Phenotypic Changes of Pancreatic Ductal Adenocarcinoma Cells. BMC Cancer 2014, 14, 181. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Xia, X.; Yang, C.; Shen, J.; Mai, J.; Kim, H.-C.; Kirui, D.; Kang, Y.; Fleming, J.B.; Koay, E.J.; et al. SMAD4 Gene Mutation Renders Pancreatic Cancer Resistance to Radiotherapy through Promotion of Autophagy. Clin Cancer Res. 2018, 24. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Shi, S.; Qin, Y.; Meng, Q.; Hua, J.; Hu, Q.; Ji, S.; Zhang, B.; Xu, J.; Yu, X.-J. Localisation of PGK1 Determines Metabolic Phenotype to Balance Metastasis and Proliferation in Patients with SMAD4-Negative Pancreatic Cancer. Gut 2020, 69, 888. [Google Scholar] [CrossRef]
- Bardeesy, N.; Cheng, K.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 Is Dispensable for Normal Pancreas Development yet Critical in Progression and Tumor Biology of Pancreas Cancer. Gene Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef] [Green Version]
- Kojima, K.; Vickers, S.M.; Adsay, N.V.; Jhala, N.C.; Kim, H.-G.; Schoeb, T.R.; Grizzle, W.E.; Klug, C.A. Inactivation of Smad4 Accelerates KrasG12D-Mediated Pancreatic Neoplasia. Cancer Res. 2007, 67, 8121–8130. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, S.; Oon, C.; Kothari, A.; Horton, W.; Link, J.; Sears, R.C.; Sherman, M.H. Acidic Fibroblast Growth Factor Underlies Microenvironmental Regulation of MYC in Pancreatic Cancer. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Nakhai, H.; Siveke, J.T.; Mendoza-Torres, L.; Schmid, R.M. Conditional Inactivation of Myc Impairs Development of the Exocrine Pancreas. Development 2008, 135, 3191–3196. [Google Scholar] [CrossRef] [Green Version]
- Lobo, V.J.S.-A.; Fernández, L.C.; Carrillo-de-Santa-Pau, E.; Richart, L.; Cobo, I.; Cendrowski, J.; Moreno, U.; del Pozo, N.; Megías, D.; Bréant, B.; et al. C-Myc Downregulation Is Required for Preacinar to Acinar Maturation and Pancreatic Homeostasis. Gut 2018, 67, 707–718. [Google Scholar] [CrossRef]
- Sodir, N.M.; Kortlever, R.M.; Barthet, V.J.A.; Campos, T.; Pellegrinet, L.; Kupczak, S.; Anastasiou, P.; Swigart, L.B.; Soucek, L.; Arends, M.J.; et al. MYC Instructs and Maintains Pancreatic Adenocarcinoma Phenotype. Cancer Discov. 2020. [Google Scholar] [CrossRef] [Green Version]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Swigart, L.B.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e14. [Google Scholar] [CrossRef] [PubMed]
- Muthalagu, N.; Monteverde, T.; Raffo-Iraolagoitia, X.; Wiesheu, R.; Whyte, D.; Hedley, A.; Laing, S.; Kruspig, B.; Upstill-Goddard, R.; Shaw, R.; et al. Repression of the Type I Interferon Pathway Underlies MYC- and KRAS-Dependent Evasion of NK and B Cells in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2020, 10, 872–887. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, P.; Cañamero, M.; del Pozo, N.; Madriles, F.; Zapata, A.; Real, F.X. Gata6 Is Required for Complete Acinar Differentiation and Maintenance of the Exocrine Pancreas in Adult Mice. Gut 2013, 62, 1481–1488. [Google Scholar] [CrossRef]
- Kwei, K.A.; Bashyam, M.D.; Kao, J.; Ratheesh, R.; Reddy, E.C.; Kim, Y.H.; Montgomery, K.; Giacomini, C.P.; Choi, Y.-L.; Chatterjee, S.; et al. Genomic Profiling Identifies GATA6 as a Candidate Oncogene Amplified in Pancreatobiliary Cancer. Plos Genet. 2008, 4, e1000081. [Google Scholar] [CrossRef]
- MARTINELLI, P.; Madriles, F.; Cañamero, M.; Pau, E.C.S.; Pozo, N.D.; Guerra, C.; Real, F.X. The Acinar Regulator Gata6 Suppresses KrasG12V-Driven Pancreatic Tumorigenesis in Mice. Gut 2015. [Google Scholar] [CrossRef]
- O’Kane, G.M.; Grünwald, B.T.; Jang, G.-H.; Masoomian, M.; Picardo, S.; Grant, R.C.; Denroche, R.E.; Zhang, A.; Wang, Y.; Lam, B.; et al. GATA6 Expression Distinguishes Classical and Basal-like Subtypes in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4901–4910. [Google Scholar] [CrossRef] [Green Version]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of Pancreatic Ductal Adenocarcinoma and Their Differing Responses to Therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- MARTINELLI, P.; Carrillo-de-Santa-Pau, E.; Cox, T.; Jr., B.S.; Dusetti, N.; Greenhalf, W.; Rinaldi, L.; Costello, E.; Ghaneh, P.; Malats, N.; et al. GATA6 Regulates EMT and Tumour Dissemination, and Is a Marker of Response to Adjuvant Chemotherapy in Pancreatic Cancer. Gut 2017, 66, 1665–1676. [Google Scholar] [CrossRef] [Green Version]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell 2018, 33, 512–526.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Figura, G.; Fukuda, A.; Roy, N.; Liku, M.E.; IV, J.P.M.; Kim, G.E.; Russ, H.A.; Firpo, M.A.; Mulvihill, S.J.; Dawson, D.W.; et al. The Chromatin Regulator Brg1 Suppresses Formation of Intraductal Papillary Mucinous Neoplasm and Pancreatic Ductal Adenocarcinoma. Nat. Cell Biol. 2014, 16, 255–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuda, M.; Fukuda, A.; Roy, N.; Hiramatsu, Y.; Leonhardt, L.; Kakiuchi, N.; Hoyer, K.; Ogawa, S.; Goto, N.; Ikuta, K.; et al. The BRG1/SOX9 Axis Is Critical for Acinar Cell–Derived Pancreatic Tumorigenesis. J. Clin. Investig. 2018, 128, 3475–3489. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Malik, S.; Villanueva, K.E.; Urano, A.; Lu, X.; von Figura, G.; Seeley, E.S.; Dawson, D.W.; Collisson, E.A.; Hebrok, M. Brg1 Promotes Both Tumor-Suppressive and Oncogenic Activities at Distinct Stages of Pancreatic Cancer Formation. Genes Dev. 2015, 29, 658–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livshits, G.; Alonso-Curbelo, D.; Morris, J.P.; Koche, R.; Saborowski, M.; Wilkinson, J.E.; Lowe, S.W. Arid1a Restrains Kras-Dependent Changes in Acinar Cell Identity. eLife 2018, 7, 1766. [Google Scholar] [CrossRef]
- Wang, W.; Friedland, S.C.; Guo, B.; O’Dell, M.R.; Alexander, W.B.; Whitney-Miller, C.L.; Agostini-Vulaj, D.; Huber, A.R.; Myers, J.R.; Ashton, J.M.; et al. ARID1A, a SWI/SNF Subunit, Is Critical to Acinar Cell Homeostasis and Regeneration and Is a Barrier to Transformation and Epithelial-Mesenchymal Transition in the Pancreas. Gut 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, C.; Yu, S.; Jia, C.; Yan, J.; Lu, Z.; Chen, J. Loss of ARID1A Expression Correlates With Tumor Differentiation and Tumor Progression Stage in Pancreatic Ductal Adenocarcinoma. Technol. Cancer Res. Treat. 2018, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, Y.; Fukuda, A.; Ogawa, S.; Maruno, T.; Takada, Y.; Tsuda, M.; Hiramatsu, Y.; Araki, O.; Nagao, M.; Yoshikawa, T.; et al. ARID1A Maintains Differentiation of Pancreatic Ductal Cells and Inhibits Development of Pancreatic Ductal Adenocarcinoma in Mice. Gastroenterology 2018, 155, 194–209.e2. [Google Scholar] [CrossRef] [Green Version]
- Helming, K.C.; Wang, X.; Wilson, B.G.; Vazquez, F.; Haswell, J.R.; Manchester, H.E.; Kim, Y.; Kryukov, G.V.; Ghandi, M.; Aguirre, A.J.; et al. ARID1B Is a Specific Vulnerability in ARID1A-Mutant Cancers. Nat. Med. 2014, 20, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, G.R.; Rahal, R.; Buxton, F.; Xiang, K.; McAllister, G.; Frias, E.; Bagdasarian, L.; Huber, J.; Lindeman, A.; Chen, D.; et al. Functional Epigenetics Approach Identifies BRM/SMARCA2 as a Critical Synthetic Lethal Target in BRG1-Deficient Cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 3128–3133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.-M.; et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, S.; Shimada, S.; Akiyama, Y.; Ishikawa, Y.; Ogura, T.; Ogawa, K.; Ono, H.; Mitsunori, Y.; Ban, D.; Kudo, A.; et al. Loss of KDM6A Characterizes a Poor Prognostic Subtype of Human Pancreatic Cancer and Potentiates HDAC Inhibitor Lethality. Int. J. Cancer 2019, 145, 192–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [Green Version]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual Microdissection Identifies Distinct Tumor- and Stroma-Specific Subtypes of Pancreatic Ductal Adenocarcinoma. Nat. Genet. 2015, 1–13. [Google Scholar] [CrossRef]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013.e3. [Google Scholar] [CrossRef] [Green Version]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant Metastasis Occurs Late during the Genetic Evolution of Pancreatic Cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited Heterogeneity of Known Driver Gene Mutations among the Metastases of Individual Patients with Pancreatic Cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Lemire, M.; Zhang, A.; Chan-Seng-Yue, M.; Wilson, G.; Grant, R.C.; Merico, D.; Lungu, I.; et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 2019, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; Mclaren, S.; Lin, M.-L.; et al. The Patterns and Dynamics of Genomic Instability in Metastatic Pancreatic Cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-D.; Jin, K.; Chen, X.-Y.; Lv, J.-Q.; Ji, K.-W. Clinicopathological Significance of SMAD4 Loss in Pancreatic Ductal Adenocarcinomas: A Systematic Review and Meta-Analysis. Oncotarget 2017, 8, 16704–16711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic Reprogramming during Pancreatic Cancer Progression Links Anabolic Glucose Metabolism to Distant Metastasis. Nat. Genet. 2017, 49, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.; Marayati, R.; Moffitt, R.; Yeh, J.J. Hexokinase 2 Promotes Tumor Growth and Metastasis by Regulating Lactate Production in Pancreatic Cancer. Oncotarget 2017, 8, 56081–56094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couch, F.J.; Johnson, M.R.; Rabe, K.G.; Brune, K.; de Andrade, M.; Goggins, M.; Rothenmund, H.; Gallinger, S.; Klein, A.; Petersen, G.M.; et al. The Prevalence of BRCA2 Mutations in Familial Pancreatic Cancer. Cancer Epidemiol. Prev. Biomark. 2007, 16, 342–346. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; Hruban, R.H.; Kamiyama, M.; Borges, M.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Palmisano, E.; Brune, K.; Jaffee, E.M.; et al. Exomic Sequencing Identifies PALB2 as a Pancreatic Cancer Susceptibility Gene. Science 2009, 324, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, N.J.; Jiao, Y.; Yu, J.; Kopelovich, L.; Petersen, G.M.; Bondy, M.L.; Gallinger, S.; Schwartz, A.G.; Syngal, S.; Cote, M.L.; et al. ATM Mutations in Patients with Hereditary Pancreatic Cancer. Cancer Discov. 2012, 2, 41–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.P.; Brune, K.A.; Petersen, G.M.; Goggins, M.; Tersmette, A.C.; Offerhaus, G.J.A.; Griffin, C.; Cameron, J.L.; Yeo, C.J.; Kern, S.; et al. Prospective Risk of Pancreatic Cancer in Familial Pancreatic Cancer Kindreds. Cancer Res. 2004, 64, 2634–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, G.M.; Amundadottir, L.; Fuchs, C.S.; Kraft, P.; Stolzenberg-Solomon, R.Z.; Jacobs, K.B.; Arslan, A.A.; Bueno-de-Mesquita, H.B.; Gallinger, S.; Gross, M.; et al. A Genome-Wide Association Study Identifies Pancreatic Cancer Susceptibility Loci on Chromosomes 13q22.1, 1q32.1 and 5p15.33. Nat. Genet. 2010, 42, 224–228. [Google Scholar] [CrossRef] [Green Version]
- Flandez, M.; Cendrowski, J.; Cañamero, M.; Salas, A.; del Pozo, N.; Schoonjans, K.; Real, F.X. Nr5a2 Heterozygosity Sensitises to, and Cooperates with, Inflammation in KRas(G12V)-Driven Pancreatic Tumourigenesis. Gut 2014, 63, 647–655. [Google Scholar] [CrossRef]
- Cobo, I.; Martinelli, P.; Flández, M.; Bakiri, L.; Zhang, M.; Carrillo-de-Santa-Pau, E.; Jia, J.; Lobo, V.J.S.-A.; Megías, D.; Felipe, I.; et al. Transcriptional Regulation by NR5A2 Links Differentiation and Inflammation in the Pancreas. Nature 2018, 554, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Amundadottir, L.; Kraft, P.; Stolzenberg-Solomon, R.Z.; Fuchs, C.S.; Petersen, G.M.; Arslan, A.A.; Bueno-de-Mesquita, H.B.; Gross, M.; Helzlsouer, K.; Jacobs, E.J.; et al. Genome-Wide Association Study Identifies Variants in the ABO Locus Associated with Susceptibility to Pancreatic Cancer. Nat. Genet. 2009, 41, 986–990. [Google Scholar] [CrossRef] [Green Version]
- Vera, R.; Dotor, E.; Feliu, J.; González, E.; Laquente, B.; Macarulla, T.; Martínez, E.; Maurel, J.; Salgado, M.; Manzano, J.L. SEOM Clinical Guideline for the Treatment of Pancreatic Cancer (2016). Clin. Transl. Oncol. 2016, 18, 1172–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Cutsem, E.V.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Ko, A.H.; Bekaii-Saab, T.; Ziffle, J.V.; Mirzoeva, O.M.; Joseph, N.M.; Talasaz, A.; Kuhn, P.; Tempero, M.A.; Collisson, E.A.; Kelley, R.K.; et al. A Multicenter, Open-Label Phase II Clinical Trial of Combined MEK plus EGFR Inhibition for Chemotherapy-Refractory Advanced Pancreatic Adenocarcinoma. Clin. Cancer Res. 2016, 22, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Garcia, P.L.; Miller, A.L.; Kreitzburg, K.M.; Council, L.N.; Gamblin, T.L.; Christein, J.D.; Heslin, M.J.; Arnoletti, J.P.; Richardson, J.H.; Chen, D.; et al. The BET Bromodomain Inhibitor JQ1 Suppresses Growth of Pancreatic Ductal Adenocarcinoma in Patient-Derived Xenograft Models. Oncogene 2015, 1–13. [Google Scholar] [CrossRef]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sanchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.A.; Vangala, D.; et al. Combined Inhibition of BET Family Proteins and Histone Deacetylases as a Potential Epigenetics-Based Therapy for Pancreatic Ductal Adenocarcinoma. Nature Med. 2015. [Google Scholar] [CrossRef] [Green Version]
- Richart, L.; Carrillo-de-Santa-Pau, E.; Río-Machín, A.; de Andrés, M.P.; Cigudosa, J.C.; Lobo, V.J.S.-A.; Real, F.X. BPTF Is Required for C-MYC Transcriptional Activity and in Vivo Tumorigenesis. Nat. Commun. 2016, 7, 10153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabacaoglu, D.; Ciecielski, K.J.; Ruess, D.A.; Algül, H. Immune Checkpoint Inhibition for Pancreatic Ductal Adenocarcinoma: Current Limitations and Future Options. Front. Immunol. 2018, 9, 2913–2924. [Google Scholar] [CrossRef]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 Antagonist, in Combination with Pembrolizumab and Chemotherapy for Pancreatic Cancer: The COMBAT Trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes with Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting Focal Adhesion Kinase Renders Pancreatic Cancers Responsive to Checkpoint Immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog Signaling Enhances Delivery of Chemotherapy in a Mouse Model of Pancreatic Cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagathihalli, N.S.; Castellanos, J.A.; Shi, C.; Beesetty, Y.; Reyzer, M.L.; Caprioli, R.; Chen, X.; Walsh, A.J.; Skala, M.C.; Moses, H.L.; et al. Signal Transducer and Activator of Transcription 3, Mediated Remodeling of the Tumor Microenvironment Results in Enhanced Tumor Drug Delivery in a Mouse Model of Pancreatic Cancer. Gastroenterology 2015, 149, 1932–1943.e9. [Google Scholar] [CrossRef] [Green Version]

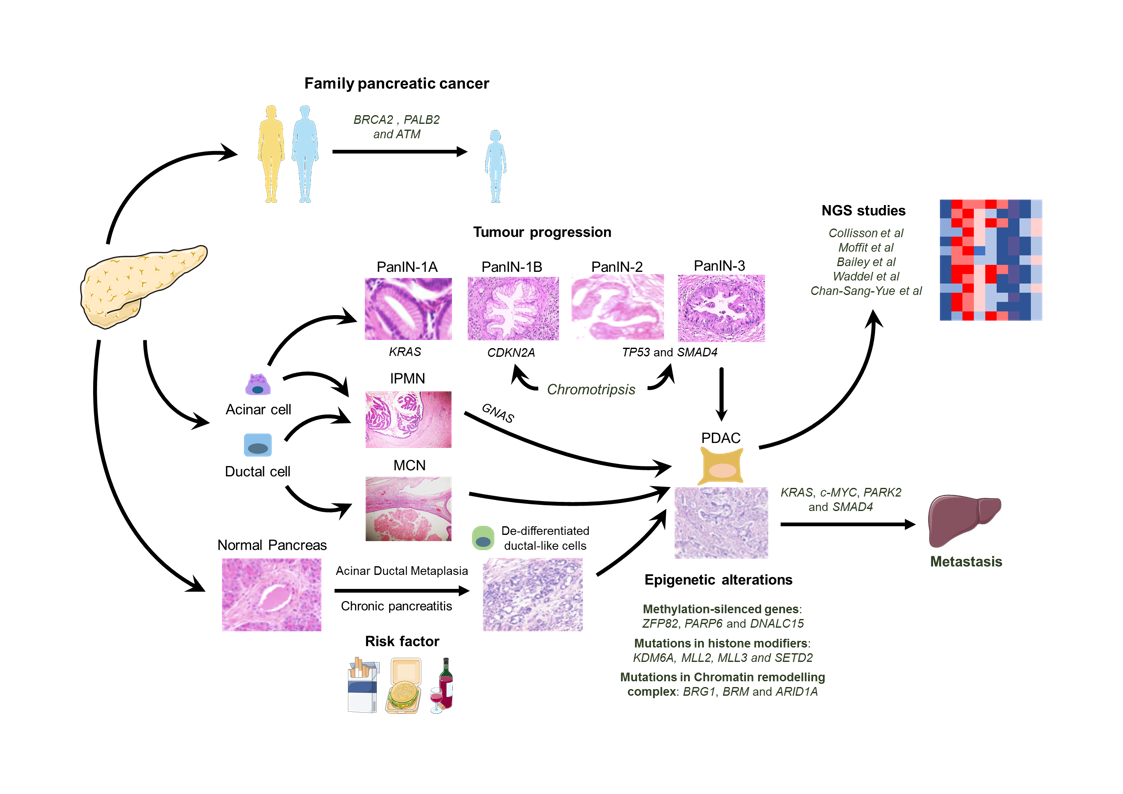{kind=link}
| Benign | Precursors | Malignant |
|---|---|---|
| Serous cystadenoma | Pancreatic intraepithelial neoplasia: PanIN 1, 2 or 3 | Serous adenocarcinoma Pancreatic ductal adenocarcinoma (PDA) |
| Mucinous cystic neoplasia (MCN) with low- or high-grade dysplasia (Mucinous cystadenoma) | Mucinous cystic neoplasia (MCN) with invasive carcinoma (PDA) | |
| Intraductal papillary mucinous neoplasm (IPMN) | Intraductal papillary mucinous neoplasm with invasive carcinoma (PDA, colloid carcinoma, etc.) | |
| Pancreatic intraductal oncocytic papillary neoplas (IOPN) | IOPN with associated invasive carcinoma | |
| Intraductal tubular papillary neoplasia (ITPN) | ITPN with invasive carcinoma | |
| Acinar cell carcinoma (ACC) | ||
| Pancreatoblastoma | ||
| Pseudopapillary solid neoplasm | Solid pseudopapillary neoplasm with recurrence | |
| Mature teratoma | Immature teratoma |
| Collisson et al. | Moffit et al. | Bailey et al. | Puleo et al. | Chan-Sang-Yue et al. | Waddell et al. |
|---|---|---|---|---|---|
| Classic Mesenchymal Exocrine | Classic Basal | Progenitor Squamous ADEX | Pure classical Pure basal-like | Classic A and B Basal-like A and B | Stable with local rearrangements Scattered rearrangements Unstable |
| Immunogenic | Immune Classical | ||||
| Normal/activated stroma | Stroma activated Desmoplastic |
| Syndrome | Inheritance Mode | Gene | PDA Risk (Mean Age) |
|---|---|---|---|
| Lynch syndrome | Autosomal dominant | MSH2 (2p), MLH1 (3p) | 1.3–4% (70 years) |
| Familial breast cancer (BRCA2) and Fanconi anemia (FANCC and FANCG) | Autosomal dominant | BRACA2 (13q); PALB2 (16p); FANCG (9p); BRCA1 (17q) | 3.5–10% |
| X Family | Autosomal domina | PALLADIN (4q) | Unknown incidence |
| Familial melanoma syndrome | Autosomal dominant | CDKN2A (9p) | 10–17% |
| Hereditary pancreatitis | Autosomal dominant or recessive | PRSS1 (7q) SPINK1 (5q) | 25–40% (60 years) |
| Peutz–Jeghers | Autosomal dominant | STK11 | 30–60% (70 years) |
| Familial pancreatic cancer | Autosomal dominant | Unknown SNP alterations postulated (telomerase, NR5A2, 13q22.1, 15q14) Locus 9q34 of the blood group | 9–38% (80 years) Group AB0 (group 0 phenotype has less risk than blood groups A and B) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez Gil, Y.; Jiménez Sánchez, P.; Muñoz Velasco, R.; García García, A.; Sánchez-Arévalo Lobo, V.J. Molecular Alterations in Pancreatic Cancer: Transfer to the Clinic. Int. J. Mol. Sci. 2021, 22, 2077. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042077
Rodríguez Gil Y, Jiménez Sánchez P, Muñoz Velasco R, García García A, Sánchez-Arévalo Lobo VJ. Molecular Alterations in Pancreatic Cancer: Transfer to the Clinic. International Journal of Molecular Sciences. 2021; 22(4):2077. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042077
Chicago/Turabian StyleRodríguez Gil, Yolanda, Paula Jiménez Sánchez, Raúl Muñoz Velasco, Ana García García, and Víctor Javier Sánchez-Arévalo Lobo. 2021. "Molecular Alterations in Pancreatic Cancer: Transfer to the Clinic" International Journal of Molecular Sciences 22, no. 4: 2077. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042077