
How Is the Fidelity of Proteins Ensured in Terms of Both Quality and Quantity at the Endoplasmic Reticulum? Mechanistic Insights into E3 Ubiquitin Ligases


Abstract
:1. Introduction
2. Ubiquitination
2.1. A Conceptual Overview of Ubiquitination
2.2. Understanding the Catalytic Mechanisms of E3 Ubiquitin Ligases
2.3. E4 Ubiuqitin Ligases and Ubiquitin Chain Elongation
2.4. Ubiquitin Code and Its Biological Significance
3. “Protein Quality Control” and “Protein Quantity Control” at the ER: ERAD
3.1. Substrate Recognition
3.2. Retrotranslocation
3.3. Ubiquitination and E3 Ubiquitin Ligases
3.3.1. HRD1
3.3.2. RNF45/gp78/AMFR
3.3.3. MARCHF6/TEB4/RNF176
3.3.4. RNF5/RMA1
3.4. Delivery and Degradation
4. “Protein Quality Control” at the ER: ER-Phagy
4.1. Macro-ER-Phagy
4.1.1. A Conceptual Overview of Macro-ER-Phagy
4.1.2. Macro-ER-Phagy and Protein Quality Control
4.2. Micro-ER-Phagy
4.3. Vesicular Delivery
5. Brief Linking of “Protein Quality Control” at the ER to Neurodegenerations
6. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bedford, L.; Lowe, J.; Dick, L.R.; Mayer, R.J.; Brownell, J.E. Ubiquitin-like protein conjugation and the ubiquitin–proteasome system as drug targets. Nat. Rev. Drug Discov. 2011, 10, 29–46. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clague, M.J.; Liu, H.; Urbé, S. Governance of endocytic trafficking and signaling by reversible ubiquitylation. Dev. Cell 2012, 23, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Wright, J.D.; Mace, P.D.; Day, C.L. Noncovalent ubiquitin interactions regulate the catalytic activity of ubiquitin writers. Trends Biochem. Sci. 2016, 41, 924–937. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Shabek, N. Ubiquitin ligases: Structure, function, and regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef]
- Mattern, M.; Sutherland, J.; Kadimisetty, K.; Barrio, R.; Rodriguez, M.S. Using ubiquitin binders to decipher the ubiquitin code. Trends Biochem. Sci. 2019, 44, 599–615. [Google Scholar] [CrossRef]
- Oh, E.; Akopian, D.; Rape, M. Principles of ubiquitin-dependent signaling. Annu. Rev. Cell Dev. Biol. 2018, 34, 137–162. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Song, L.; Luo, Z.-Q. Post-translational regulation of ubiquitin signaling. J. Cell Biol. 2019, 218, 1776–1786. [Google Scholar] [CrossRef]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Heaton, S.M.; Borg, N.A.; Dixit, V.M. Ubiquitin in the activation and attenuation of innate antiviral immunity. J. Exp. Med. 2016, 213, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Gilberto, S.; Peter, M. Dynamic ubiquitin signaling in cell cycle regulation. J. Cell Biol. 2017, 216, 2259–2271. [Google Scholar] [CrossRef] [PubMed]
- Han, H.G.; Moon, H.W.; Jeon, Y.J. ISG15 in cancer: Beyond ubiquitin-like protein. Cancer Lett. 2018, 438, 52–62. [Google Scholar] [CrossRef]
- Kang, J.A.; Jeon, Y.J. Emerging roles of USP18: From biology to pathophysiology. Int. J. Mol. Sci. 2020, 21, 6825. [Google Scholar] [CrossRef] [PubMed]
- Jeon, Y.J.; Yoo, H.M.; Chung, C.H. ISG15 and immune diseases. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2010, 1802, 485–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Brodsky, J.L. Protein quality control in the secretory pathway. J. Cell Biol. 2019, 218, 3171–3187. [Google Scholar] [CrossRef] [Green Version]
- Fregno, I.; Molinari, M. Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Sloper-Mould, K.E.; Jemc, J.C.; Pickart, C.M.; Hicke, L. Distinct functional surface regions on ubiquitin. J. Biol. Chem. 2001, 276, 30483–30489. [Google Scholar] [CrossRef] [Green Version]
- Özkaynak, E.; Finley, D.; Varshavsky, A. The yeast ubiquitin gene: Head-to-tail repeats encoding a polyubiquitin precursor protein. Nature 1984, 312, 663–666. [Google Scholar] [CrossRef]
- Finley, D.; Bartel, B.; Varshavsky, A. The tails of ubiquitin precursors are ribosomal proteins whose fusion to ubiquitin facilitates ribosome biogenesis. Nature 1989, 338, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Rape, M. Ubiquitylation at the crossroads of development and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 59–70. [Google Scholar] [CrossRef]
- Kliza, K.; Husnjak, K. Resolving the complexity of ubiquitin networks. Front. Mol. Biosci. 2020, 7, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, M.W.; Wuebbens, M.M.; Rajagopalan, K.; Schindelin, H. Mechanism of ubiquitin activation revealed by the structure of a bacterial MoeB–MoaD complex. Nature 2001, 414, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Walden, H.; Podgorski, M.S.; Schulman, B.A. Insights into the ubiquitin transfer cascade from the structure of the activating enzyme for NEDD8. Nature 2003, 422, 330–334. [Google Scholar] [CrossRef]
- Lois, L.M.; Lima, C.D. Structures of the SUMO E1 provide mechanistic insights into SUMO activation and E2 recruitment to E1. EMBO J. 2005, 24, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Schindelin, H. Structural insights into E1-catalyzed ubiquitin activation and transfer to conjugating enzymes. Cell 2008, 134, 268–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.T.; Paydar, A.; Zhuang, M.; Waddell, M.B.; Holton, J.M.; Schulman, B.A. Structural basis for recruitment of Ubc12 by an E2 binding domain in NEDD8′s E1. Mol. Cell 2005, 17, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Walden, H.; Podgorski, M.S.; Huang, D.T.; Miller, D.W.; Howard, R.J.; Minor, D.L., Jr.; Holton, J.M.; Schulman, B.A. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol. Cell 2003, 12, 1427–1437. [Google Scholar] [CrossRef]
- Schulman, B.A.; Harper, J.W. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Burroughs, A.M.; Jaffee, M.; Iyer, L.M.; Aravind, L. Anatomy of the E2 ligase fold: Implications for enzymology and evolution of ubiquitin/Ub-like protein conjugation. J. Struct. Biol. 2008, 162, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Wijk, S.J.; Timmers, H.M. The family of ubiquitin-conjugating enzymes (E2s): Deciding between life and death of proteins. FASEB J. 2010, 24, 981–993. [Google Scholar] [CrossRef]
- Buetow, L.; Huang, D.T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016, 17, 626. [Google Scholar] [CrossRef] [Green Version]
- Kravtsova-Ivantsiv, Y.; Ciechanover, A. Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 2012, 125, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, K.-C.; Wood, N.T.; Knebel, A.; Rafie, K.; Stanley, M.; Mabbitt, P.D.; Sundaramoorthy, R.; Hofmann, K.; van Aalten, D.M.; Virdee, S. Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nature 2018, 556, 381–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadwell, K.; Coscoy, L. Ubiquitination on nonlysine residues by a viral E3 ubiquitin ligase. Science 2005, 309, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, Y.; Okuda-Shimizu, Y.; Hendershot, L.M. Ubiquitylation of an ERAD substrate occurs on multiple types of amino acids. Mol. Cell 2010, 40, 917–926. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Herr, R.A.; Hansen, T.H. Ubiquitination of substrates by esterification. Traffic 2012, 13, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Bhogaraju, S.; Kalayil, S.; Liu, Y.; Bonn, F.; Colby, T.; Matic, I.; Dikic, I. Phosphoribosylation of ubiquitin promotes serine ubiquitination and impairs conventional ubiquitination. Cell 2016, 167, 1636–1649.e1613. [Google Scholar] [CrossRef]
- Qiu, J.; Sheedlo, M.J.; Yu, K.; Tan, Y.; Nakayasu, E.S.; Das, C.; Liu, X.; Luo, Z.-Q. Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature 2016, 533, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Mevissen, T.E.; Komander, D. Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [Green Version]
- Clague, M.J.; Barsukov, I.; Coulson, J.M.; Liu, H.; Rigden, D.J.; Urbé, S. Deubiquitylases from genes to organism. Physiol. Rev. 2013, 93, 1289–1315. [Google Scholar] [CrossRef]
- Rehman, S.A.A.; Kristariyanto, Y.A.; Choi, S.-Y.; Nkosi, P.J.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 is a member of an evolutionarily conserved and structurally distinct new family of deubiquitinating enzymes. Mol. Cell 2016, 63, 146–155. [Google Scholar]
- Yau, R.; Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 2016, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 339–434. [Google Scholar] [CrossRef] [PubMed]
- Ohi, M.D.; Vander Kooi, C.W.; Rosenberg, J.A.; Chazin, W.J.; Gould, K.L. Structural insights into the U-box, a domain associated with multi-ubiquitination. Nat. Struct. Mol. Biol. 2003, 10, 250–255. [Google Scholar] [CrossRef]
- Dou, H.; Buetow, L.; Sibbet, G.J.; Cameron, K.; Huang, D.T. BIRC7–E2 ubiquitin conjugate structure reveals the mechanism of ubiquitin transfer by a RING dimer. Nat. Struct. Mol. Biol. 2012, 19, 876. [Google Scholar] [CrossRef] [Green Version]
- Plechanovová, A.; Jaffray, E.G.; Tatham, M.H.; Naismith, J.H.; Hay, R.T. Structure of a RING E3 ligase and ubiquitin-loaded E2 primed for catalysis. Nature 2012, 489, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Pruneda, J.N.; Littlefield, P.J.; Soss, S.E.; Nordquist, K.A.; Chazin, W.J.; Brzovic, P.S.; Klevit, R.E. Structure of an E3: E2∼ Ub complex reveals an allosteric mechanism shared among RING/U-box ligases. Mol. Cell 2012, 47, 933–942. [Google Scholar] [CrossRef] [Green Version]
- Zheng, N.; Wang, P.; Jeffrey, P.D.; Pavletich, N.P. Structure of a c-Cbl–UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell 2000, 102, 533–539. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.; Bonvin, A.M.; Winkler, G.S.; van Schaik, F.M.; Timmers, H.T.M.; Boelens, R. Structural model of the UbcH5B/CNOT4 complex revealed by combining NMR, mutagenesis, and docking approaches. Structure 2004, 12, 633–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buetow, L.; Gabrielsen, M.; Anthony, N.G.; Dou, H.; Patel, A.; Aitkenhead, H.; Sibbet, G.J.; Smith, B.O.; Huang, D.T. Activation of a primed RING E3-E2–ubiquitin complex by non-covalent ubiquitin. Mol. Cell 2015, 58, 297–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mace, P.D.; Linke, K.; Feltham, R.; Schumacher, F.-R.; Smith, C.A.; Vaux, D.L.; Silke, J.; Day, C.L. Structures of the cIAP2 RING domain reveal conformational changes associated with ubiquitin-conjugating enzyme (E2) recruitment. J. Biol. Chem. 2008, 283, 31633–31640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Q.; Lin, S.-C.; Lamothe, B.; Lu, M.; Lo, Y.-C.; Hura, G.; Zheng, L.; Rich, R.L.; Campos, A.D.; Myszka, D.G. E2 interaction and dimerization in the crystal structure of TRAF6. Nat. Struct. Mol. Biol. 2009, 16, 658. [Google Scholar] [CrossRef] [Green Version]
- Plechanovová, A.; Jaffray, E.G.; McMahon, S.A.; Johnson, K.A.; Navrátilová, I.; Naismith, J.H.; Hay, R.T. Mechanism of ubiquitylation by dimeric RING ligase RNF4. Nat. Struct. Mol. Biol. 2011, 18, 1052. [Google Scholar] [CrossRef]
- Brzovic, P.S.; Rajagopal, P.; Hoyt, D.W.; King, M.-C.; Klevit, R.E. Structure of a BRCA1–BARD1 heterodimeric RING–RING complex. Nat. Struct. Biol. 2001, 8, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, G.; van der Stoop, P.; Weichenrieder, O.; Perrakis, A.; van Lohuizen, M.; Sixma, T.K. Structure and E3-ligase activity of the Ring–Ring complex of Polycomb proteins Bmi1 and Ring1b. EMBO J. 2006, 25, 2465–2474. [Google Scholar] [CrossRef]
- Badciong, J.C.; Haas, A.L. MdmX is a RING finger ubiquitin ligase capable of synergistically enhancing Mdm2 ubiquitination. J. Biol. Chem. 2002, 277, 49668–49675. [Google Scholar] [CrossRef] [Green Version]
- Linares, L.K.; Hengstermann, A.; Ciechanover, A.; Müller, S.; Scheffner, M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc. Natl. Acad. Sci. USA 2003, 100, 12009–12014. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Cao, R.; Wang, M.; Myers, M.P.; Zhang, Y.; Xu, R.-M. Structure of a Bmi-1-Ring1B polycomb group ubiquitin ligase complex. J. Biol. Chem. 2006, 281, 20643–20649. [Google Scholar] [CrossRef] [Green Version]
- Micale, L.; Chaignat, E.; Fusco, C.; Reymond, A.; Merla, G. The tripartite motif: Structure and function. Adv. Exp. Med. Biol. 2012, 770, 11. [Google Scholar] [PubMed]
- Lydeard, J.R.; Schulman, B.A.; Harper, J.W. Building and remodelling Cullin–RING E3 ubiquitin ligases. EMBO Rep. 2013, 14, 1050–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of cullin–RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Barford, D. Insights into the anaphase-promoting complex: A molecular machine that regulates mitosis. Curr. Opin. Struct. Biol. 2014, 29, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Kumar, S. Mammalian HECT ubiquitin-protein ligases: Biological and pathophysiological aspects. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2014, 1843, 61–74. [Google Scholar] [CrossRef]
- Rotin, D.; Kumar, S. Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2009, 10, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, D.M.; Lissounov, A.; Brzovic, P.S.; Klevit, R.E. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 2011, 474, 105–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, J.J.; Monteferrario, D.; Noordermeer, S.M.; Van Dijk, W.J.; Van Der Reijden, B.A.; Sixma, T.K. The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J. 2012, 31, 3833–3844. [Google Scholar] [CrossRef] [Green Version]
- Stieglitz, B.; Morris-Davies, A.C.; Koliopoulos, M.G.; Christodoulou, E.; Rittinger, K. LUBAC synthesizes linear ubiquitin chains via a thioester intermediate. EMBO Rep. 2012, 13, 840–846. [Google Scholar] [CrossRef]
- Spratt, D.E.; Walden, H.; Shaw, G.S. RBR E3 ubiquitin ligases: New structures, new insights, new questions. Biochem. J. 2014, 458, 421–437. [Google Scholar] [CrossRef] [Green Version]
- Chaugule, V.K.; Burchell, L.; Barber, K.R.; Sidhu, A.; Leslie, S.J.; Shaw, G.S.; Walden, H. Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J. 2011, 30, 2853–2867. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Kirisako, T.; Kamei, K.; Murata, S.; Kato, M.; Fukumoto, H.; Kanie, M.; Sano, S.; Tokunaga, F.; Tanaka, K.; Iwai, K. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006, 25, 4877–4887. [Google Scholar] [CrossRef]
- Aguilera, M.; Oliveros, M.; Martínez-Padrón, M.; Barbas, J.A.; Ferrús, A. Ariadne-1: A vital Drosophila gene is required in development and defines a new conserved family of ring-finger proteins. Genetics 2000, 155, 1231–1244. [Google Scholar] [PubMed]
- Ikeda, F.; Deribe, Y.L.; Skånland, S.S.; Stieglitz, B.; Grabbe, C.; Franz-Wachtel, M.; Van Wijk, S.J.; Goswami, P.; Nagy, V.; Terzic, J. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 2011, 471, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, F.; Nakagawa, T.; Nakahara, M.; Saeki, Y.; Taniguchi, M.; Sakata, S.-I.; Tanaka, K.; Nakano, H.; Iwai, K. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature 2011, 471, 633–636. [Google Scholar] [CrossRef]
- Walden, H.; Rittinger, K. RBR ligase–mediated ubiquitin transfer: A tale with many twists and turns. Nat. Struct. Mol. Biol. 2018, 25, 440–445. [Google Scholar] [CrossRef]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 enzymes: More than just middle men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef] [Green Version]
- Koegl, M.; Hoppe, T.; Schlenker, S.; Ulrich, H.D.; Mayer, T.U.; Jentsch, S. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 1999, 96, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, T. Multiubiquitylation by E4 enzymes:‘one size’doesn’t fit all. Trends Biochem. Sci. 2005, 30, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Lopata, A.; Kniss, A.; Löhr, F.; Rogov, V.V.; Dötsch, V. Ubiquitination in the ERAD Process. Int. J. Mol. Sci. 2020, 21, 5369. [Google Scholar] [CrossRef]
- Antoniou, N.; Lagopati, N.; Balourdas, D.I.; Nikolaou, M.; Papalampros, A.; Vasileiou, P.V.; Myrianthopoulos, V.; Kotsinas, A.; Shiloh, Y.; Liontos, M. The role of E3, E4 ubiquitin ligase (UBE4B) in human pathologies. Cancers 2020, 12, 62. [Google Scholar] [CrossRef] [Green Version]
- Pant, V.; Lozano, G. Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev. 2014, 28, 1739–1751. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Pop, M.S.; Kulikov, R.; Love, I.M.; Kung, A.L.; Grossman, S.R. CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proc. Natl. Acad. Sci. USA 2009, 106, 16275–16280. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, S.; Yada, M.; Matsumoto, M.; Ishida, N.; Nakayama, K.-I. U box proteins as a new family of ubiquitin-protein ligases. J. Biol. Chem. 2001, 276, 33111–33120. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.S.; Ma, P.C.; Ota, I.M.; Varshavsky, A. A proteolytic pathway that recognizes ubiquitin as a degradation signal. J. Biol. Chem. 1995, 270, 17442–17456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lv, Y.; Zhang, Y.; Gao, H. Regulation of p53 level by UBE4B in breast cancer. PLoS ONE 2014, 9, e90154. [Google Scholar]
- Contino, G.; Amati, F.; Pucci, S.; Pontieri, E.; Pichiorri, F.; Novelli, A.; Botta, A.; Mango, R.; Nardone, A.M.; Sangiuolo, F.C. Expression analysis of the gene encoding for the U-box-type ubiquitin ligase UBE4A in human tissues. Gene 2004, 328, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, M.; Ozaki, T.; Miyazaki, K.; Hayashi, S.; Furuya, K.; Watanabe, K.-i.; Nakagawa, T.; Hanamoto, T.; Todo, S.; Nakagawara, A. UFD2a mediates the proteasomal turnover of p73 without promoting p73 ubiquitination. Oncogene 2005, 24, 7156–7169. [Google Scholar] [CrossRef] [Green Version]
- Carén, H.; Holmstrand, A.; Sjöberg, R.-M.; Martinsson, T. The two human homologues of yeast UFD2 ubiquitination factor, UBE4A and UBE4B, are located in common neuroblastoma deletion regions and are subject to mutations in tumours. Eur. J. Cancer 2006, 42, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Morreale, F.E.; Walden, H. Types of ubiquitin ligases. Cell 2016, 165, 248–248.e241. [Google Scholar] [CrossRef]
- Grossman, S.R.; Deato, M.E.; Brignone, C.; Chan, H.M.; Kung, A.L.; Tagami, H.; Nakatani, Y.; Livingston, D.M. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science 2003, 300, 342–344. [Google Scholar] [CrossRef]
- Arany, Z.n.; Sellers, W.R.; Livingston, D.M.; Eckner, R. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell 1994, 77, 799–800. [Google Scholar] [CrossRef]
- Lundblad, J.R.; Kwok, R.P.; Laurance, M.E.; Harter, M.L.; Goodman, R.H. Adenoviral ElA-associated protein p300 as a functional homologue of the transcriptional co-activator CBP. Nature 1995, 374, 85–88. [Google Scholar] [CrossRef]
- Ballinger, C.A.; Connell, P.; Wu, Y.; Hu, Z.; Thompson, L.J.; Yin, L.-Y.; Patterson, C. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol. 1999, 19, 4535–4545. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Soda, M.; Hatakeyama, S.; Akagi, T.; Hashikawa, T.; Nakayama, K.-I.; Takahashi, R. CHIP is associated with Parkin, a gene responsible for familial Parkinson’s disease, and enhances its ubiquitin ligase activity. Mol. Cell 2002, 10, 55–67. [Google Scholar] [CrossRef]
- Sui, G.; Affar, E.B.; Shi, Y.; Brignone, C.; Wall, N.R.; Yin, P.; Donohoe, M.; Luke, M.P.; Calvo, D.; Grossman, S.R. Yin Yang 1 is a negative regulator of p53. Cell 2004, 117, 859–872. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-S.; Galvin, K.M.; See, R.H.; Eckner, R.; Livingston, D.; Moran, E.; Shi, Y. Relief of YY1 transcriptional repression by adenovirus E1A is mediated by E1A-associated protein p300. Genes Dev. 1995, 9, 1188–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.B.; Mallampalli, R.K. Masking of a nuclear signal motif by monoubiquitination leads to mislocalization and degradation of the regulatory enzyme cytidylyltransferase. Mol. Cell. Biol. 2009, 29, 3062–3075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braten, O.; Livneh, I.; Ziv, T.; Admon, A.; Kehat, I.; Caspi, L.H.; Gonen, H.; Bercovich, B.; Godzik, A.; Jahandideh, S. Numerous proteins with unique characteristics are degraded by the 26S proteasome following monoubiquitination. Proc. Natl. Acad. Sci. USA 2016, 113, E4639–E4647. [Google Scholar] [CrossRef] [Green Version]
- Ohtake, F.; Tsuchiya, H. The emerging complexity of ubiquitin architecture. J. Biochem. 2017, 161, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Dwane, L.; Gallagher, W.M.; Chonghaile, T.N.; O’Connor, D.P. The emerging role of non-traditional ubiquitination in oncogenic pathways. J. Biol. Chem. 2017, 292, 3543–3551. [Google Scholar] [CrossRef] [Green Version]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.T.; Ciechanover, A. The ubiquitin code in the ubiquitin-proteasome system and autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [Green Version]
- Alfano, C.; Faggiano, S.; Pastore, A. The ball and chain of polyubiquitin structures. Trends Biochem. Sci. 2016, 41, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Thrower, J.S.; Hoffman, L.; Rechsteiner, M.; Pickart, C.M. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000, 19, 94–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, N.-L.; Hill, C.P. Defining polyubiquitin chain topology. Nat. Struct. Biol. 2001, 8, 650–652. [Google Scholar] [CrossRef] [PubMed]
- Hicke, L.; Dunn, R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu. Rev. Cell Dev. Biol. 2003, 19, 141–172. [Google Scholar] [CrossRef]
- Huang, T.T.; D’Andrea, A.D. Regulation of DNA repair by ubiquitylation. Nat. Rev. Mol. Cell Biol. 2006, 7, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, H.-J.; Rape, M. Enhanced protein degradation by branched ubiquitin chains. Cell 2014, 157, 910–921. [Google Scholar] [CrossRef] [Green Version]
- Kristariyanto, Y.A.; Rehman, S.A.A.; Campbell, D.G.; Morrice, N.A.; Johnson, C.; Toth, R.; Kulathu, Y. K29-selective ubiquitin binding domain reveals structural basis of specificity and heterotypic nature of k29 polyubiquitin. Mol. Cell 2015, 58, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, M.A.; Swatek, K.N.; Hospenthal, M.K.; Komander, D. Ubiquitin linkage-specific affimers reveal insights into K6-linked ubiquitin signaling. Mol. Cell 2017, 68, 233–246. e235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yau, R.G.; Doerner, K.; Castellanos, E.R.; Haakonsen, D.L.; Werner, A.; Wang, N.; Yang, X.W.; Martinez-Martin, N.; Matsumoto, M.L.; Dixit, V.M. Assembly and function of heterotypic ubiquitin chains in cell-cycle and protein quality control. Cell 2017, 171, 918–933.e920. [Google Scholar] [CrossRef] [Green Version]
- Emmerich, C.H.; Bakshi, S.; Kelsall, I.R.; Ortiz-Guerrero, J.; Shpiro, N.; Cohen, P. Lys63/Met1-hybrid ubiquitin chains are commonly formed during the activation of innate immune signalling. Biochem. Biophys. Res. Commun. 2016, 474, 452–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmerich, C.H.; Ordureau, A.; Strickson, S.; Arthur, J.S.C.; Pedrioli, P.G.; Komander, D.; Cohen, P. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc. Natl. Acad. Sci. USA 2013, 110, 15247–15252. [Google Scholar] [CrossRef] [Green Version]
- Wertz, I.E.; Newton, K.; Seshasayee, D.; Kusam, S.; Lam, C.; Zhang, J.; Popovych, N.; Helgason, E.; Schoeffler, A.; Jeet, S. Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature 2015, 528, 370–375. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Nam, S.M.; Jeon, Y.J. Proteostasis in the endoplasmic reticulum: Road to cure. Cancers 2019, 11, 1793. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.W.; Han, H.G.; Jeon, Y.J. Protein quality control in the endoplasmic reticulum and cancer. Int. J. Mol. Sci. 2018, 19, 3020. [Google Scholar] [CrossRef] [Green Version]
- Yoo, Y.S.; Han, H.G.; Jeon, Y.J. Unfolded protein response of the endoplasmic reticulum in tumor progression and immunogenicity. Oxidative Med. Cell. Longev. 2017, 2017, 2969271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodsky, J.L.; Wojcikiewicz, R.J. Substrate-specific mediators of ER associated degradation (ERAD). Curr. Opin. Cell Biol. 2009, 21, 516–521. [Google Scholar] [CrossRef] [Green Version]
- Hampton, R.Y. ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 2002, 14, 476–482. [Google Scholar] [CrossRef]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef]
- Adle, D.J.; Wei, W.; Smith, N.; Bies, J.J.; Lee, J. Cadmium-mediated rescue from ER-associated degradation induces expression of its exporter. Proc. Natl. Acad. Sci. USA 2009, 106, 10189–10194. [Google Scholar] [CrossRef] [Green Version]
- Morito, D.; Nagata, K. Pathogenic hijacking of ER-associated degradation: Is ERAD flexible? Mol. Cell 2015, 59, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Christianson, J.C.; Ye, Y. Cleaning up in the endoplasmic reticulum: Ubiquitin in charge. Nat. Struct. Mol. Biol. 2014, 21, 325. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Kopito, R.R.; Christianson, J.C. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol. 2013, 5, a013185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Ng, D.T. Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell Biol. 2015, 16, 742–752. [Google Scholar] [CrossRef]
- Dougan, S.K.; Hu, C.-C.A.; Paquet, M.-E.; Greenblatt, M.B.; Kim, J.; Lilley, B.N.; Watson, N.; Ploegh, H.L. Derlin-2-deficient mice reveal an essential role for protein dislocation in chondrocytes. Mol. Cell. Biol. 2011, 31, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Francisco, A.B.; Singh, R.; Li, S.; Vani, A.K.; Yang, L.; Munroe, R.J.; Diaferia, G.; Cardano, M.; Biunno, I.; Qi, L. Deficiency of suppressor enhancer Lin12 1 like (SEL1L) in mice leads to systemic endoplasmic reticulum stress and embryonic lethality. J. Biol. Chem. 2010, 285, 13694–13703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggiano, A.; Foresti, O.; Carvalho, P. ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 869–879. [Google Scholar] [CrossRef] [Green Version]
- Helenius, A.; Aebi, M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004, 73, 1019–1049. [Google Scholar] [CrossRef]
- Gonzalez, D.S.; Karaveg, K.; Vandersall-Nairn, A.S.; Lal, A.; Moremen, K.W. Identification, expression, and characterization of a cDNA encoding human endoplasmic reticulum mannosidase I, the enzyme that catalyzes the first mannose trimming step in mammalian Asn-linked oligosaccharide biosynthesis. J. Biol. Chem. 1999, 274, 21375–21386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, L.O.; Herscovics, A. Cloning and expression of a specific human α1, 2-mannosidase that trims Man9GlcNAc2 to Man8GlcNAc2 isomer B during N-glycan biosynthesis. Glycobiology 1999, 9, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Olivari, S.; Cali, T.; Salo, K.E.; Paganetti, P.; Ruddock, L.W.; Molinari, M. EDEM1 regulates ER-associated degradation by accelerating de-mannosylation of folding-defective polypeptides and by inhibiting their covalent aggregation. Biochem. Biophys. Res. Commun. 2006, 349, 1278–1284. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Tremblay, L.O.; Sleno, B.; Kamiya, Y.; Wada, I.; Nagata, K.; Kato, K.; Herscovics, A. EDEM1 accelerates the trimming of α1, 2-linked mannose on the C branch of N-glycans. Glycobiology 2010, 20, 567–575. [Google Scholar] [CrossRef] [Green Version]
- Hirao, K.; Natsuka, Y.; Tamura, T.; Wada, I.; Morito, D.; Natsuka, S.; Romero, P.; Sleno, B.; Tremblay, L.O.; Herscovics, A. EDEM3, a soluble EDEM homolog, enhances glycoprotein endoplasmic reticulum-associated degradation and mannose trimming. J. Biol. Chem. 2006, 281, 9650–9658. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Kamiya, Y.; Kamiya, D.; Kato, K.; Nagata, K. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem. 2009, 284, 17061–17068. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; You, Z.; Tremblay, L.O.; Nagata, K.; Herscovics, A. Stimulation of ERAD of misfolded null Hong Kong α1-antitrypsin by Golgi α1, 2-mannosidases. Biochem. Biophys. Res. Commun. 2007, 362, 626–632. [Google Scholar] [CrossRef]
- Lederkremer, G.Z. Glycoprotein folding, quality control and ER-associated degradation. Curr. Opin. Struct. Biol. 2009, 19, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Ninagawa, S.; Okada, T.; Sumitomo, Y.; Kamiya, Y.; Kato, K.; Horimoto, S.; Ishikawa, T.; Takeda, S.; Sakuma, T.; Yamamoto, T. EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol. 2014, 206, 347–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernasconi, R.; Pertel, T.; Luban, J.; Molinari, M. A dual task for the Xbp1-responsive OS-9 variants in the mammalian endoplasmic reticulum: Inhibiting secretion of misfolded protein conformers and enhancing their disposal. J. Biol. Chem. 2008, 283, 16446–16454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1–SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, N.; Wada, I.; Nagasawa, K.; Moriyama, T.; Okawa, K.; Nagata, K. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J. Biol. Chem. 2008, 283, 20914–20924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plemper, R.K.; Böhmler, S.; Bordallo, J.; Sommer, T.; Wolf, D.H. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature 1997, 388, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Ushioda, R.; Hoseki, J.; Nagata, K. Glycosylation-independent ERAD pathway serves as a backup system under ER stress. Mol. Biol. Cell 2013, 24, 3155–3163. [Google Scholar] [CrossRef] [PubMed]
- Shenkman, M.; Groisman, B.; Ron, E.; Avezov, E.; Hendershot, L.M.; Lederkremer, G.Z. A shared endoplasmic reticulum-associated degradation pathway involving the EDEM1 protein for glycosylated and nonglycosylated proteins. J. Biol. Chem. 2013, 288, 2167–2178. [Google Scholar] [CrossRef] [Green Version]
- Grubb, S.; Guo, L.; Fisher, E.A.; Brodsky, J.L. Protein disulfide isomerases contribute differentially to the endoplasmic reticulum–associated degradation of apolipoprotein B and other substrates. Mol. Biol. Cell 2012, 23, 520–532. [Google Scholar] [CrossRef]
- Hampton, R.Y.; Sommer, T. Finding the will and the way of ERAD substrate retrotranslocation. Curr. Opin. Cell Biol. 2012, 24, 460–466. [Google Scholar] [CrossRef] [PubMed]
- DeLaBarre, B.; Brunger, A.T. Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat. Struct. Mol. Biol. 2003, 10, 856–863. [Google Scholar] [CrossRef]
- Meyer, H.; Bug, M.; Bremer, S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 2012, 14, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Huyton, T.; Pye, V.E.; Briggs, L.C.; Flynn, T.C.; Beuron, F.; Kondo, H.; Ma, J.; Zhang, X.; Freemont, P.S. The crystal structure of murine p97/VCP at 3.6 Å. J. Struct. Biol. 2003, 144, 337–348. [Google Scholar] [CrossRef]
- Dreveny, I.; Kondo, H.; Uchiyama, K.; Shaw, A.; Zhang, X.; Freemont, P.S. Structural basis of the interaction between the AAA ATPase p97/VCP and its adaptor protein p47. EMBO J. 2004, 23, 1030–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinovich, E.; Kerem, A.; Fröhlich, K.-U.; Diamant, N.; Bar-Nun, S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol. Cell. Biol. 2002, 22, 626–634. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Yin, C.; Doong, H.; Fang, S.; Peterhoff, C.; Nixon, R.A.; Monteiro, M.J. Characterization of erasin (UBXD2): A new ER protein that promotes ER-associated protein degradation. J. Cell Sci. 2006, 119, 4011–4024. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Otsuka, T.; Ohsaki, Y.; Cheng, J.; Taniguchi, T.; Hashimoto, H.; Taniguchi, H.; Fujimoto, T. Derlin-1 and UBXD8 are engaged in dislocation and degradation of lipidated ApoB-100 at lipid droplets. Mol. Biol. Cell 2012, 23, 800–810. [Google Scholar] [CrossRef]
- Ballar, P.; Shen, Y.; Yang, H.; Fang, S. The role of a novel p97/valosin-containing protein-interacting motif of gp78 in endoplasmic reticulum-associated degradation. J. Biol. Chem. 2006, 281, 35359–35368. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Shibata, Y.; Yun, C.; Ron, D.; Rapoport, T.A. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 2004, 429, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, E.J.; Olzmann, J.A.; Kopito, R.R. Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant α-1 antitrypsin from the endoplasmic reticulum. Nat. Struct. Mol. Biol. 2011, 18, 1147. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, M.; Iwase, I.; Yamasaki, Y.; Takai, T.; Wu, Y.; Kanemoto, S.; Matsuhisa, K.; Asada, R.; Okuma, Y.; Watanabe, T. Genome-wide identification and gene expression profiling of ubiquitin ligases for endoplasmic reticulum protein degradation. Sci. Rep. 2016, 6, 30955. [Google Scholar] [CrossRef] [PubMed]
- Nadav, E.; Shmueli, A.; Barr, H.; Gonen, H.; Ciechanover, A.; Reiss, Y. A novel mammalian endoplasmic reticulum ubiquitin ligase homologous to the yeast Hrd1. Biochem. Biophys. Res. Commun. 2003, 303, 91–97. [Google Scholar] [CrossRef]
- Kikkert, M.; Doolman, R.; Dai, M.; Avner, R.; Hassink, G.; Van Voorden, S.; Thanedar, S.; Roitelman, J.; Chau, V.; Wiertz, E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 3525–3534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, S.; Ferrone, M.; Yang, C.; Jensen, J.P.; Tiwari, S.; Weissman, A.M. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2001, 98, 14422–14427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassink, G.; Kikkert, M.; Voorden, S.v.; Lee, S.-J.; Spaapen, R.; LAAR, T.v.; Coleman, C.S.; Bartee, E.; Früh, K.; Chau, V. TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem. J. 2005, 388, 647–655. [Google Scholar] [CrossRef] [Green Version]
- Younger, J.M.; Chen, L.; Ren, H.-Y.; Rosser, M.F.; Turnbull, E.L.; Fan, C.-Y.; Patterson, C.; Cyr, D.M. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 2006, 126, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Jeon, Y.J.; Khelifa, S.; Ratnikov, B.; Scott, D.A.; Feng, Y.; Parisi, F.; Ruller, C.; Lau, E.; Kim, H.; Brill, L.M. Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress-inducing chemotherapies. Cancer Cell 2015, 27, 354–369. [Google Scholar] [CrossRef] [Green Version]
- Tomati, V.; Sondo, E.; Armirotti, A.; Caci, E.; Pesce, E.; Marini, M.; Gianotti, A.; Jeon, Y.J.; Cilli, M.; Pistorio, A. Genetic inhibition of the ubiquitin ligase Rnf5 attenuates phenotypes associated to F508del cystic fibrosis mutation. Sci. Rep. 2015, 5, 12138. [Google Scholar] [CrossRef]
- Meacham, G.C.; Patterson, C.; Zhang, W.; Younger, J.M.; Cyr, D.M. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 2001, 3, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Chiba, T.; Tokunaga, F.; Kawasaki, H.; Iwai, K.; Suzuki, T.; Ito, Y.; Matsuoka, K.; Yoshida, M.; Tanaka, K. E3 ubiquitin ligase that recognizes sugar chains. Nature 2002, 418, 438–442. [Google Scholar] [CrossRef]
- Yoshida, Y.; Tokunaga, F.; Chiba, T.; Iwai, K.; Tanaka, K.; Tai, T. Fbs2 is a new member of the E3 ubiquitin ligase family that recognizes sugar chains. J. Biol. Chem. 2003, 278, 43877–43884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magadan, J.G.; Perez-Victoria, F.J.; Sougrat, R.; Ye, Y.; Strebel, K.; Bonifacino, J.S. Multilayered mechanism of CD4 downregulation by HIV-1 Vpu involving distinct ER retention and ERAD targeting steps. PLoS Pathog 2010, 6, e1000869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Shen, S.; Song, S.; He, S.; Cui, Y.; Xing, G.; Wang, J.; Yin, Y.; Fan, L.; He, F. The E3 ligase Smurf1 regulates Wolfram syndrome protein stability at the endoplasmic reticulum. J. Biol. Chem. 2011, 286, 18037–18047. [Google Scholar] [CrossRef] [Green Version]
- Fry, W.H.; Simion, C.; Sweeney, C.; Carraway, K.L. Quantity control of the ErbB3 receptor tyrosine kinase at the endoplasmic reticulum. Mol. Cell. Biol. 2011, 31, 3009–3018. [Google Scholar] [CrossRef] [Green Version]
- Morito, D.; Hirao, K.; Oda, Y.; Hosokawa, N.; Tokunaga, F.; Cyr, D.M.; Tanaka, K.; Iwai, K.; Nagata, K. Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTRΔF508. Mol. Biol. Cell 2008, 19, 1328–1336. [Google Scholar] [CrossRef] [Green Version]
- Jo, Y.; Lee, P.C.; Sguigna, P.V.; DeBose-Boyd, R.A. Sterol-induced degradation of HMG CoA reductase depends on interplay of two Insigs and two ubiquitin ligases, gp78 and Trc8. Proc. Natl. Acad. Sci. USA 2011, 108, 20503–20508. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Xu, Y.; Liu, Y.; Ye, Y. gp78 functions downstream of Hrd1 to promote degradation of misfolded proteins of the endoplasmic reticulum. Mol. Biol. Cell 2015, 26, 4438–4450. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Park, J.H.; Chung, C.H. Interferon-stimulated gene 15 in the control of cellular responses to genotoxic stress. Mol. Cells 2017, 40, 83. [Google Scholar] [CrossRef] [Green Version]
- Leto, D.E.; Morgens, D.W.; Zhang, L.; Walczak, C.P.; Elias, J.E.; Bassik, M.C.; Kopito, R.R. Genome-wide CRISPR analysis identifies substrate-specific conjugation modules in ER-associated degradation. Mol. Cell 2019, 73, 377–389.e311. [Google Scholar] [CrossRef] [Green Version]
- Blount, J.R.; Burr, A.A.; Denuc, A.; Marfany, G.; Todi, S.V. Ubiquitin-specific protease 25 functions in Endoplasmic Reticulum-associated degradation. PLoS ONE 2012, 7, e36542. [Google Scholar] [CrossRef] [Green Version]
- Ishikura, S.; Weissman, A.M.; Bonifacino, J.S. Serine residues in the cytosolic tail of the T-cell antigen receptor α-chain mediate ubiquitination and endoplasmic reticulum-associated degradation of the unassembled protein. J. Biol. Chem. 2010, 285, 23916–23924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.; Tsai, B.; Arvan, P. New insights into the physiological role of endoplasmic reticulum-associated degradation. Trends Cell Biol. 2017, 27, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Qi, L. Quality control in the endoplasmic reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Somlo, D.R.; Kim, G.H.; Prescianotto-Baschong, C.; Sun, S.; Beuret, N.; Long, Q.; Rutishauser, J.; Arvan, P.; Spiess, M. ER-associated degradation is required for vasopressin prohormone processing and systemic water homeostasis. J. Clin. Investig. 2017, 127, 3897–3912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.H.; Shi, G.; Somlo, D.R.; Haataja, L.; Song, S.; Long, Q.; Nillni, E.A.; Low, M.J.; Arvan, P.; Myers, M.G. Hypothalamic ER–associated degradation regulates POMC maturation, feeding, and age-associated obesity. J. Clin. Investig. 2018, 128, 1125–1140. [Google Scholar] [CrossRef]
- Ji, Y.; Kim, H.; Yang, L.; Sha, H.; Roman, C.A.; Long, Q.; Qi, L. The Sel1L-Hrd1 endoplasmic reticulum-associated degradation complex manages a key checkpoint in B cell development. Cell Rep. 2016, 16, 2630–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Shi, G.; Sha, H.; Ji, Y.; Han, X.; Shu, X.; Ma, H.; Inoue, T.; Gao, B.; Kim, H. IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 2015, 17, 1546–1555. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Lourie, R.; Cohen, S.B.; Ji, Y.; Goodrich, J.K.; Poole, A.C.; Ley, R.E.; Denkers, E.Y.; McGuckin, M.A.; Long, Q. Epithelial Sel1L is required for the maintenance of intestinal homeostasis. Mol. Biol. Cell 2016, 27, 483–490. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Sun, S.; Wang, H.; Liu, M.; Long, Q.; Yin, L.; Kersten, S.; Zhang, K.; Qi, L. Hepatic Sel1L-Hrd1 ER-associated degradation (ERAD) manages FGF21 levels and systemic metabolism via CREBH. EMBO J. 2018, 37, e99277. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Qi, L. ER-associated degradation in health and disease–from substrate to organism. J. Cell Sci. 2019, 132, jcs232850. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.G.; Swarbrick, G.M.; Bays, N.W.; Cronin, S.R.; Wilhovsky, S.; Seelig, L.; Kim, C.; Hampton, R.Y. Endoplasmic reticulum degradation requires lumen to cytosol signaling: Transmembrane control of Hrd1p by Hrd3p. J. Cell Biol. 2000, 151, 69–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, P.; Goder, V.; Rapoport, T.A. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 2006, 126, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Gauss, R.; Jarosch, E.; Sommer, T.; Hirsch, C. A complex of Yos9p and the HRD ligase integrates endoplasmic reticulum quality control into the degradation machinery. Nat. Cell Biol. 2006, 8, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Shi, G.; Han, X.; Francisco, A.B.; Ji, Y.; Mendonça, N.; Liu, X.; Locasale, J.W.; Simpson, K.W.; Duhamel, G.E. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. USA 2014, 111, E582–E591. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.M.; Inoue, T.; Banks, L.; Tsai, B. The ERdj5-Sel1L complex facilitates cholera toxin retrotranslocation. Mol. Biol. Cell 2013, 24, 785–795. [Google Scholar] [CrossRef]
- Mueller, B.; Lilley, B.N.; Ploegh, H.L. SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J. Cell Biol. 2006, 175, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Mueller, B.; Klemm, E.J.; Spooner, E.; Claessen, J.H.; Ploegh, H.L. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc. Natl. Acad. Sci. USA 2008, 105, 12325–12330. [Google Scholar] [CrossRef] [Green Version]
- Iida, Y.; Fujimori, T.; Okawa, K.; Nagata, K.; Wada, I.; Hosokawa, N. SEL1L protein critically determines the stability of the HRD1-SEL1L endoplasmic reticulum-associated degradation (ERAD) complex to optimize the degradation kinetics of ERAD substrates. J. Biol. Chem. 2011, 286, 16929–16939. [Google Scholar] [CrossRef] [Green Version]
- Klemm, E.J.; Spooner, E.; Ploegh, H.L. Dual role of ancient ubiquitous protein 1 (AUP1) in lipid droplet accumulation and endoplasmic reticulum (ER) protein quality control. J. Biol. Chem. 2011, 286, 37602–37614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christianson, J.C.; Olzmann, J.A.; Shaler, T.A.; Sowa, M.E.; Bennett, E.J.; Richter, C.M.; Tyler, R.E.; Greenblatt, E.J.; Harper, J.W.; Kopito, R.R. Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 2012, 14, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehnert, M.; Sommer, T.; Jarosch, E. Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat. Cell Biol. 2014, 16, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Wahlman, J.; DeMartino, G.N.; Skach, W.R.; Bulleid, N.J.; Brodsky, J.L.; Johnson, A.E. Real-time fluorescence detection of ERAD substrate retrotranslocation in a mammalian in vitro system. Cell 2007, 129, 943–955. [Google Scholar] [CrossRef]
- Brodsky, J.L. Cleaning up: ER-associated degradation to the rescue. Cell 2012, 151, 1163–1167. [Google Scholar] [CrossRef] [Green Version]
- Baldridge, R.D.; Rapoport, T.A. Autoubiquitination of the Hrd1 ligase triggers protein retrotranslocation in ERAD. Cell 2016, 166, 394–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, H.; Yagishita, N.; Aratani, S.; Saito-Fujita, T.; Morota, S.; Yamano, Y.; Hansson, M.J.; Inazu, M.; Kokuba, H.; Sudo, K. The E3 ligase synoviolin controls body weight and mitochondrial biogenesis through negative regulation of PGC-1β. EMBO J. 2015, 34, 1042–1055. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, S.; Yagishita, N.; Sasaki, T.; Nakazawa, M.; Kato, Y.; Yamadera, T.; Bae, E.; Toriyama, S.; Ikeda, R.; Zhang, L. Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase ‘Synoviolin’. EMBO J. 2007, 26, 113–122. [Google Scholar] [CrossRef]
- Kong, S.; Yang, Y.; Xu, Y.; Wang, Y.; Zhang, Y.; Melo-Cardenas, J.; Xu, X.; Gao, B.; Thorp, E.B.; Zhang, D.D. Endoplasmic reticulum-resident E3 ubiquitin ligase Hrd1 controls B-cell immunity through degradation of the death receptor CD95/Fas. Proc. Natl. Acad. Sci. USA 2016, 113, 10394–10399. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Kong, S.; Zhang, Y.; Melo-Cardenas, J.; Gao, B.; Zhang, Y.; Zhang, D.D.; Zhang, B.; Song, J.; Thorp, E. The endoplasmic reticulum–resident E3 ubiquitin ligase Hrd1 controls a critical checkpoint in B cell development in mice. J. Biol. Chem. 2018, 293, 12934–12944. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, N.; Liu, T.; Ji, Y.; Reinert, R.B.; Torres, M.; Li, X.; Zhang, M.; Tang, C.-H.A.; Hu, C.-C.A.; Liu, C. Sel1L-Hrd1 ER-associated degradation maintains β cell identity via TGF-β signaling. J. Clin. Investig. 2020, 130, 3499–3510. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Marvin, J.M.; Tatsis, N.; Eisenlohr, L.C. Cutting Edge: Selective role of ubiquitin in MHC class I antigen presentation. J. Immunol. 2011, 186, 1904–1908. [Google Scholar] [CrossRef]
- Burr, M.L.; Cano, F.; Svobodova, S.; Boyle, L.H.; Boname, J.M.; Lehner, P.J. HRD1 and UBE2J1 target misfolded MHC class I heavy chains for endoplasmic reticulum-associated degradation. Proc. Natl. Acad. Sci. USA 2011, 108, 2034–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burr, M.L.; van den Boomen, D.J.; Bye, H.; Antrobus, R.; Wiertz, E.J.; Lehner, P.J. MHC class I molecules are preferentially ubiquitinated on endoplasmic reticulum luminal residues during HRD1 ubiquitin E3 ligase-mediated dislocation. Proc. Natl. Acad. Sci. USA 2013, 110, 14290–14295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Qiu, Q.; Gao, B.; Kong, S.; Lin, Z.; Fang, D. Hrd1-mediated BLIMP-1 ubiquitination promotes dendritic cell MHCII expression for CD4 T cell priming during inflammation. J. Exp. Med. 2014, 211, 2467–2479. [Google Scholar] [CrossRef] [Green Version]
- Piskurich, J.F.; Lin, K.-I.; Lin, Y.; Wang, Y.; Ting, J.P.-Y.; Calame, K. BLIMP-1 mediates extinction of major histocompatibility class II transactivator expression in plasma cells. Nat. Immunol. 2000, 1, 526–532. [Google Scholar] [CrossRef]
- Xu, Y.; Fang, D. Endoplasmic reticulum-associated degradation and beyond: The multitasking roles for HRD1 in immune regulation and autoimmunity. J. Autoimmun. 2020, 109, 102423. [Google Scholar] [CrossRef]
- Gao, B.; Lee, S.M.; Chen, A.; Zhang, J.; Zhang, D.D.; Kannan, K.; Ortmann, R.A.; Fang, D. Synoviolin promotes IRE1 ubiquitination and degradation in synovial fibroblasts from mice with collagen-induced arthritis. EMBO Rep. 2008, 9, 480–485. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014, 28, 708–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PONTING, C.P. Proteins of the endoplasmic-reticulum-associated degradation pathway: Domain detection and function prediction. Biochem. J. 2000, 351, 527–535. [Google Scholar] [CrossRef]
- Song, B.-L.; Sever, N.; DeBose-Boyd, R.A. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol. Cell 2005, 19, 829–840. [Google Scholar] [CrossRef]
- Chen, B.; Mariano, J.; Tsai, Y.C.; Chan, A.H.; Cohen, M.; Weissman, A.M. The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc. Natl. Acad. Sci. USA 2006, 103, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Das, R.; Mariano, J.; Tsai, Y.C.; Kalathur, R.C.; Kostova, Z.; Li, J.; Tarasov, S.G.; McFeeters, R.L.; Altieri, A.S.; Ji, X. Allosteric activation of E2-RING finger-mediated ubiquitylation by a structurally defined specific E2-binding region of gp78. Mol. Cell 2009, 34, 674–685. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Soetandyo, N.; Lee, J.-g.; Liu, L.; Xu, Y.; Clemons Jr, W.M.; Ye, Y. USP13 antagonizes gp78 to maintain functionality of a chaperone in ER-associated degradation. eLife 2014, 3, e01369. [Google Scholar] [CrossRef]
- Jo, Y.; DeBose-Boyd, R.A. Control of cholesterol synthesis through regulated ER-associated degradation of HMG CoA reductase. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 185–198. [Google Scholar] [CrossRef] [Green Version]
- Wangeline, M.A.; Vashistha, N.; Hampton, R.Y. Proteostatic tactics in the strategy of sterol regulation. Annu. Rev. Cell Dev. Biol. 2017, 33, 467–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, X.; Cui, Y.; Tang, Y.; Chen, W.; Li, S.; Yu, H.; Pan, Y.; Wang, C. The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 2014, 41, 919–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, Z.; Wang, H.; Fan, H.; Zhu, X.; Zhou, J.; Fei, E.; Wang, G. Gp78, an ER associated E3, promotes SOD1 and ataxin-3 degradation. Hum. Mol. Genet. 2009, 18, 4268–4281. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Liu, C.; Zhong, Y.; Luo, S.; Monteiro, M.J.; Fang, S. Huntingtin interacts with the cue domain of gp78 and inhibits gp78 binding to ubiquitin and p97/VCP. PLoS ONE 2010, 5, e8905. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Choe, V.; Cheng, H.; Tsai, Y.C.; Weissman, A.M.; Luo, S.; Rao, H. Ubiquitin ligase gp78 targets unglycosylated prion protein PrP for ubiquitylation and degradation. PLoS ONE 2014, 9, e92290. [Google Scholar]
- Foresti, O.; Ruggiano, A.; Hannibal-Bach, H.K.; Ejsing, C.S.; Carvalho, P. Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. eLife 2013, 2, e00953. [Google Scholar] [CrossRef] [PubMed]
- Zavacki, A.M.; e Drigo, R.A.; Freitas, B.C.; Chung, M.; Harney, J.W.; Egri, P.; Wittmann, G.; Fekete, C.; Gereben, B.; Bianco, A.C. The E3 ubiquitin ligase TEB4 mediates degradation of type 2 iodothyronine deiodinase. Mol. Cell. Biol. 2009, 29, 5339–5347. [Google Scholar] [CrossRef] [Green Version]
- Zelcer, N.; Sharpe, L.J.; Loregger, A.; Kristiana, I.; Cook, E.C.; Phan, L.; Stevenson, J.; Brown, A.J. The E3 ubiquitin ligase MARCH6 degrades squalene monooxygenase and affects 3-hydroxy-3-methyl-glutaryl coenzyme A reductase and the cholesterol synthesis pathway. Mol. Cell. Biol. 2014, 34, 1262–1270. [Google Scholar] [CrossRef] [Green Version]
- Zattas, D.; Berk, J.M.; Kreft, S.G.; Hochstrasser, M. A conserved C-terminal element in the yeast Doa10 and human MARCH6 ubiquitin ligases required for selective substrate degradation. J. Biol. Chem. 2016, 291, 12105–12118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meacham, G.C.; Lu, Z.; King, S.; Sorscher, E.; Tousson, A.; Cyr, D.M. The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999, 18, 1492–1505. [Google Scholar] [CrossRef]
- Fisher, E.A.; Zhou, M.; Mitchell, D.M.; Wu, X.; Omura, S.; Wang, H.; Goldberg, A.L.; Ginsberg, H.N. The degradation of apolipoprotein B100 is mediated by the ubiquitin-proteasome pathway and involves heat shock protein 70. J. Biol. Chem. 1997, 272, 20427–20434. [Google Scholar] [CrossRef] [Green Version]
- Younger, J.M.; Ren, H.-Y.; Chen, L.; Fan, C.-Y.; Fields, A.; Patterson, C.; Cyr, D.M. A foldable CFTRΔF508 biogenic intermediate accumulates upon inhibition of the Hsc70–CHIP E3 ubiquitin ligase. J. Cell Biol. 2004, 167, 1075–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.-h.; Kimura, T.; Momohara, S.; Takeuchi, M.; Tani, T.; Kimata, Y.; Kadokura, H.; Kohno, K. A novel ER J-protein DNAJB12 accelerates ER-associated degradation of membrane proteins including CFTR. Cell Struct. Funct. 2010, 35, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Grove, D.E.; Fan, C.-Y.; Ren, H.Y.; Cyr, D.M. The endoplasmic reticulum–associated Hsp40 DNAJB12 and Hsc70 cooperate to facilitate RMA1 E3–dependent degradation of nascent CFTRΔF508. Mol. Biol. Cell 2011, 22, 301–314. [Google Scholar] [CrossRef] [PubMed]
- van den Boomen, D.J.; Volkmar, N.; Lehner, P.J. Ubiquitin-mediated regulation of sterol homeostasis. Curr. Opin. Cell Biol. 2020, 65, 103–111. [Google Scholar] [CrossRef]
- Scott, N.A.; Sharpe, L.J.; Brown, A.J. The E3 ubiquitin ligase MARCHF6 as a metabolic integrator in cholesterol synthesis and beyond. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2020, 1866, 158837. [Google Scholar] [CrossRef]
- Chua, N.K.; Hart-Smith, G.; Brown, A.J. Non-canonical ubiquitination of the cholesterol-regulated degron of squalene monooxygenase. J. Biol. Chem. 2019, 294, 8134–8147. [Google Scholar] [CrossRef] [Green Version]
- Chua, N.K.; Howe, V.; Jatana, N.; Thukral, L.; Brown, A.J. A conserved degron containing an amphipathic helix regulates the cholesterol-mediated turnover of human squalene monooxygenase, a rate-limiting enzyme in cholesterol synthesis. J. Biol. Chem. 2017, 292, 19959–19973. [Google Scholar] [CrossRef] [Green Version]
- Howe, V.; Chua, N.K.; Stevenson, J.; Brown, A.J. The regulatory domain of squalene monooxygenase contains a re-entrant loop and senses cholesterol via a conformational change. J. Biol. Chem. 2015, 290, 27533–27544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, K.T.; Lee, C.-S.; Mun, S.-H.; Truong, N.T.; Park, S.K.; Hwang, C.-S. N-terminal acetylation and the N-end rule pathway control degradation of the lipid droplet protein PLIN2. J. Biol. Chem. 2019, 294, 379–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, M.L.; Krus, K.L.; Kaushik, S.; Dang, D.; Chopra, R.; Qi, L.; Shakkottai, V.G.; Cuervo, A.M.; Lieberman, A.P. Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat. Commun. 2018, 9, 3671. [Google Scholar] [CrossRef]
- Wang, L.; Dong, H.; Soroka, C.J.; Wei, N.; Boyer, J.L.; Hochstrasser, M. Degradation of the bile salt export pump at endoplasmic reticulum in progressive familial intrahepatic cholestasis type II. Hepatology 2008, 48, 1558–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, K.T.; Mun, S.-H.; Lee, C.-S.; Hwang, C.-S. Control of protein degradation by N-terminal acetylation and the N-end rule pathway. Exp. Mol. Med. 2018, 50, 1–8. [Google Scholar] [CrossRef]
- Loregger, A.; Cook, E.C.L.; Nelson, J.K.; Moeton, M.; Sharpe, L.J.; Engberg, S.; Karimova, M.; Lambert, G.; Brown, A.J.; Zelcer, N. A MARCH6 and IDOL E3 ubiquitin ligase circuit uncouples cholesterol synthesis from lipoprotein uptake in hepatocytes. Mol. Cell. Biol. 2016, 36, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, L.J.; Howe, V.; Scott, N.A.; Luu, W.; Phan, L.; Berk, J.M.; Hochstrasser, M.; Brown, A.J. Cholesterol increases protein levels of the E3 ligase MARCH6 and thereby stimulates protein degradation. J. Biol. Chem. 2019, 294, 2436–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, N.; Harada, K.; Kato, M.; Hirose, S. Ubiquitin-specific protease 19 regulates the stability of the E3 ubiquitin ligase MARCH6. Exp. Cell Res. 2014, 328, 207–216. [Google Scholar] [CrossRef]
- Tcherpakov, M.; Delaunay, A.; Toth, J.; Kadoya, T.; Petroski, M.D.; Ze’ev, A.R. Regulation of endoplasmic reticulum-associated degradation by RNF5-dependent ubiquitination of JNK-associated membrane protein (JAMP). J. Biol. Chem. 2009, 284, 12099–12109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.; Ahn, J.; Liu, C.; Tanabe, K.; Apodaca, J.; Suzuki, T.; Rao, H. The Png1–Rad23 complex regulates glycoprotein turnover. J. Cell Biol. 2006, 172, 211–219. [Google Scholar] [CrossRef]
- Li, G.; Zhao, G.; Zhou, X.; Schindelin, H.; Lennarz, W.J. The AAA ATPase p97 links peptide N-glycanase to the endoplasmic reticulum-associated E3 ligase autocrine motility factor receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 8348–8353. [Google Scholar] [CrossRef] [Green Version]
- Ernst, R.; Mueller, B.; Ploegh, H.L.; Schlieker, C. The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol. Cell 2009, 36, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Sowa, M.E.; Bennett, E.J.; Gygi, S.P.; Harper, J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell 2009, 138, 389–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Li, L.; Ye, Y. Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J. Cell Biol. 2006, 174, 963–971. [Google Scholar] [CrossRef]
- Zhong, X.; Pittman, R.N. Ataxin-3 binds VCP/p97 and regulates retrotranslocation of ERAD substrates. Hum. Mol. Genet. 2006, 15, 2409–2420. [Google Scholar] [CrossRef]
- Xu, Y.; Cai, M.; Yang, Y.; Huang, L.; Ye, Y. SGTA recognizes a noncanonical ubiquitin-like domain in the Bag6-Ubl4A-Trc35 complex to promote endoplasmic reticulum-associated degradation. Cell Rep. 2012, 2, 1633–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Liu, Y.; Lee, J.-G.; Ye, Y. A ubiquitin-like domain recruits an oligomeric chaperone to a retrotranslocation complex in endoplasmic reticulum-associated degradation. J. Biol. Chem. 2013, 288, 18068–18076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Liu, Y.; Soetandyo, N.; Baek, K.; Hegde, R.; Ye, Y. A ubiquitin ligase-associated chaperone holdase maintains polypeptides in soluble states for proteasome degradation. Mol. Cell 2011, 42, 758–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, e423. [Google Scholar] [CrossRef] [Green Version]
- Bernales, S.; Schuck, S.; Walter, P. ER-phagy: Selective autophagy of the endoplasmic reticulum. Autophagy 2007, 3, 285–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grumati, P.; Dikic, I.; Stolz, A. ER-phagy at a glance. J. Cell Sci. 2018, 131, jcs217364. [Google Scholar] [CrossRef] [Green Version]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef]
- Fumagalli, F.; Noack, J.; Bergmann, T.J.; Cebollero, E.; Pisoni, G.B.; Fasana, E.; Fregno, I.; Galli, C.; Loi, M.; Soldà, T. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol. 2016, 18, 1173–1184. [Google Scholar] [CrossRef] [Green Version]
- Grumati, P.; Morozzi, G.; Hölper, S.; Mari, M.; Harwardt, M.-L.I.; Yan, R.; Müller, S.; Reggiori, F.; Heilemann, M.; Dikic, I. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. eLife 2017, 6, e25555. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.D.; Harley, M.E.; Kemp, A.J.; Wills, J.; Lee, M.; Arends, M.; von Kriegsheim, A.; Behrends, C.; Wilkinson, S. CCPG1 is a non-canonical autophagy cargo receptor essential for ER-phagy and pancreatic ER proteostasis. Dev. Cell 2018, 44, 217–232.e211. [Google Scholar] [CrossRef]
- An, H.; Ordureau, A.; Paulo, J.A.; Shoemaker, C.J.; Denic, V.; Harper, J.W. TEX264 is an endoplasmic reticulum-resident ATG8-interacting protein critical for ER remodeling during nutrient stress. Mol. Cell 2019, 74, 891–908.e810. [Google Scholar] [CrossRef]
- Chen, Q.; Xiao, Y.; Chai, P.; Zheng, P.; Teng, J.; Chen, J. ATL3 is a tubular ER-phagy receptor for GABARAP-mediated selective autophagy. Curr. Biol. 2019, 29, 846–855.e846. [Google Scholar] [CrossRef] [Green Version]
- Chino, H.; Hatta, T.; Natsume, T.; Mizushima, N. Intrinsically disordered protein TEX264 mediates ER-phagy. Mol. Cell 2019, 74, 909–921.e906. [Google Scholar] [CrossRef]
- Wilkinson, S. ER-phagy: Shaping up and destressing the endoplasmic reticulum. FEBS J. 2019, 286, 2645–2663. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, S. Emerging principles of selective ER autophagy. J. Mol. Biol. 2020, 432, 185–205. [Google Scholar] [CrossRef]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Kishi-Itakura, C.; Koyama-Honda, I.; Itakura, E.; Mizushima, N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell Sci. 2014, 127, 4089–4102. [Google Scholar] [CrossRef] [Green Version]
- Chino, H.; Mizushima, N. ER-Phagy: Quality control and turnover of endoplasmic reticulum. Trends Cell Biol. 2020, 30, 384–398. [Google Scholar] [CrossRef] [PubMed]
- De Leonibus, C.; Cinque, L.; Settembre, C. Emerging lysosomal pathways for quality control at the endoplasmic reticulum. FEBS Lett. 2019, 593, 2319–2329. [Google Scholar] [CrossRef] [Green Version]
- Stolz, A.; Grumati, P. The various shades of ER-phagy. FEBS J. 2019, 286, 4642–4649. [Google Scholar] [CrossRef]
- Schuck, S.; Gallagher, C.M.; Walter, P. ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J. Cell Sci. 2014, 127, 4078–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fregno, I.; Fasana, E.; Bergmann, T.J.; Raimondi, A.; Loi, M.; Soldà, T.; Galli, C.; D’Antuono, R.; Morone, D.; Danieli, A. ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J. 2018, 37, e99259. [Google Scholar] [CrossRef]
- Bolender, R.P.; Weibel, E.R. A morphometric study of the removal of phenobarbital-induced membranes from hepatocytes after cessation of treatment. J. Cell Biol. 1973, 56, 746–761. [Google Scholar] [CrossRef] [PubMed]
- Zurek, N.; Sparks, L.; Voeltz, G. Reticulon short hairpin transmembrane domains are used to shape ER tubules. Traffic 2011, 12, 28–41. [Google Scholar] [CrossRef] [Green Version]
- Rismanchi, N.; Soderblom, C.; Stadler, J.; Zhu, P.-P.; Blackstone, C. Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum. Mol. Genet. 2008, 17, 1591–1604. [Google Scholar] [CrossRef]
- Bhaskara, R.M.; Grumati, P.; Garcia-Pardo, J.; Kalayil, S.; Covarrubias-Pinto, A.; Chen, W.; Kudryashev, M.; Dikic, I.; Hummer, G. Curvature induction and membrane remodeling by FAM134B reticulon homology domain assist selective ER-phagy. Nat. Commun. 2019, 10, 2370. [Google Scholar] [CrossRef] [Green Version]
- Brady, J.P.; Claridge, J.K.; Smith, P.G.; Schnell, J.R. A conserved amphipathic helix is required for membrane tubule formation by Yop1p. Proc. Natl. Acad. Sci. USA 2015, 112, E639–E648. [Google Scholar] [CrossRef] [Green Version]
- Ishida, Y.; Kubota, H.; Yamamoto, A.; Kitamura, A.; Bächinger, H.P.; Nagata, K. Type I collagen in Hsp47-null cells is aggregated in endoplasmic reticulum and deficient in N-propeptide processing and fibrillogenesis. Mol. Biol. Cell 2006, 17, 2346–2355. [Google Scholar] [CrossRef]
- Forrester, A.; De Leonibus, C.; Grumati, P.; Fasana, E.; Piemontese, M.; Staiano, L.; Fregno, I.; Raimondi, A.; Marazza, A.; Bruno, G. A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM 134B complex. EMBO J. 2019, 38, e99847. [Google Scholar] [CrossRef]
- Gelsthorpe, M.E.; Baumann, N.; Millard, E.; Gale, S.E.; Langmade, S.J.; Schaffer, J.E.; Ory, D.S. Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J. Biol. Chem. 2008, 283, 8229–8236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienkowski, R.S.; Gotkin, M.G. Control of collagen deposition in mammalian lung. Proc. Soc. Exp. Biol. Med. 1995, 209, 118–140. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.S.; Strittmatter, S.M. The reticulons: A family of proteins with diverse functions. Genome Biol. 2007, 8, 234. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, C.N.; Williams, J.M.; Knupp, J.; Arunagiri, A.; Arvan, P.; Tsai, B. Cells deploy a two-pronged strategy to rectify misfolded proinsulin aggregates. Mol. Cell 2019, 75, 442–456.e444. [Google Scholar] [CrossRef]
- Busch, D.J.; Houser, J.R.; Hayden, C.C.; Sherman, M.B.; Lafer, E.M.; Stachowiak, J.C. Intrinsically disordered proteins drive membrane curvature. Nat. Commun. 2015, 6, 7875. [Google Scholar] [CrossRef] [Green Version]
- Snead, W.T.; Hayden, C.C.; Gadok, A.K.; Zhao, C.; Lafer, E.M.; Rangamani, P.; Stachowiak, J.C. Membrane fission by protein crowding. Proc. Natl. Acad. Sci. USA 2017, 114, E3258–E3267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Ni, H.-M.; Guo, F.; Ding, Y.; Shi, Y.-H.; Lahiri, P.; Fröhlich, L.F.; Rülicke, T.; Smole, C.; Schmidt, V.C. Sequestosome 1/p62 protein is associated with autophagic removal of excess hepatic endoplasmic reticulum in mice. J. Biol. Chem. 2016, 291, 18663–18674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, C.H.; Kim, H.Y.; Heo, A.J.; Lee, S.H.; Lee, M.J.; Kim, S.B.; Srinivasrao, G.; Mun, S.R.; Cha-Molstad, H.; Ciechanover, A. The N-Degron pathway mediates ER-phagy. Mol. Cell 2019, 75, 1058–1072.e1059. [Google Scholar] [CrossRef]
- Roberts, P.; Moshitch-Moshkovitz, S.; Kvam, E.; O’Toole, E.; Winey, M.; Goldfarb, D.S. Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae. Mol. Biol. Cell 2003, 14, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, J.A.; Schessner, J.P.; Bircham, P.W.; Tsuji, T.; Funaya, C.; Pajonk, O.; Schaeff, K.; Ruffini, G.; Papagiannidis, D.; Knop, M. ESCRT machinery mediates selective microautophagy of endoplasmic reticulum in yeast. EMBO J. 2020, 39, e102586. [Google Scholar] [CrossRef]
- Loi, M.; Raimondi, A.; Morone, D.; Molinari, M. ESCRT-III-driven piecemeal micro-ER-phagy remodels the ER during recovery from ER stress. Nat. Commun. 2019, 10, 5058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurokawa, K.; Nakano, A. The ER exit sites are specialized ER zones for the transport of cargo proteins from the ER to the Golgi apparatus. J. Biochem. 2019, 165, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Omari, S.; Makareeva, E.; Roberts-Pilgrim, A.; Mirigian, L.; Jarnik, M.; Ott, C.; Lippincott-Schwartz, J.; Leikin, S. Noncanonical autophagy at ER exit sites regulates procollagen turnover. Proc. Natl. Acad. Sci. USA 2018, 115, E10099–E10108. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamimoto, T.; Shoji, S.; Hidvegi, T.; Mizushima, N.; Umebayashi, K.; Perlmutter, D.H.; Yoshimori, T. Intracellular inclusions containing mutant α1-antitrypsin Z are propagated in the absence of autophagic activity. J. Biol. Chem. 2006, 281, 4467–4476. [Google Scholar] [CrossRef] [Green Version]
- Kroeger, H.; Miranda, E.; MacLeod, I.; Pérez, J.; Crowther, D.C.; Marciniak, S.J.; Lomas, D.A. Endoplasmic reticulum-associated degradation (ERAD) and autophagy cooperate to degrade polymerogenic mutant serpins. J. Biol. Chem. 2009, 284, 22793–22802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidvegi, T.; Ewing, M.; Hale, P.; Dippold, C.; Beckett, C.; Kemp, C.; Maurice, N.; Mukherjee, A.; Goldbach, C.; Watkins, S. An autophagy-enhancing drug promotes degradation of mutant α1-antitrypsin Z and reduces hepatic fibrosis. Science 2010, 329, 229–232. [Google Scholar] [CrossRef]
- Colla, E. Linking the endoplasmic reticulum to Parkinson’s disease and alpha-synucleinopathy. Front. Neurosci. 2019, 13, 560. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.A.d.; Manaa, W.E.; Duplan, E.; Checler, F. The Endoplasmic Reticulum Stress/Unfolded Protein Response and Their Contributions to Parkinson’s Disease Physiopathology. Cells 2020, 9, 2495. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.A.; Lieberman, A.P. The intersection of lysosomal and endoplasmic reticulum calcium with autophagy defects in lysosomal diseases. Neurosci. Lett. 2019, 697, 10–16. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, G.; Fratila, O.; Buhas, C.; Judea-Pusta, C.T.; Negrut, N.; Bustea, C.; Bungau, S. Cross-talks among GBA mutations, glucocerebrosidase, and α-synuclein in GBA-associated Parkinson’s disease and their targeted therapeutic approaches: A comprehensive review. Transl. Neurodegener. 2021, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Ghemrawi, R.; Khair, M. Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 6127. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Del Tredici, K.; Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 2013, 9, 13. [Google Scholar] [CrossRef]
- Smith, W.W.; Jiang, H.; Pei, Z.; Tanaka, Y.; Morita, H.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum. Mol. Genet. 2005, 14, 3801–3811. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F. α-Synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Falarti, E.; Bodei, S.; Sigala, S.; Battistin, L.; Spillantini, M.; Missale, C.; Spano, P. Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson’s disease. J. Neurochem. 2011, 116, 588–605. [Google Scholar] [CrossRef]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Khurana, V.; Auluck, P.K.; Tardiff, D.F.; Mazzulli, J.R.; Soldner, F.; Baru, V.; Lou, Y.; Freyzon, Y.; Cho, S. Identification and rescue of α-synuclein toxicity in Parkinson patient–derived neurons. Science 2013, 342, 983–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J. Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Kim, B.C.; Nguyen, D.-T.T.; Samidurai, M.; Choi, S.-M. Levodopa (L-DOPA) attenuates endoplasmic reticulum stress response and cell death signaling through DRD2 in SH-SY5Y neuronal cells under α-synuclein-induced toxicity. Neuroscience 2017, 358, 336–348. [Google Scholar] [CrossRef]
- Bellani, S.; Mescola, A.; Ronzitti, G.; Tsushima, H.; Tilve, S.; Canale, C.; Valtorta, F.; Chieregatti, E. GRP78 clustering at the cell surface of neurons transduces the action of exogenous alpha-synuclein. Cell Death Differ. 2014, 21, 1971–1983. [Google Scholar] [CrossRef]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.-C.; McCaffery, J.M. The Parkinson’s disease protein α-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef] [Green Version]
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. α-Synuclein–induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936. [Google Scholar] [CrossRef] [Green Version]
- Betzer, C.; Lassen, L.B.; Olsen, A.; Kofoed, R.H.; Reimer, L.; Gregersen, E.; Zheng, J.; Calì, T.; Gai, W.P.; Chen, T. Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation. EMBO Rep. 2018, 19, e44617. [Google Scholar] [CrossRef]
- Jung, E.-M.; Yoo, Y.-M.; Park, S.Y.; Ahn, C.; Jeon, B.-H.; Hong, E.-J.; Kim, W.-Y.; Jeung, E.-B. Calbindin-D 9k Is a Novel Risk Gene for Neurodegenerative Disease. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2020, 54, 438–456. [Google Scholar]
- Shimura, H.; Hattori, N.; Kubo, S.-i.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Soda, M.; Takahashi, R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J. Biol. Chem. 2000, 275, 35661–35664. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Soda, M.; Inoue, H.; Hattori, N.; Mizuno, Y.; Takahashi, R. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 2001, 105, 891–902. [Google Scholar] [CrossRef] [Green Version]
- Rana, A.; Rera, M.; Walker, D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 8638–8643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omura, T.; Kaneko, M.; Okuma, Y.; Orba, Y.; Nagashima, K.; Takahashi, R.; Fujitani, N.; Matsumura, S.; Hata, A.; Kubota, K. A ubiquitin ligase HRD1 promotes the degradation of Pael receptor, a substrate of Parkin. J. Neurochem. 2006, 99, 1456–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of lysosomal storage disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Korkotian, E.; Schwarz, A.; Pelled, D.; Schwarzmann, G.; Segal, M.; Futerman, A.H. Elevation of intracellular glucosylceramide levels results in an increase in endoplasmic reticulum density and in functional calcium stores in cultured neurons. J. Biol. Chem. 1999, 274, 21673–21678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd-Evans, E.; Pelled, D.; Riebeling, C.; Bodennec, J.; de-Morgan, A.; Waller, H.; Schiffmann, R.; Futerman, A.H. Glucosylceramide and glucosylsphingosine modulate calcium mobilization from brain microsomes via different mechanisms. J. Biol. Chem. 2003, 278, 23594–23599. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Liou, B.; Ran, H.; Skelton, M.R.; Williams, M.T.; Vorhees, C.V.; Kitatani, K.; Hannun, Y.A.; Witte, D.P.; Xu, Y.-H. Neuronopathic Gaucher disease in the mouse: Viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 2010, 19, 1088–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, B.; Peng, Y.; Li, R.; Inskeep, V.; Zhang, W.; Quinn, B.; Dasgupta, N.; Blackwood, R.; Setchell, K.D.; Fleming, S. Modulating ryanodine receptors with dantrolene attenuates neuronopathic phenotype in Gaucher disease mice. Hum. Mol. Genet. 2016, 25, 5126–5141. [Google Scholar] [CrossRef]
- Zimmer, K.P.; le Coutre, P.; Aerts, H.M.; Harzer, K.; Fukuda, M.; O’Brien, J.S.; Naim, H.Y. Intracellular transport of acid β-glucosidase and lysosome-associated membrane proteins is affected in Gaucher’s disease (G202R mutation). J. Pathol. 1999, 188, 407–414. [Google Scholar] [CrossRef]
- Sawkar, A.R.; Cheng, W.-C.; Beutler, E.; Wong, C.-H.; Balch, W.E.; Kelly, J.W. Chemical chaperones increase the cellular activity of N370S β-glucosidase: A therapeutic strategy for Gaucher disease. Proc. Natl. Acad. Sci. USA 2002, 99, 15428–15433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, I.; Horowitz, M. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum. Mol. Genet. 2005, 14, 2387–2398. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preston, G.M.; Brodsky, J.L. The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem. J. 2017, 474, 445–469. [Google Scholar] [CrossRef] [Green Version]
- Anding, A.L.; Baehrecke, E.H. Cleaning house: Selective autophagy of organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]




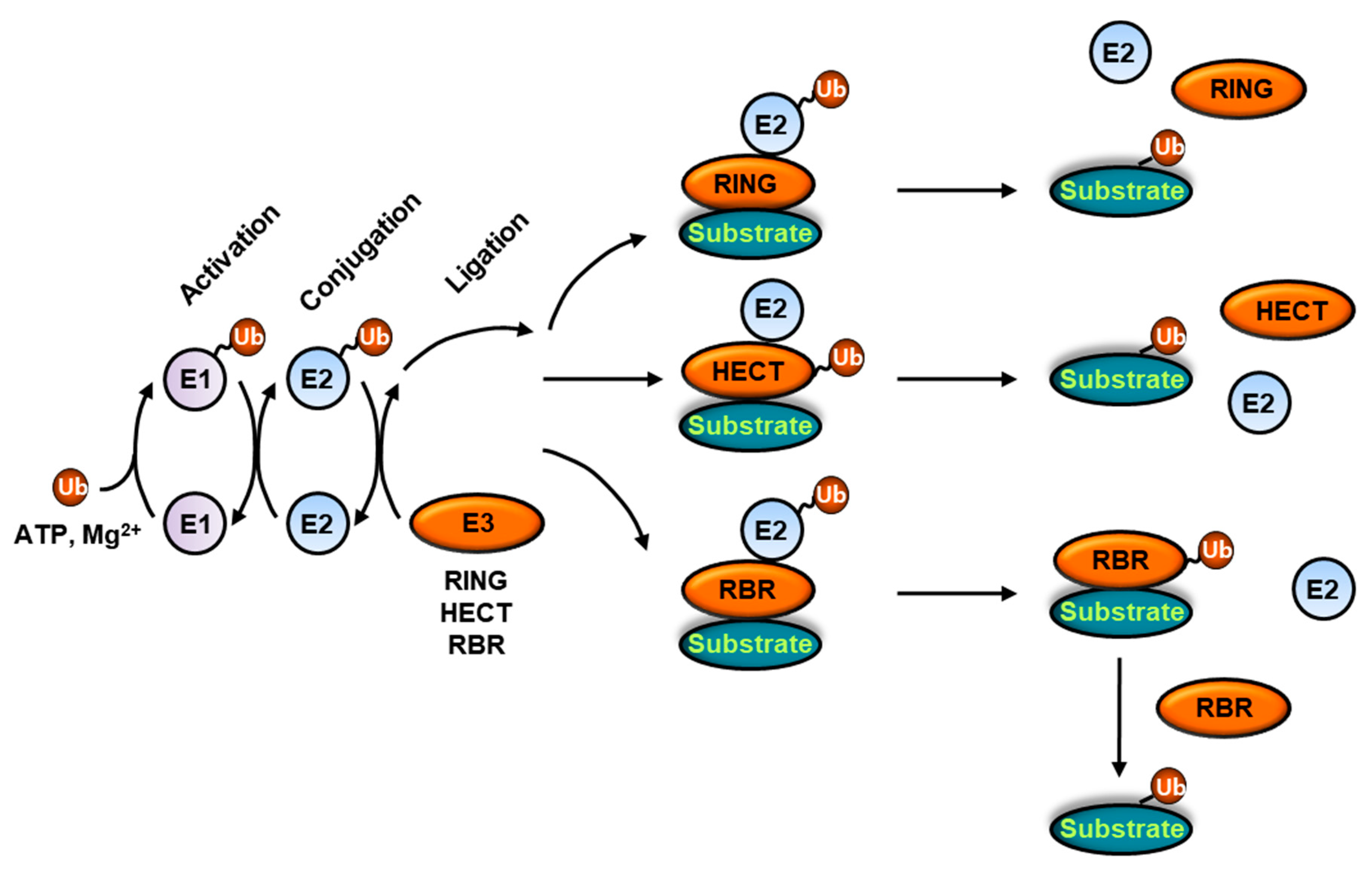{kind=link}
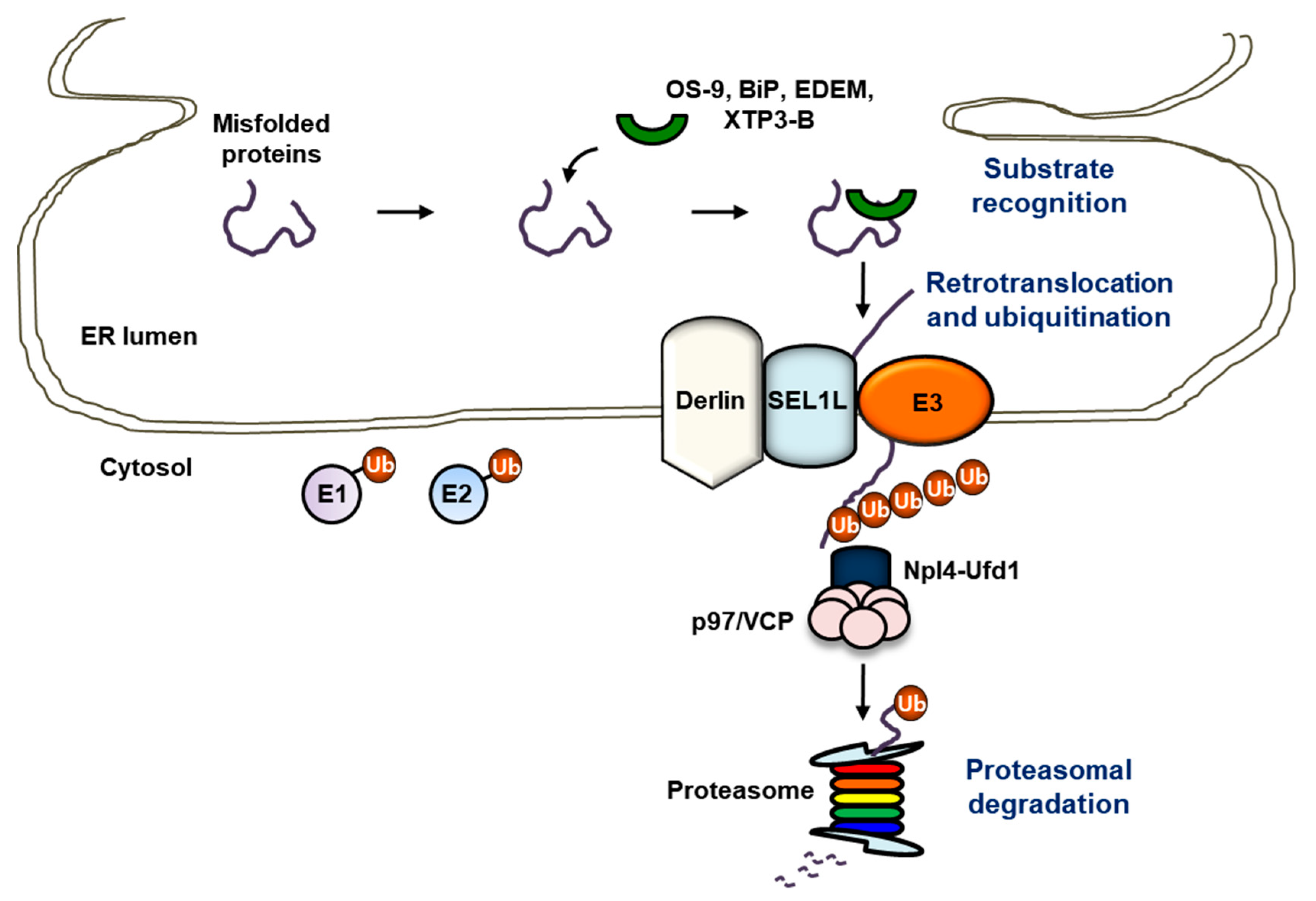{kind=link}
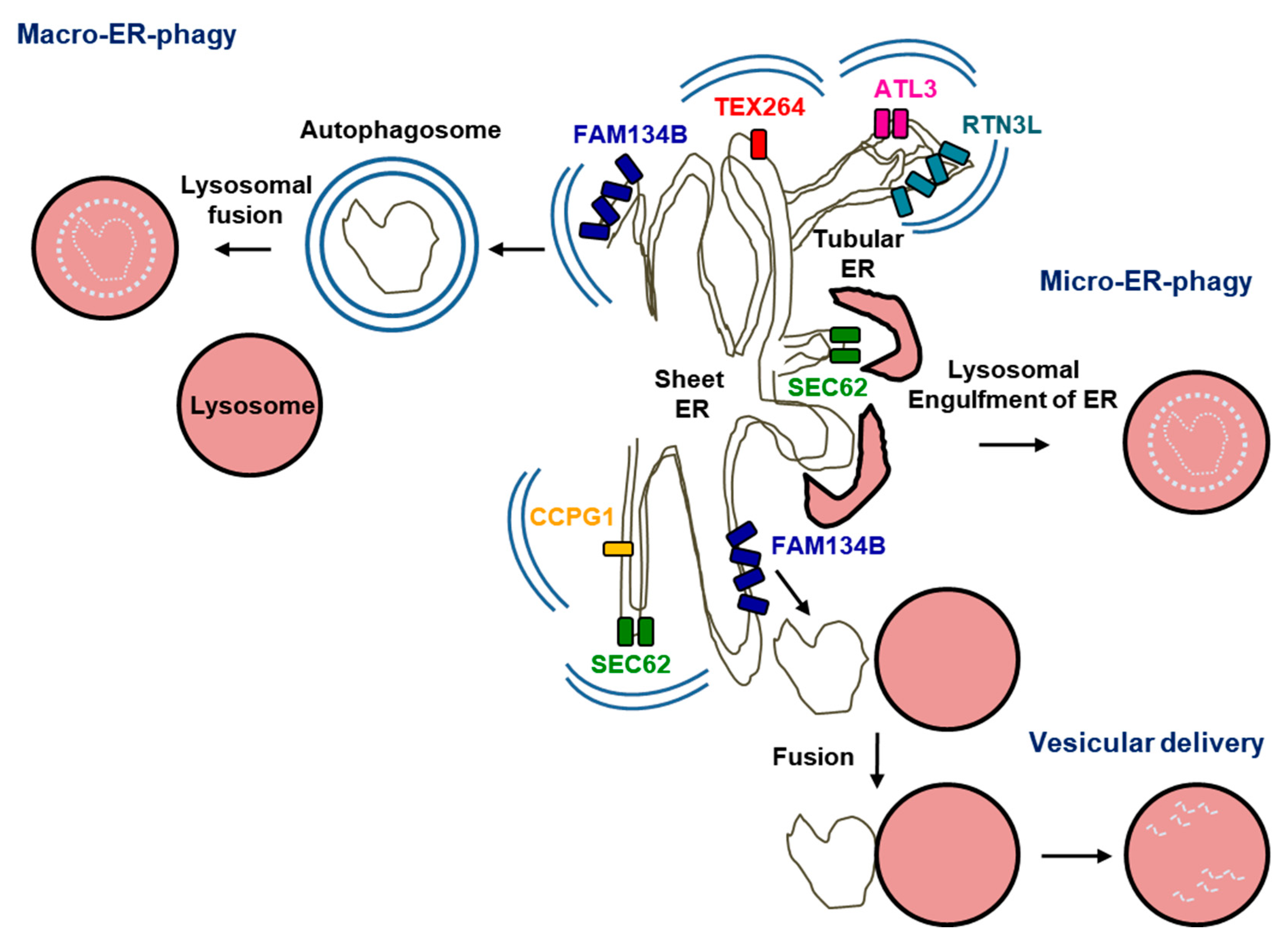{kind=link}
| E3 Ubiquitin Ligases (Mammalian) | Substrates | Notes |
|---|---|---|
| HRD1 | pre-BCR, BLIMP1, Fas, IRE1, NRF2, p53, PGC1β, AVP, TGF-β receptor I, TCR-α, MHC I | Homolog of yeast Hrd1 |
| MARCHF6 | SQLE, HMGCR, PLIN2, BSEP | Homolog of yeast Doa10 |
| gp78 | CFTR, HMGCR, SOD1, ataxin-3, huntingtin, prion | Homolog of yeast Hrd1 |
| RNF5 | CFTR, JAMP | Integral ER membrane-resident |
| CHIP | CFTR, Pael-receptor | Cytoplasmic |
| PARKIN | Pael-receptor, GCase | Cytoplasmic |
| Smurf1 | WFS1 | Cytoplasmic/nuclear |
| Nrdp1 | ErbB3 | Cytoplasmic |
| SCFFbx2 | TCR-α, CFTR | Cytoplasmic/ER membrane-associated |
| SCFFbx6 | TCR-α | Cytoplasmic/ER membrane-associated |
| SCFβ-TrCP | CD4, Tetherin | Cytoplasmic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, J.A.; Jeon, Y.J. How Is the Fidelity of Proteins Ensured in Terms of Both Quality and Quantity at the Endoplasmic Reticulum? Mechanistic Insights into E3 Ubiquitin Ligases. Int. J. Mol. Sci. 2021, 22, 2078. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042078
Kang JA, Jeon YJ. How Is the Fidelity of Proteins Ensured in Terms of Both Quality and Quantity at the Endoplasmic Reticulum? Mechanistic Insights into E3 Ubiquitin Ligases. International Journal of Molecular Sciences. 2021; 22(4):2078. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042078
Chicago/Turabian StyleKang, Ji An, and Young Joo Jeon. 2021. "How Is the Fidelity of Proteins Ensured in Terms of Both Quality and Quantity at the Endoplasmic Reticulum? Mechanistic Insights into E3 Ubiquitin Ligases" International Journal of Molecular Sciences 22, no. 4: 2078. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22042078