
Progress in the Development of Eukaryotic Elongation Factor 2 Kinase (eEF2K) Natural Product and Synthetic Small Molecule Inhibitors for Cancer Chemotherapy

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Physiological Function of eEF2K
3. The Role of eEF2K in Cancer
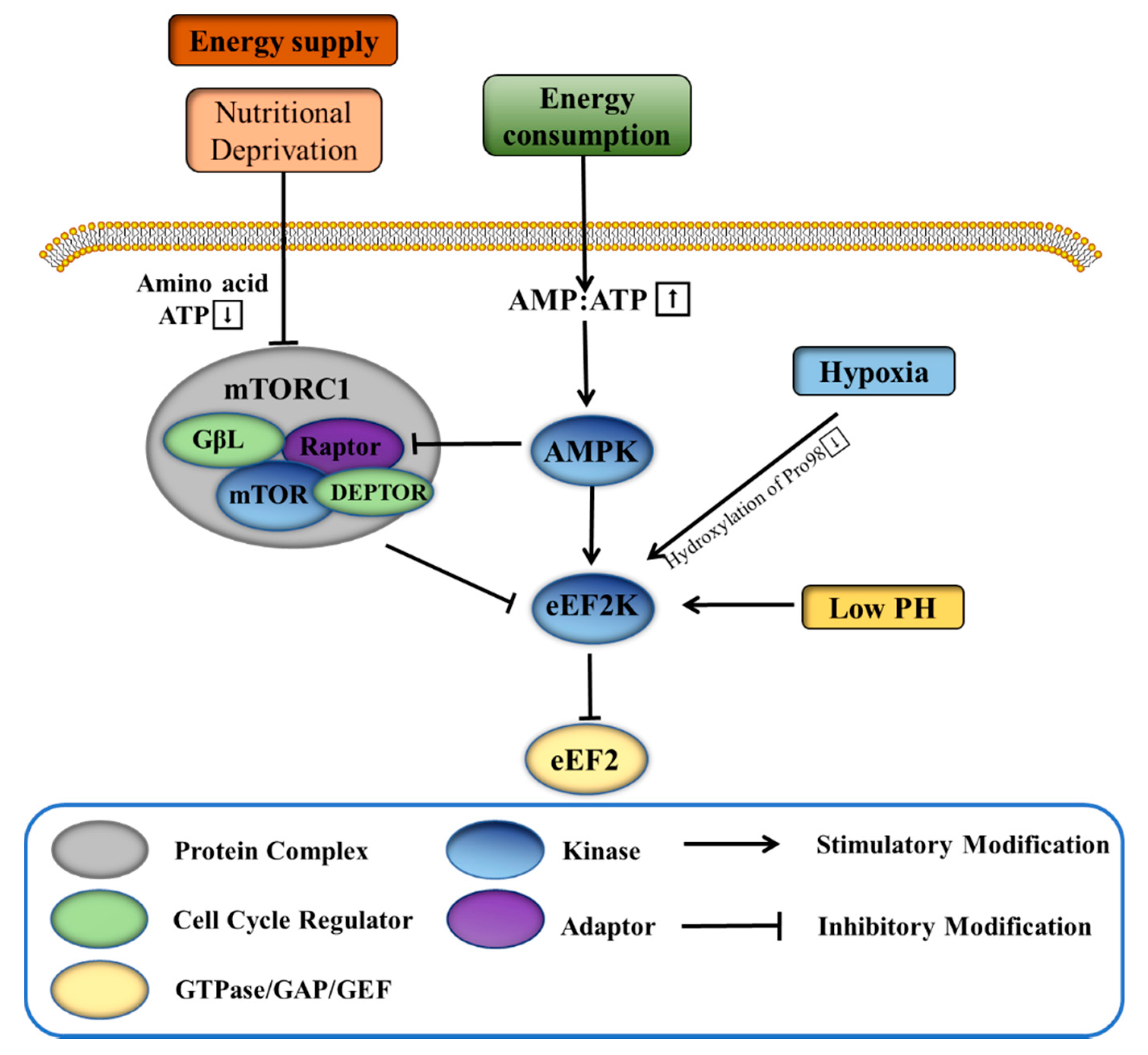
3.1. eEF2K Helps Tumor Cells to Cope with Harsh Environments
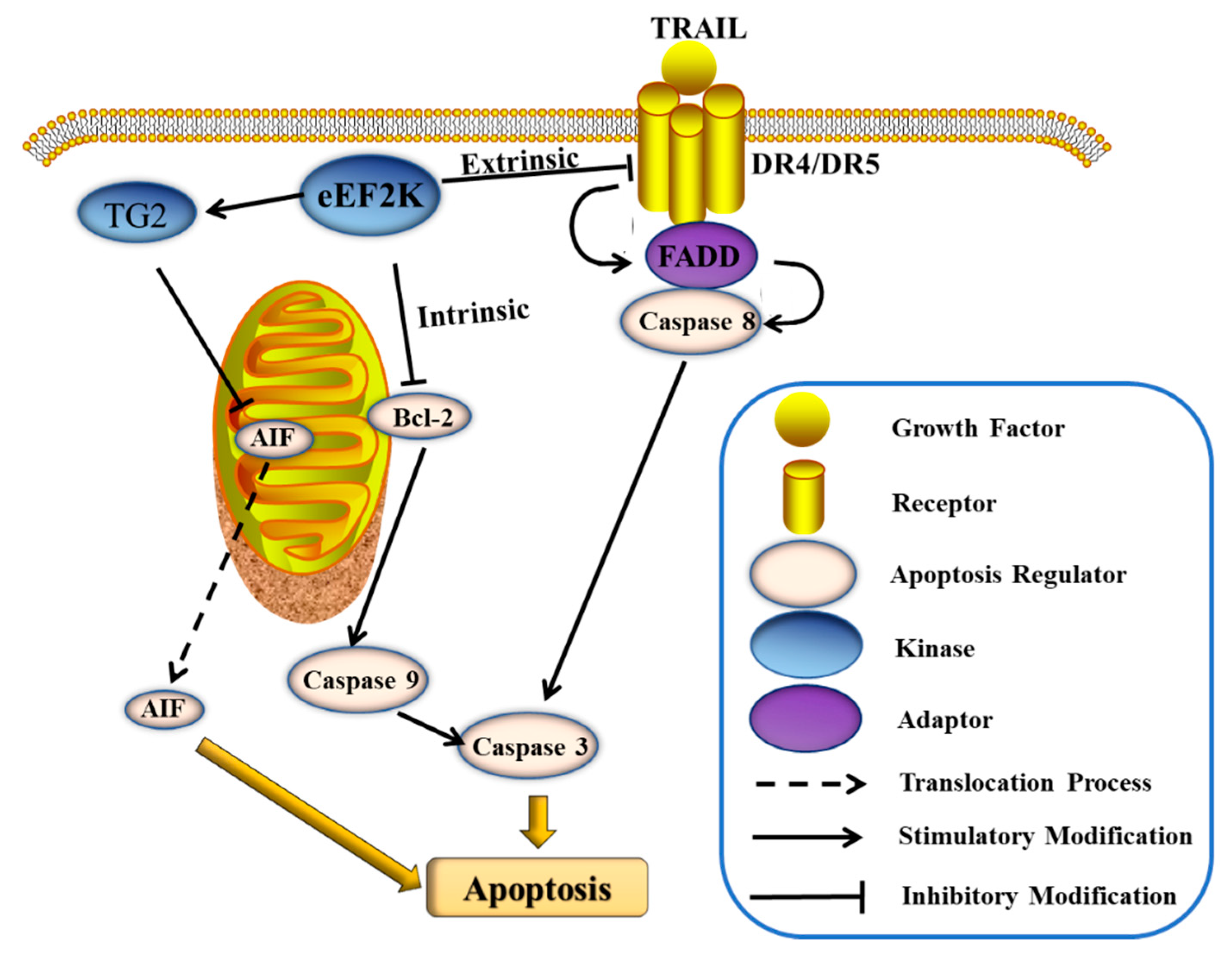
3.2. eEF2K Inhibits Cell Apoptosis
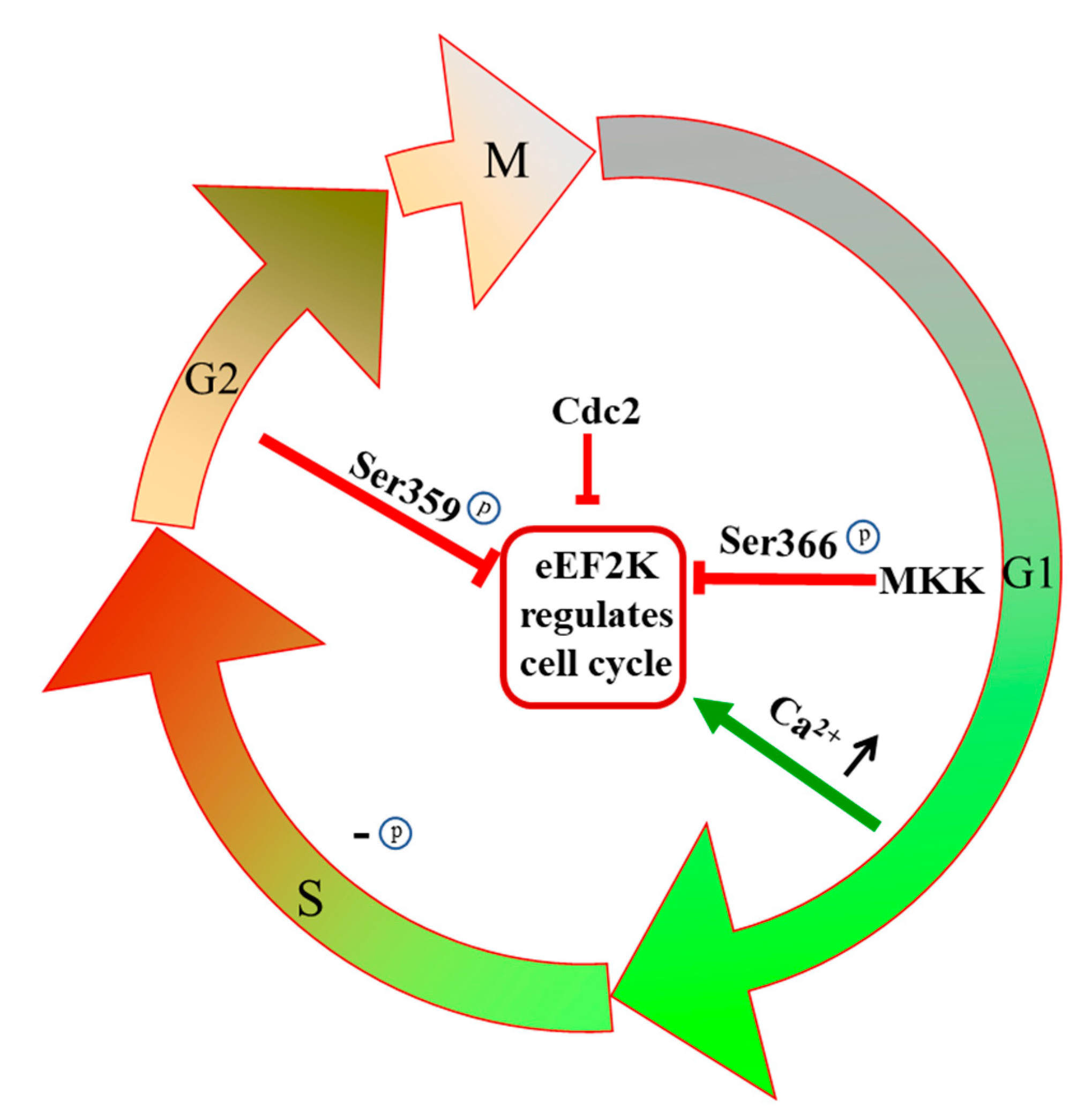
3.3. eEF2K Regulates the Cell Cycle
3.4. eEF2K Regulates Cell Autophagy
3.5. eEF2K Promotes Tumor Angiogenesis, Metastasis, and Invasion
4. Natural Product and Synthetic Small Molecule Inhibitors of eEF2K
4.1. Discovery and Development of Single Target eEF2K Inhibitors
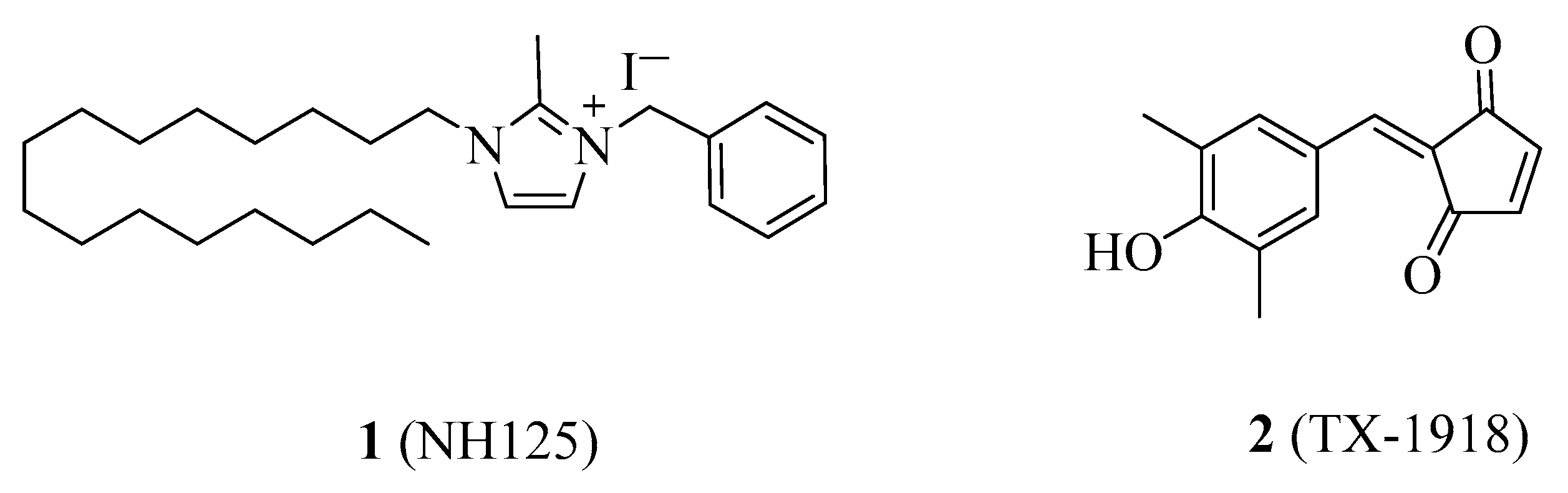
4.1.1. NH125
4.1.2. TX-1918
4.1.3. A-484954 and Its Derivatives
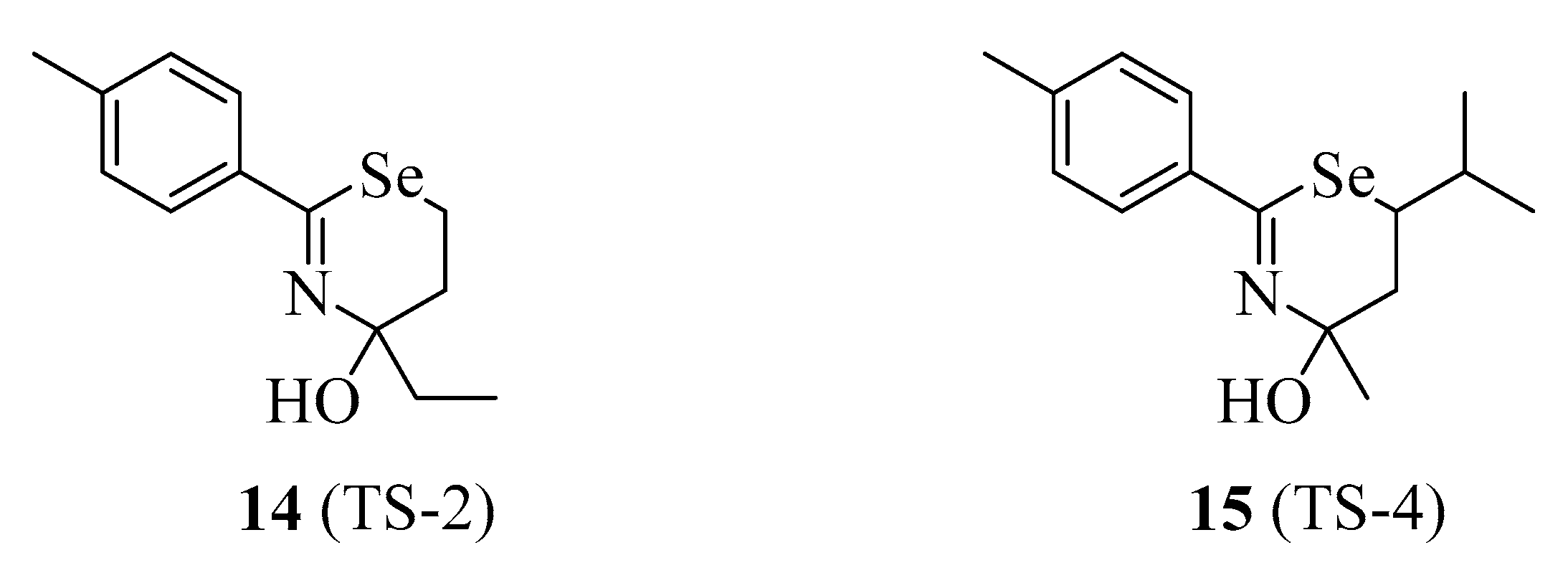
4.1.4. TS-2 and TS-4
4.1.5. Thieno[2,3-b]pyridines
4.1.6. β-Phenylalanines
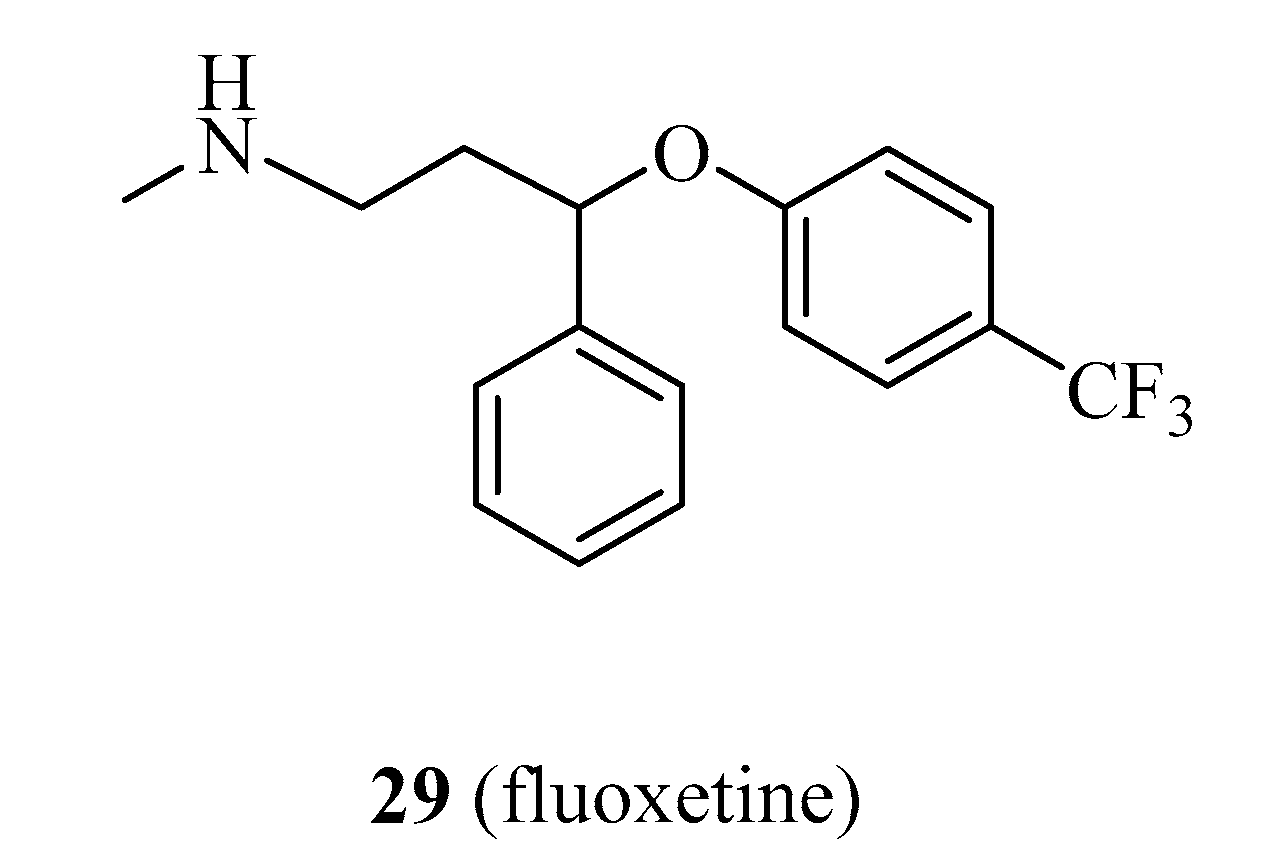
4.1.7. Fluoxetine
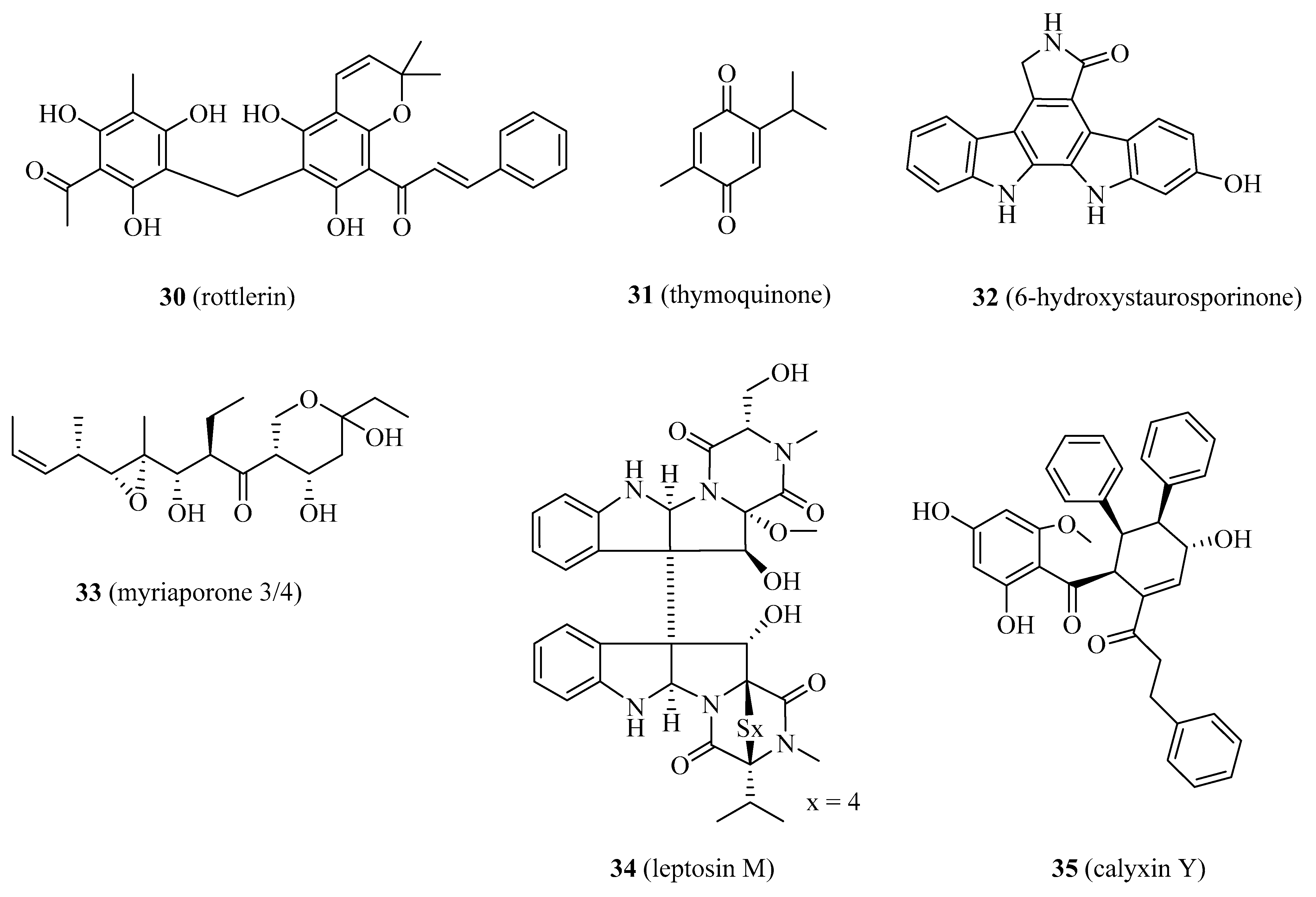
4.1.8. Rottlerin
4.1.9. Thymoquinone
4.1.10. 6-hydroxystaurosporinone
4.1.11. Myriaporone 3/4
4.1.12. Leptosin M
4.1.13. Calyxin Y
4.2. Discovery and Development of Multi-Target Inhibitors of eEF2K
4.2.1. Inhibitors Targeting PLK1/eEF2K
4.2.2. Inhibitors Targeting GLP-1R/eEF2K
4.2.3. Inhibitors Targeting the Protein/Protein Interaction of Hsp90 and eEF2K
5. The Role of Natural Product and Synthetic Small Molecule Activators of Eef2k
6. Conclusions and Future Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Clin. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, S.A. Chapter Three—Acquired Resistance to Drugs Targeting Tyrosine Kinases. Adv. Cancer. Res. 2018, 138, 71–98. [Google Scholar] [PubMed]
- Cote, G.P.; Luo, X.; Murphy, M.B.; Egelhoff, T.T. Mapping of the novel protein kinase catalytic domain of dictyostelium myosin II heavy chain kinase A. J. Biol. Chem. 1997, 272, 6846–6849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenney, J.W.; Moore, C.E.; Wang, X.; Proud, C.G. Eukaryotic elongation factor 2 kinase, an unusual enzyme with multiple roles. Adv. Biol. Regul. 2014, 55, 15–27. [Google Scholar] [CrossRef]
- Proud, C.G. Regulation and roles of elongation factor 2 kinase. Biochem. Soc. Trans. 2015, 43, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Matsushita, M.; Nairn, A.C.; Kuriyan, J. Crystal structure of the atypical protein kinase domain of a TRP channel with phosphotransferase activity. Mol. Cell 2001, 7, 1047–1057. [Google Scholar] [CrossRef]
- Delaidelli, A.; Khan, D.; Leprivier, G.; Pfister, S.M.; Taylor, M.D.; Maris, J.M.; Sorensen, P. OS5-173 Inhibition of eEF2K as a novel therapeutic strategy in neuroblastoma and medulloblastoma. Can. J. Neurol. Sci. 2016, 43, S3. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.C.; Voisin, V.; Wang, S.; Wang, D.Y.; Jones, R.A.; Datti, A.; Uehling, D.; Al-Awar, R.; Egan, S.E.; Bader, G.D. Combined deletion of Pten and p53 in mammary epithelium accelerates triple-negative breast cancer with dependency on eEF2K. EMBO Mol. Med. 2015, 6, 1542–1560. [Google Scholar] [CrossRef]
- Gassart, A.D.; Martinon, F. Translating the anticancer properties of eEF2K. Cell Cycle 2017, 16, 299–300. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bialkowska, A.; Rusovici, R.; Chanchevalap, S.; Shim, H.; Katz, J.P.; Yang, V.W.; Yun, C.C. Lysophosphatidic acid facilitates proliferation of colon cancer cells via induction of Krüppel-like factor 5. J. Biol. Chem. 2007, 282, 15541–15549. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Li, Y.; Xu, S.; Lu, J.; Zhu, Z.; Chen, S.; Tan, Y.; He, P.; Xu, J.; Proud, C.G.; et al. Eukaryotic elongation factor 2 kinase promotes angiogenesis in hepatocellular carcinoma via PI3K/Akt and STAT3. Int. J. Cancer. 2020, 146, 1383–1395. [Google Scholar] [CrossRef] [PubMed]
- Horman, S.; Beauloye, C.; Vertommen, D.; Vanoverschelde, J.L.; Hue, L.; Rider, M.H. Myocardial ischemia and increased heart work modulate the phosphorylation state of eukaryotic elongation factor-2. J. Biol. Chem. 2003, 278, 41970–41976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakas, D.; Ozpolat, B. Eukaryotic elongation factor-2 kinase (eEF2K) signaling in tumor and microenvironment as a novel molecular target. J. Mol. Med. 2020, 98, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Ashour, A.A.; Abdel-Aziz, A.A.H.; Mansour, A.M.; Alpay, S.N.; Huo, L.F.; Ozpolat, B. Targeting elongation factor-2 kinase (eEF-2K) induces apoptosis in human pancreatic cancer cells. Apoptosis 2014, 19, 241–258. [Google Scholar] [CrossRef]
- Moore, C.E.; Wang, X.; Xie, J.; Pickford, J.; Barron, J.; Regufe da Mota, S.; Versele, M.; Proud, C.G. Elongation factor 2 kinase promotes cell survival by inhibiting protein synthesis without inducing autophagy. Cell Signal. 2016, 28, 284–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, W.; Williams, M.; Terada, N.; Alessi, D.R.; Proud, C.G. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001, 20, 4370–4379. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.M. Regulation of translation by hydrogen peroxide. Antioxid. Redox. Signal. 2011, 15, 191–203. [Google Scholar] [CrossRef]
- Spahn, C.M.; Gomez-Lorenzo, M.G.; Grassucci, R.A.; Jørgensen, R.; Andersen, G.R.; Beckmann, R.; Penczek, P.A.; Ballesta, J.P.; Frank, J. Domain movements of elongation factor eEF2 and the eukaryotic 80S ribosome facilitate tRNA translocation. EMBO J. 2004, 23, 1008–1019. [Google Scholar] [CrossRef] [Green Version]
- Hizli, A.A.; Chi, Y.; Swanger, J.; Carter, J.H.; Liao, Y.; Welcker, M.; Ryazanov, A.G.; Clurman, B.E. Phosphorylation of eukaryotic elongation factor 2 (eEF2) by cyclin A-cyclin-dependent kinase 2 regulates its inhibition by eEF2 kinase. Mol. Cell. Biol. 2013, 33, 596–604. [Google Scholar] [CrossRef] [Green Version]
- Kruiswijk, F.; Yuniati, L.; Magliozzi, R.; Low, T.Y.; Lim, R.; Bolder, R.; Mohammed, S.; Proud, C.G.; Heck, A.J.; Pagano, M.; et al. Coupled activation and degradation of eEF2K regulates protein synthesis in response to genotoxic stress. Sci. Signal. 2012, 5, ra40. [Google Scholar] [CrossRef] [Green Version]
- Leprivier, G.; Rotblat, B.; Khan, D.; Jan, E.; Sorensen, P.H. Stress-mediated translational control in cancer cells. Biochim. Biophys. Acta 2015, 1849, 845–860. [Google Scholar] [CrossRef]
- Russnes, H.G.; Caldas, C. eEF2K-a new target in breast cancers with combined inactivation of p53 and PTEN. EMBO Mol. Med. 2014, 6, 1512–1514. [Google Scholar] [CrossRef] [PubMed]
- Hamurcu, Z.; Ashour, A.; Kahraman, N.; Ozpolat, B. FOXM1 regulates expression of eukaryotic elongation factor 2 kinase and promotes proliferation, invasion and tumorgenesis of human triple negative breast cancer cells. Oncotarget 2016, 7, 16619–16635. [Google Scholar] [CrossRef] [Green Version]
- Shi, N.; Chen, X.; Liu, R.; Wang, D.; Su, M.; Wang, Q.; He, A.; Gu, H. Eukaryotic elongation factors 2 promotes tumor cell proliferation and correlates with poor prognosis in ovarian cancer. Tissue Cell 2018, 53, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, R.; Pichler, M.; Kanlikilicer, P.; Ivan, C.; Bayraktar, E.; Kahraman, N.; Aslan, B.; Oguztuzun, S.; Ulasli, M.; Arslan, A.; et al. MicroRNA 603 acts as a tumor suppressor and inhibits triple-negative breast cancer tumorigenesis by targeting elongation factor 2 kinase. Oncotarget 2017, 8, 11641–11658. [Google Scholar] [CrossRef] [Green Version]
- Bircan, H.A.; Gurbuz, N.; Pataer, A.; Caner, A.; Kahraman, N.; Bayraktar, E.; Bayraktar, R.; Erdogan, M.A.; Kabil, N.; Ozpolat, B. Elongation factor-2 kinase (eEF-2K) expression is associated with poor patient survival and promotes proliferation, invasion and tumor growth of lung cancer. Lung Cancer 2018, 124, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Leprivier, G.; Remke, M.; Rotblat, B.; Dubuc, A.; Mateo, A.R.; Kool, M.; Agnihotri, S.; El-Naggar, A.; Yu, B.; Somasekharan, S.P.; et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 2013, 153, 1064–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewell, J.L.; Guan, K.L. Nutrient signaling to mTOR and cell growth. Trends Biochem. Sci. 2013, 38, 233–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryazanov, A.G.; Shestakova, E.A.; Natapov, P.G. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature 1988, 334, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. mTORC1 regulates the efficiency and cellular capacity for protein synthesis. Biochem. Soc. Trans. 2013, 41, 923–926. [Google Scholar] [CrossRef] [Green Version]
- Proud, C.G. Signalling to translation: How signal transduction pathways control the protein synthetic machinery. Biochem. J. 2007, 403, 217–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faller, W.J.; Jackson, T.J.; Knight, J.R.; Ridgway, R.A.; Jamieson, T.; Karim, S.A.; Jones, C.; Radulescu, S.; Huels, D.J.; Myant, K.B.; et al. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 2015, 517, 497–500. [Google Scholar] [CrossRef] [Green Version]
- Connolly, E.; Braunstein, S.; Formenti, S.; Schneider, R.J. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol. Cell. Biol. 2006, 26, 3955–3965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, C.E.J.; Mikolajek, H.; Sergio, R.D.M.; Wang, X.; Kenney, J.W.; Werner, J.R.M.; Proud, C.G.J.M.; Biology, C. Elongation Factor 2 Kinase Is Regulated by Proline Hydroxylation and Protects Cells during Hypoxia. Mol. Cell. Biol. 2015, 35, 1788–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Ren, X.; Yuan, Y.; Shan, Y.; Li, L.; Chen, X.; Zhang, L.; Takahashi, Y.; Yang, J.W.; Han, B.; et al. eEF-2 kinase is a critical regulator of Warburg effect through controlling PP2A-A synthesis. Oncogene 2016, 35, 6293–6308. [Google Scholar] [CrossRef]
- Helmlinger, G.; Schell, A.; Dellian, M.; Forbes, N.S.; Jain, R.K. Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin. Cancer Res. 2002, 8, 1284–1291. [Google Scholar]
- Shime, H.; Yabu, M.; Akazawa, T.; Kodama, K.; Matsumoto, M.; Seya, T.; Inoue, N. Tumor-secreted lactic acid promotes IL-23/IL-17 proinflammatory pathway. J. Immunol. 2008, 180, 7175–7183. [Google Scholar] [CrossRef]
- Dorovkov, M.V.; Pavur, K.S.; Petrov, A.G. Regulation of elongation factor-2 kinase by pH. Biochemistry 2002, 41, 13444–13450. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Mikolajek, H.; Pigott, C.R.; Hooper, K.J.; Mellows, T.; Moore, C.E.; Mohammed, H.; Werner, J.M.; Thomas, G.J.; Proud, C.G. Molecular mechanism for the control of eukaryotic elongation factor 2 kinase by pH: Role in cancer cell survival. Mol. Cell. Biol. 2015, 35, 1805–1824. [Google Scholar] [CrossRef] [Green Version]
- Theodoropoulos, G.E.; Gazouli, M.; Vaiopoulou, A.; Leandrou, M.; Nikouli, S.; Vassou, E.; Kouraklis, G.; Nikiteas, N. Polymorphisms of Caspase 8 and Caspase 9 gene and colorectal cancer susceptibility and prognosis. Int. J. Colorectal. Dis. 2011, 26, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jun, D.Y.; Han, C.R.; Kim, Y.H.J.B.B.R.C. Protein tyrosine kinase p56lck-deficiency confers hypersusceptibility to rho-fluorophenylalanine (pFPhe)-induced apoptosis by augmenting mitochondrial apoptotic pathway in human Jurkat T cells. Biochem. Biophys. Res. Commun. 2008, 377, 280–285. [Google Scholar] [CrossRef]
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Bellail, A.C.; Tse, M.C.L.; Song, J.H.; Phuphanich, S.; Olson, J.J.; Sun, S.Y.; Hao, C.H. DR5-mediated DISC controls caspase-8 cleavage and initiation of apoptosis in human glioblastomas. J. Cell. Mol. Med. 2010, 14, 1303–1317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cheng, Y.; Zhang, L.; Ren, X.C.; Huber-Keener, K.J.; Lee, S.; Yun, J.; Wang, H.G.; Yang, J.M. Inhibition of eEF-2 kinase sensitizes human glioma cells to TRAIL and down-regulates Bcl-xL expression. Biochem. Biophys. Res. Commun. 2011, 414, 129–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Loo, G.; Saelens, X.; van Gurp, M.; MacFarlane, M.; Martin, S.J.; Vandenabeele, P. The role of mitochondrial factors in apoptosis: A Russian roulette with more than one bullet. Cell Death Differ. 2002, 9, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Tekedereli, I.; Alpay, S.N.; Tavares, C.D.J.; Cobanoglu, Z.E.; Kaoud, T.S.; Sahin, I.; Sood, A.K.; Lopez-Berestein, G.; Dalby, K.N.; Ozpolat, B. Targeted Silencing of Elongation Factor 2 Kinase Suppresses Growth and Sensitizes Tumors to Doxorubicin in an Orthotopic Model of Breast Cancer. PLoS ONE 2012, 7, e41171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmer, T.G.; Ward, M.D.; Yurkow, E.J.; Vyas, V.H.; Kearney, T.J.; Hait, W.N. Activity and regulation by growth factors of calmodulin-dependent protein kinase III (elongation factor 2-kinase) in human breast cancer. Br. J. Cancer 1999, 79, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Roberts, E.C.; Hammond, K.; Traish, A.M.; Resing, K.A.; Ahn, N.G. Identification of G2/M targets for the MAP kinase pathway by functional proteomics. Proteomics 2010, 6, 4541–4553. [Google Scholar] [CrossRef] [PubMed]
- Ratan, R.R.; Maxfield, F.R.; Shelanski, M.L. Long-lasting and rapid calcium changes during mitosis. J. Cell Biol. 1988, 107, 993–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santella, L. The role of calcium in the cell cycle: Facts and hypotheses. Biochem. Biophys. Res. Commun. 1998, 244, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Gutzkow, K.B.; Låhne, H.U.; Naderi, S.; Torgersen, K.M.; Skålhegg, B.; Koketsu, M.; Uehara, Y.; Blomhoff, H.K. Cyclic AMP inhibits translation of cyclin D3 in T lymphocytes at the level of elongation by inducing eEF2-phosphorylation. Cell. Signal. 2003, 15, 871–881. [Google Scholar] [CrossRef]
- Pyr Dit Ruys, S.; Wang, X.; Smith, E.M.; Herinckx, G.; Hussain, N.; Rider, M.H.; Vertommen, D.; Proud, C.G. Identification of autophosphorylation sites in eukaryotic elongation factor-2 kinase. Biochem. J. 2012, 442, 681–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.M.; Proud, C.G. cdc2–cyclin B regulates eEF2 kinase activity in a cell cycle- and amino acid-dependent manner. EMBO J. 2008, 27, 1005–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.; Sandhu, S.; Mcarthur, G. Cell cycle control as a promising target in melanoma. Curr. Opin. Oncol. 2015, 27, 141–150. [Google Scholar] [CrossRef]
- Petroni, G.; Formenti, S.C.; Chen-Kiang, S.; Galluzzi, L. Immunomodulation by anticancer cell cycle inhibitors. Nat. Rev. Immunol. 2020, 20, 669–679. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, H.; Ren, X.; Niu, T.; Hait, W.N.; Yang, J. Cytoprotective Effect of the Elongation Factor-2 Kinase-Mediated Autophagy in Breast Cancer Cells Subjected to Growth Factor Inhibition. PLoS ONE 2010, 5, e9715. [Google Scholar] [CrossRef]
- Jung, S.; Jeong, H.; Yu, S.W. Autophagy as a decisive process for cell death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [Green Version]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Ren, X.; Zhang, Y.; Patel, R.; Sharma, A.; Wu, H.; Robertson, G.P.; Yan, L.; Rubin, E.; Yang, J.M. eEF-2 kinase dictates cross-talk between autophagy and apoptosis induced by Akt Inhibition, thereby modulating cytotoxicity of novel Akt inhibitor MK-2206. Cancer Res. 2011, 71, 2654–2663. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.Y.; Tian, Y.; Liu, L.; Zhan, J.H.; Hou, X.; Chen, X.; Zhou, T.; Huang, Y.; Zhang, L. Inhibiting eEF-2 kinase-mediated autophagy enhanced the cytocidal effect of AKT inhibitor on human nasopharyngeal carcinoma. Drug Des. Dev. Ther. 2018, 12, 2655–2663. [Google Scholar] [CrossRef] [Green Version]
- Py, B.F.; Boyce, M.; Yuan, J. A critical role of eEF-2K in mediating autophagy in response to multiple cellular stresses. Autophagy 2009, 5, 393–396. [Google Scholar] [CrossRef] [Green Version]
- Boyce, M.; Py, B.F.; Ryazanov, A.G.; Minden, J.S.; Long, K.; Ma, D.; Yuan, J. A pharmacoproteomic approach implicates eukaryotic elongation factor 2 kinase in ER stress-induced cell death. Cell Death Differ. 2008, 15, 589–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Zhu, H.; Liu, D.X.; Niu, T.K.; Ren, X.; Patel, R.; Hait, W.N.; Yang, J.M. Silencing of elongation factor-2 kinase potentiates the effect of 2-deoxy-D-glucose against human glioma cells through blunting of autophagy. Cancer Res. 2009, 69, 2453–2460. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.M.; Liu, X.Y.; Sham, K.W.; Lai, J.M.; Cheng, C.H. Silencing of EEF2K (eukaryotic elongation factor-2 kinase) reveals AMPK-ULK1-dependent autophagy in colon cancer cells. Autophagy 2014, 10, 1495–1508. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Xu, Z.Q.; Zong, Y.P.; Ou, B.C.; Shen, X.H.; Feng, H.; Zheng, M.H.; Zhao, J.K.; Lu, A.G. CXCL5 induces tumor angiogenesis via enhancing the expression of FOXD1 mediated by the AKT/NF-κB pathway in colorectal cancer. Cell Death Dis. 2019, 10, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshi, T.; Watanabe Miyano, S.; Watanabe, H.; Sonobe, R.M.K.; Seki, Y.; Ohta, E.; Nomoto, K.; Matsui, J.; Funahashi, Y. Lenvatinib induces death of human hepatocellular carcinoma cells harboring an activated FGF signaling pathway through inhibition of FGFR–MAPK cascades. Biochem. Biophys. Res. Commun. 2019, 513, 1–7. [Google Scholar] [CrossRef]
- Zhu, H.; Song, H.; Chen, G.; Yang, X.; Liu, J.; Ge, Y.; Lu, J.; Qin, Q.; Zhang, C.; Xu, L.; et al. eEF2K promotes progression and radioresistance of esophageal squamous cell carcinoma. Radiother. Oncol. 2017, 124, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Xu, X.; Liu, Q.; Luo, F.; Shi, J.; He, X. MicroRNA-877 acts as a tumor suppressor by directly targeting eEF2K in renal cell carcinoma. Oncol. Lett. 2016, 11, 1474–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashour, A.A.; Gurbuz, N.; Alpay, S.N.; Abdel-Aziz, A.A.H.; Mansour, A.M.; Huo, L.; Ozpolat, B. Elongation factor-2 kinase regulates TG2/β1 integrin/Src/uPAR pathway and epithelial-mesenchymal transition mediating pancreatic cancer cells invasion. J. Cell. Mol. Med. 2014, 18, 2235–2251. [Google Scholar] [CrossRef]
- Bayraktar, R.; Ivan, C.; Bayraktar, E.; Kanlikilicer, P.; Kabil, N.N.; Kahraman, N.; Mokhlis, H.A.; Karakas, D.; Rodriguez-Aguayo, C.; Arslan, A.; et al. Dual Suppressive Effect of miR-34a on the FOXM1/eEF2-Kinase Axis Regulates Triple-Negative Breast Cancer Growth and Invasion. Clin. Cancer Res. 2018, 24, 4225–4241. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Shen, K.; Lenchine, R.V.; Gethings, L.A.; Trim, P.J.; Snel, M.F.; Zhou, Y.; Kenney, J.W.; Kamei, M.; Kochetkova, M.; et al. Eukaryotic elongation factor 2 kinase upregulates the expression of proteins implicated in cell migration and cancer cell metastasis. Int. J. Cancer 2018, 142, 1865–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Will, N.; Lee, K.; Hajredini, F.; Giles, D.H.; Abzalimov, R.R.; Clarkson, M.; Dalby, K.N.; Ghose, R. Structural Dynamics of the Activation of Elongation Factor 2 Kinase by Ca(2+)-Calmodulin. J. Mol. Biol. 2018, 430, 2802–2821. [Google Scholar] [CrossRef]
- Alexey, R.G.; Michael, D.W. Identification of a new class of protein kinases represented by eukaryotic elongation factor-2 kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 4884–4889. [Google Scholar]
- Yamamoto, K.; Kitayama, T.; Ishida, N.; Watanabe, T.; Tanabe, H.; Takatani, M.; Okamoto, T.; Utsumi, R. Identification and Characterization of a Potent Antibacterial Agent, NH125 against Drug-resistant Bacteria. Biosci. Biotech. Biochem. 2000, 64, 919–923. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Yang, J.M.; Kinzy, T.; Utsumi, R.; Okamoto, T.; Kitayama, T.; Ortiz, P.; Hait, W. Identification and Characterization of an Inhibitor of Eukaryotic Elongation Factor 2 Kinase against Human Cancer Cell Lines. Cancer Res. 2003, 63, 6894–6899. [Google Scholar]
- Chen, Z.; Gopalakrishnan, S.M.; Bui, M.H.; Soni, N.B.; Warrior, U.; Johnson, E.F.; Donnelly, J.B.; Glaser, K.B. 1-Benzyl-3-cetyl-2-methylimidazolium iodide (NH125) induces phosphorylation of eukaryotic elongation factor-2 (eEF2). J. Biol. Chem. 2011, 286, 43951–43958. [Google Scholar] [CrossRef] [Green Version]
- Devkota, A.K.; Tavares, C.D.; Warthaka, M.; Abramczyk, O.; Marshall, K.D.; Kaoud, T.S.; Gorgulu, K.; Ozpolat, B.; Dalby, K.N. Investigating the kinetic mechanism of inhibition of elongation factor 2 kinase by NH125: Evidence of a common in vitro artifact. Biochemistry 2012, 51, 2100–2112. [Google Scholar] [CrossRef] [Green Version]
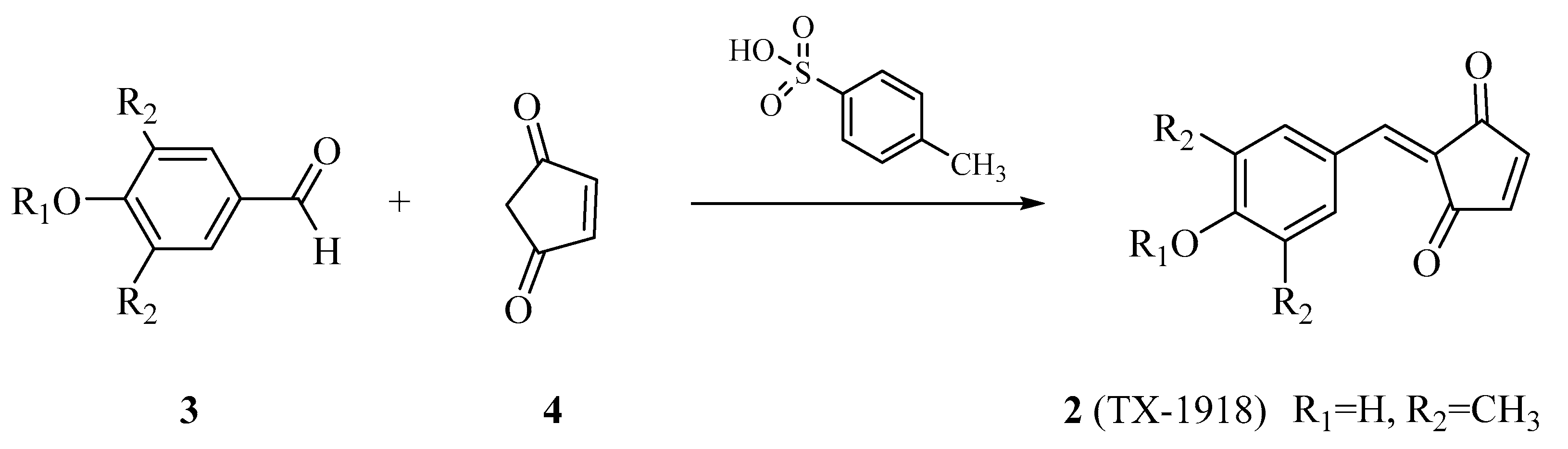
- Hori, H.; Nagasawa, H.; Ishibashi, M.; Uto, Y.; Hirata, A.; Saijo, K.; Ohkura, K.; Kirk, K.L.; Uehara, Y. TX-1123: An antitumor 2-hydroxyarylidene-4-cyclopentene-1,3-dione as a protein tyrosine kinase inhibitor having low mitochondrial toxicity. Bioorg. Med. Chem. 2002, 10, 3257–3265. [Google Scholar] [CrossRef]
- Tomoko, K.; Muneyoshi, O.; Hideyuki, Y. Mechanisms underlying the relaxation by A484954, a eukaryotic elongation factor 2 kinase inhibitor, in rat isolated mesenteric artery. J. Pharmacol. Sci. 2018, 137, 86–92. [Google Scholar]
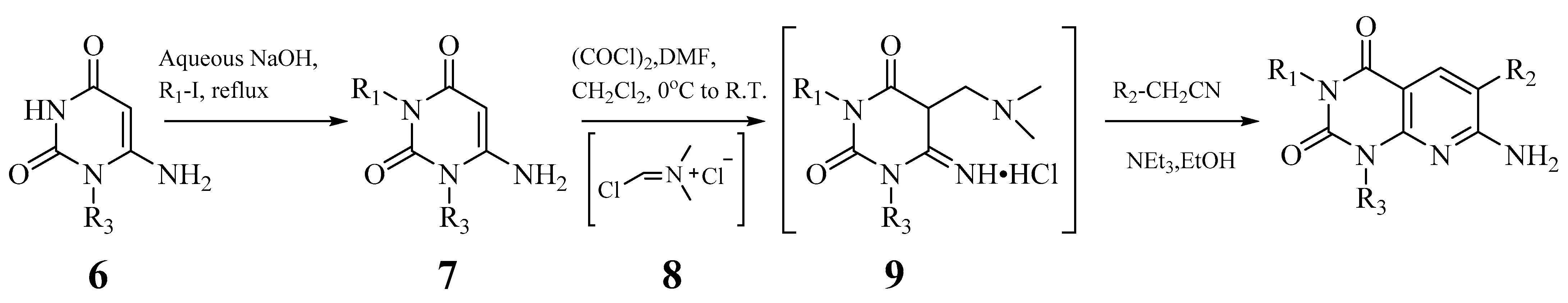
- Edupuganti, R.; Wang, Q.; Tavares, C.D.J.; Chitjian, C.A.; Bachman, J.L.; Ren, P.; Anslyn, E.V.; Dalby, K.N. Synthesis and biological evaluation of pyrido[2,3-d]pyrimidine-2,4-dione derivatives as eEF-2K inhibitors. Bioorg. Med. Chem. 2014, 22, 4910–4916. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhen, Y.; Wang, G.; Yang, G.; Fu, L.; Liu, B.; Ouyang, L. Designing an eEF2K-Targeting PROTAC small molecule that induces apoptosis in MDA-MB-231 cells. Eur. J. Med. Chem. 2020, 204, 112505. [Google Scholar] [CrossRef]
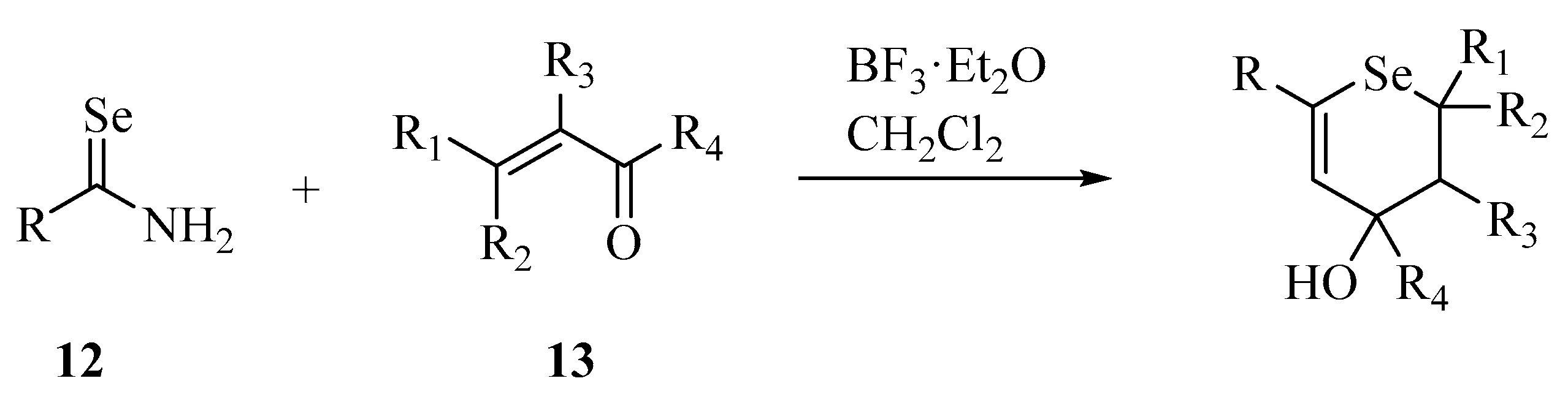
- Koketsu, M.; Senda, T.; Yoshimura, K.; Ishihara, H. Synthesis and characterization of novel 1,3-selenazine derivatives. BF3·Et2O-assisted reaction of primary selenoamides with α,β-unsaturated ketones. J. Chem. Soc. Perkin Trans. 1999, 1, 453–456. [Google Scholar] [CrossRef]
- Cho, S.I.; Koketsu, M.; Ishihara, H.; Matsushita, M.; Nairn, A.C.; Fukazawa, H.; Uehara, Y. Novel compounds, ‘1,3-selenazine derivatives’ as specific inhibitors of eukaryotic elongation factor-2 kinase. Biochim. Biophys. Acta (BBA) Gen. Subj. 2000, 1475, 207–215. [Google Scholar] [CrossRef]
- Reynisson, J.; Court, W.; O’Neill, C.; Day, J.; Patterson, L.; McDonald, E.; Workman, P.; Katan, M.; Eccles, S.A. PLC, Phospholipase C, PLC-gamma, The identification of novel PLC-γ inhibitors using virtual high throughput screening. Bioorg. Med. Chem. 2009, 17, 3169–3176. [Google Scholar] [CrossRef]
- Leung, E.; Hung, J.M.; Barker, D.; Reynisson, J. The effect of a thieno[2,3-b]pyridine PLC-γ inhibitor on the proliferation, morphology, migration and cell cycle of breast cancer cells. MedChemComm 2014, 5, 99–106. [Google Scholar] [CrossRef]
- Ostanin, K.; Hunsaker, T. Enzyme Assay and Use Thereof. US Patent 7338775B1, 4 March 2008. Current Assignee: Myrexis Inc.. [Google Scholar]
- Lockman, J.W.; Reeder, M.D.; Suzuki, K.; Ostanin, K.; Willardsen, J.A. Inhibition of eEF2-K by thieno[2,3-b]pyridine analogues. Bioorg. Med. Chem. Lett. 2010, 20, 2283–2286. [Google Scholar] [CrossRef]
- Guo, Y.; Zhao, Y.; Wang, G.; Chen, Y.; Jiang, Y.; Ouyang, L.; Liu, B. Design, synthesis and structure–activity relationship of a focused library of β-phenylalanine derivatives as novel eEF2K inhibitors with apoptosis-inducing mechanisms in breast cancer. Eur. J. Med. Chem. 2018, 143, 402–418. [Google Scholar] [CrossRef]
- Sun, D.; Zhu, L.; Zhao, Y.; Jiang, Y.; Chen, L.; Yu, Y.; Ouyang, L. Fluoxetine induces autophagic cell death via eEF2K-AMPK-mTOR-ULK complex axis in triple negative breast cancer. Cell Prolif. 2018, 51, e12402. [Google Scholar] [CrossRef] [Green Version]
- Soltoff, S.P. Rottlerin: An inappropriate and ineffective inhibitor of PKCδ. Trends Pharmacol. Sci. 2007, 28, 453–458. [Google Scholar] [CrossRef]
- Akar, U.; Ozpolat, B.; Mehta, K.; Fok, J.; Kondo, Y.; Lopez-Berestein, G. Tissue transglutaminase inhibits autophagy in pancreatic cancer cells. Mol. Cancer Res. 2007, 5, 241–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCracken, M.A.; Miraglia, L.J.; McKay, R.A.; Strobl, J.S. Protein kinase C delta is a prosurvival factor in human breast tumor cell lines. Mol. Cancer Ther. 2003, 2, 273–281. [Google Scholar] [PubMed]
- Lim, J.H.; Woo, S.M.; Min, K.J.; Park, E.J. Rottlerin induces apoptosis of HT29 colon carcinoma cells through NAG-1 upregulation via an ERK and p38 MAPK-dependent and PKC δ-independent mechanism. Chem. Biol. Interact. 2012, 197, 1–7. [Google Scholar] [CrossRef]
- Clark, A.S.; West, K.A.; Blumberg, P.M.; Dennis, P.A. Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: PKCdelta promotes cellular survival and chemotherapeutic resistance. Cancer Res. 2003, 63, 780–786. [Google Scholar]
- Ni, H.; Ergin, M.; Tibudan, S.S.; Denning, M.F.; Izban, K.F.; Alkan, S. Protein kinase C-delta is commonly expressed in multiple myeloma cells and its downregulation by rottlerin causes apoptosis. Br. J. Haematol. 2003, 121, 849–856. [Google Scholar] [CrossRef]
- Gschwendt, M.; Kittstein, W.; Marks, F. Elongation factor-2 kinase: Effective inhibition by the novel protein kinase inhibitor rottlerin and relative insensitivity towards staurosporine. FEBS Lett. 1994, 338, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Soltoff, S.P. Rottlerin Is a Mitochondrial Uncoupler That Decreases Cellular ATP Levels and Indirectly Blocks Protein Kinase Cδ Tyrosine Phosphorylation. J. Biol. Chem. 2001, 276, 37986–37992. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.Z. Rottlerin induces calcium influx and protein degradation in cultured lenses independent of effects on protein kinase C delta. Basic Clin. Pharmacol. Toxicol. 2007, 101, 459–464. [Google Scholar] [CrossRef]
- Parmer, T.G.; Ward, M.D.; Hait, W.N. Effects of rottlerin, an inhibitor of calmodulin-dependent protein kinase III, on cellular proliferation, viability, and cell cycle distribution in malignant glioma cells. Cell Growth Differ. 1997, 8, 327–334. [Google Scholar] [PubMed]
- Ohno, I.; Eibl, G.; Odinokova, I.; Edderkaoui, M.; Damoiseaux, R.D.; Yazbec, M.; Abrol, R.; Goddard, W.A.; Yokosuka, O.; Pandol, S.J.; et al. Rottlerin stimulates apoptosis in pancreatic cancer cells through interactions with proteins of the Bcl-2 family. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 298, G63–G73. [Google Scholar] [CrossRef] [Green Version]
- Mansour, M.A.; Nagi, M.N.; El-Khatib, A.S.; Al-Bekairi, A.M. Effects of thymoquinone on antioxidant enzyme activities, lipid peroxidation and DT-diaphorase in different tissues of mice: A possible mechanism of action. Cell Biochem. Funct. 2002, 20, 143–151. [Google Scholar] [CrossRef]
- Banerjee, S.; Padhye, S.; Azmi, A.; Wang, Z.; Philip, P.A.; Kucuk, O.; Sarkar, F.H.; Mohammad, R.M. Review on molecular and therapeutic potential of thymoquinone in cancer. Nutr. Cancer 2010, 62, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Asaduzzaman Khan, M.; Tania, M.; Fu, S.; Fu, J. Thymoquinone, as an anticancer molecule: From basic research to clinical investigation. Oncotarget 2017, 8, 51907–51919. [Google Scholar] [CrossRef] [Green Version]
- Ballout, F.; Monzer, A.; Fatfat, M.; Ouweini, H.E.; Jaffa, M.A.; Abdel-Samad, R.; Darwiche, N.; Abou-Kheir, W.; Gali-Muhtasib, H. Thymoquinone induces apoptosis and DNA damage in 5-Fluorouracil-resistant colorectal cancer stem/progenitor cells. Oncotarget 2020, 11, 2959–2972. [Google Scholar] [CrossRef] [PubMed]
- AlGhamdi, A.A.; Mohammed, M.R.S.; Zamzami, M.A.; Al-Malki, A.L.; Qari, M.H.; Khan, M.I.; Choudhry, H. Untargeted metabolomics identifies key metabolic pathways altered by thymoquinone in leukemic cancer cells. Nutrients 2020, 12, 1792. [Google Scholar] [CrossRef] [PubMed]
- Arafa el, S.A.; Zhu, Q.; Shah, Z.I.; Wani, G.; Barakat, B.M.; Racoma, I.; El-Mahdy, M.A.; Wani, A.A. Thymoquinone up-regulates PTEN expression and induces apoptosis in doxorubicin-resistant human breast cancer cells. Mutat. Res. 2011, 706, 28–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akter, Z.; Ahmed, F.R.; Tania, M.; Khan, M.A. Targeting inflammatory mediators: An anticancer mechanism of thymoquinone action. Curr. Med. Chem. 2021, 28, 80–92. [Google Scholar] [CrossRef]
- Ahmad, A.; Mishra, R.K.; Vyawahare, A.; Kumar, A.; Rehman, M.U.; Qamar, W.; Khan, A.Q.; Khan, R. Thymoquinone (2-Isoprpyl-5-methyl-1, 4-benzoquinone) as a chemopreventive/anticancer agent: Chemistry and biological effects. Saudi Pharm. J. 2019, 27, 1113–1126. [Google Scholar] [CrossRef]
- Kabil, N.; Bayraktar, R.; Kahraman, N.; Mokhlis, H.A.; Calin, G.A.; Lopez-Berestein, G.; Ozpolat, B. Thymoquinone inhibits cell proliferation, migration, and invasion by regulating the elongation factor 2 kinase (eEF-2K) signaling axis in triple-negative breast cancer. Breast Cancer Res. Ther. 2018, 171, 593–605. [Google Scholar] [CrossRef]
- Ishibashi, M. Isolation of bioactive natural products from myxomycetes. Med. Chem. 2005, 1, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, N.; Zhang, T.; Zhang, B.; Sajeevan, T.P.; Joseph, V.; Armstrong, L.; He, S.; Yan, X.; Naman, C.B. A systematic review of recently reported marine derived natural product kinase inhibitors. Mar. Drugs 2019, 17, 493. [Google Scholar] [CrossRef] [Green Version]
- Bronstrup, M.; Sasse, F. Natural products targeting the elongation phase of eukaryotic protein biosynthesis. Nat. Prod. Rep. 2020, 37, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Muthukumar, Y.; Roy, M.; Raja, A.; Taylor, R.E.; Sasse, F. The marine polyketide myriaporone 3/4 stalls translation by targeting the elongation phase. Chembiochem 2013, 14, 260–264. [Google Scholar] [CrossRef]
- Yamada, T.; Iwamoto, C.; Yamagaki, N.; Yamanouchi, T.; Minoura, K.; Yamori, T.; Uehara, Y.; Andoh, T.; Umemura, K.; Numata, A. Leptosins M-N1, cytotoxic metabolites from a Leptosphaeria species separated from a marine alga. Structure determination and biological activities. Tetrahedron 2002, 58, 479–487. [Google Scholar] [CrossRef]
- Zhang, C.; Lei, J.L.; Zhang, H. Calyxin Y sensitizes cisplatin-sensitive and resistant hepatocellular carcinoma cells to cisplatin through apoptotic and autophagic cell death via SCF βTrCP-mediated eEF2K degradation. Oncotarget 2017, 8, 70595–70616. [Google Scholar] [CrossRef]
- Pan, Z.; Chen, Y.; Liu, J.; Jiang, Q.; Yang, S.; Guo, L.; He, G. Design, synthesis, and biological evaluation of polo-like kinase 1/eukaryotic elongation factor 2 kinase (PLK1/EEF2K) dual inhibitors for regulating breast cancer cells apoptosis and autophagy. Eur. J. Med. Chem. 2018, 144, 517–528. [Google Scholar] [CrossRef]
- Cao, M.J.; Zhu, T.; Liu, J.T.; Ouyang, L.; Lin, H.W. New sorbicillinoid derivatives with GLP-1R and eEF2K affinities from a sponge-derived fungus Penicillium chrysogenum 581F1. Nat. Prod. Res. 2020, 34, 2880–2886. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, J.M.; Iannone, M.; Shih, W.J.; Lin, Y.; Hait, W.N. Disruption of the EF-2 kinase/Hsp90 protein complex: A possible mechanism to inhibit glioblastoma by geldanamycin. Cancer Res. 2001, 61, 4010–4016. [Google Scholar] [PubMed]
- Khaledian, B.; Taguchi, A.; Shin-Ya, K.; Kondo-Ida, L.; Kagaya, N.; Suzuki, M.; Kajino, T.; Yamaguchi, T.; Shimada, Y.; Takahashi, T. Inhibition of heat shock protein 90 destabilizes receptor tyrosine kinase ROR1 in lung adenocarcinoma. Cancer Sci. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Talaei, S.; Mellatyar, H.; Asadi, A.; Akbarzadeh, A.; Sheervalilou, R.; Zarghami, N. Spotlight on 17-AAG as an Hsp90 inhibitor for molecular targeted cancer treatment. Chem. Biol. Drug Des. 2019, 93, 760–786. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, K.C.; Haixiao, J.; Naman, C.B.; Sajeevan, T.P. Prospects of multitarget drug designing strategies by linking molecular docking and molecular dynamics to explore the protein–Ligand recognition process. Drug Dev. Res. 2020, 81, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Korner, M.; Christ, E.; Wild, D.; Reubi, J.C. Glucagon-like peptide-1 receptor overexpression in cancer and its impact on clinical applications. Front. Endocrinol. 2012, 3, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apel, A.; Zentgraf, H.; Büchler, M.W.; Herr, I. Autophagy-A double-edged sword in oncology. Int. J. Cancer 2009, 125, 991–995. [Google Scholar] [CrossRef] [PubMed]
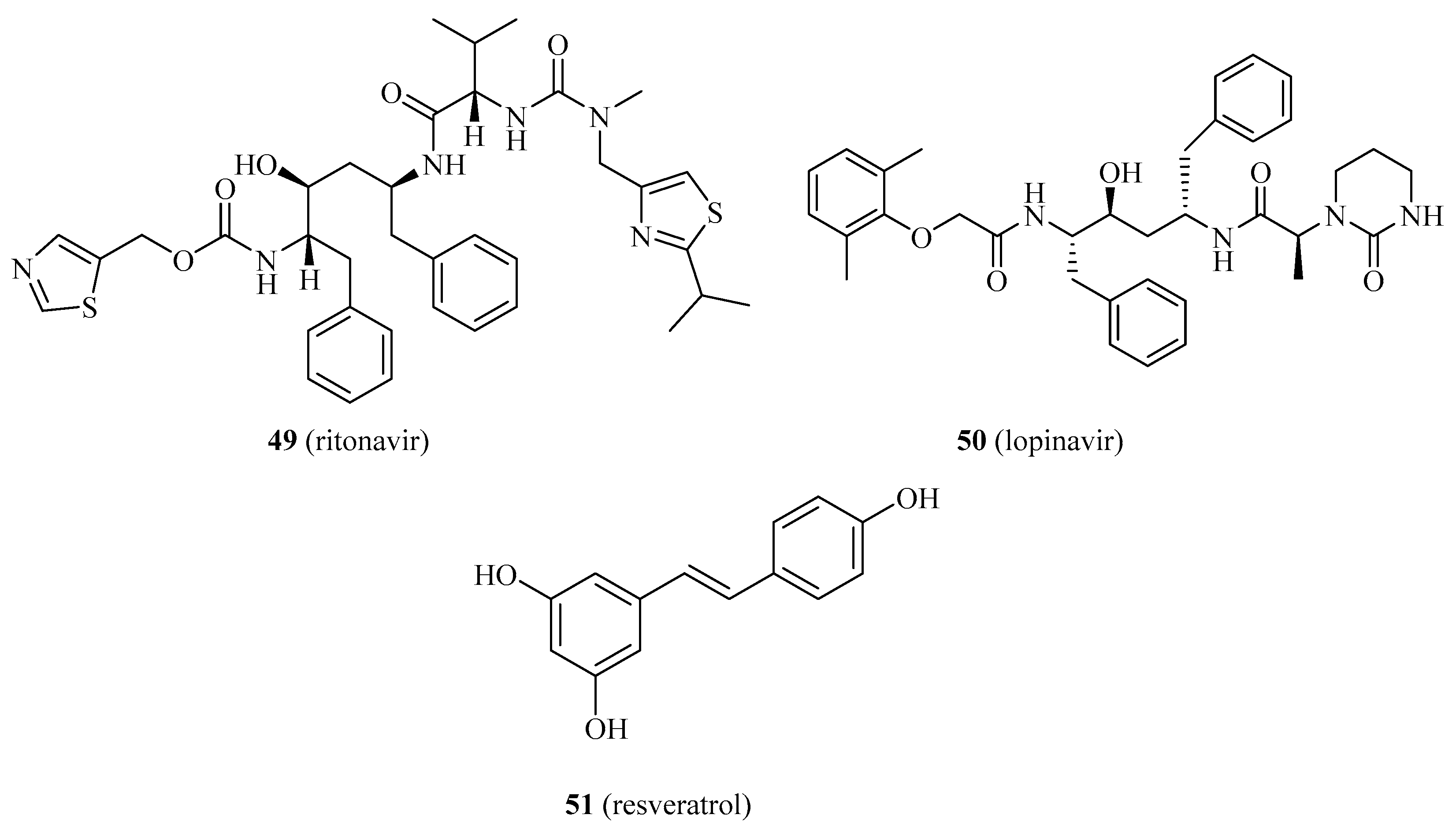
- Hong-Brown, L.Q.; Kazi, A.A.; Lang, C.H. Mechanisms mediating the effects of alcohol and HIV anti-retroviral agents on mTORC1, mTORC2 and protein synthesis in myocytes. World J. Biol. Chem. 2012, 3, 110–120. [Google Scholar] [CrossRef]
- Hong-Brown, L.Q.; Brown, C.R.; Huber, D.S.; Lang, C.H. Lopinavir impairs protein synthesis and induces eEF2 phosphorylation via the activation of AMP-activated protein kinase. J. Cell. Biochem. 2008, 105, 814–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Ong, Y.K.; Wang, Y. Role of adjunctive treatment strategies in COVID-19 and a review of international and national clinical guidelines. Mil. Med. Res. 2020, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Dace, D.S.; Ryazanov, A.G.; Kelly, J.; Apte, R.S. Resveratrol regulates pathologic angiogenesis by a eukaryotic elongation factor-2 kinase-regulated pathway. Am. J. Pathol. 2010, 177, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Feng, Y.; Tan, H.Y.; Cheung, F.; Hong, M.; Lao, L.; Nagamatsu, T. Inhibition of eukaryotic elongation factor-2 confers to tumor suppression by a herbal formulation Huanglian-Jiedu decoction in human hepatocellular carcinoma. J. Ethnopharmacol. 2015, 164, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
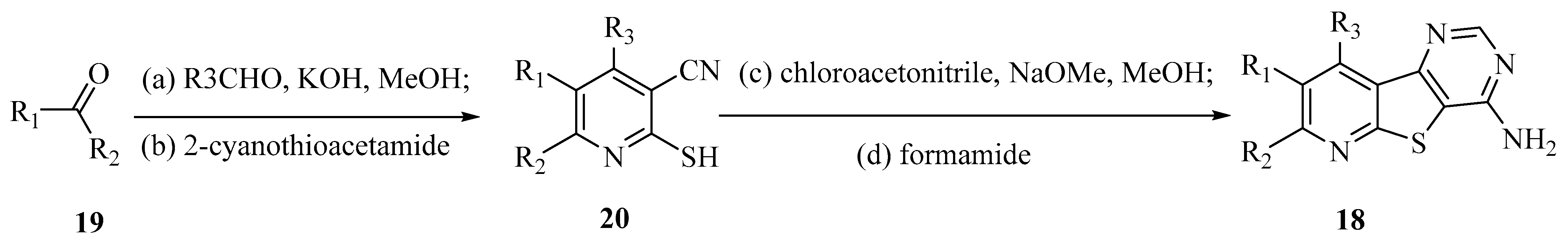{kind=link}
{kind=link}
{kind=link}
| Compound | Original Name in Published Report | Discovery Method a | Development Status for Cancer Therapy b | Reference |
|---|---|---|---|---|
| 1 | NH125 | KIO | Recent | [73,80,81,82,83] |
| 2 | TX-1918 | KIO | NA | [84] |
| 5 | A-484954 | HTS | Recent | [82,85] |
| 10 | compound 9 | KIO | NA | [86] |
| 11 | compound 11l | KIO | Recent | [87] |
| 14 | TS-2 | KIO | NA | [88,89] |
| 15 | TS-4 | KIO | NA | [88,89] |
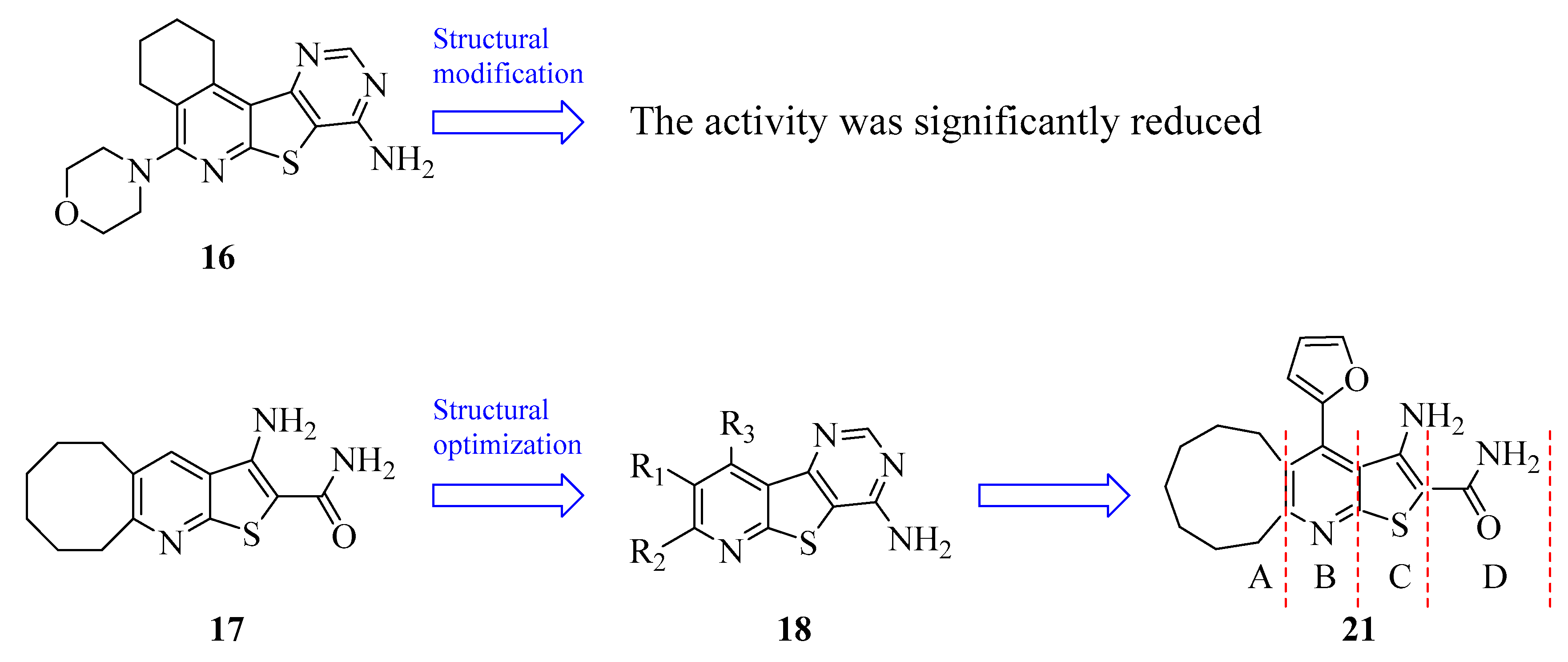
| 16 | compound 1 | HTS | NA | [90,91,92] |
| 17 | compound 2 | HTS | NA | [90,91,92] |
| 21 | compound 34 | KIO | NA | [93] |
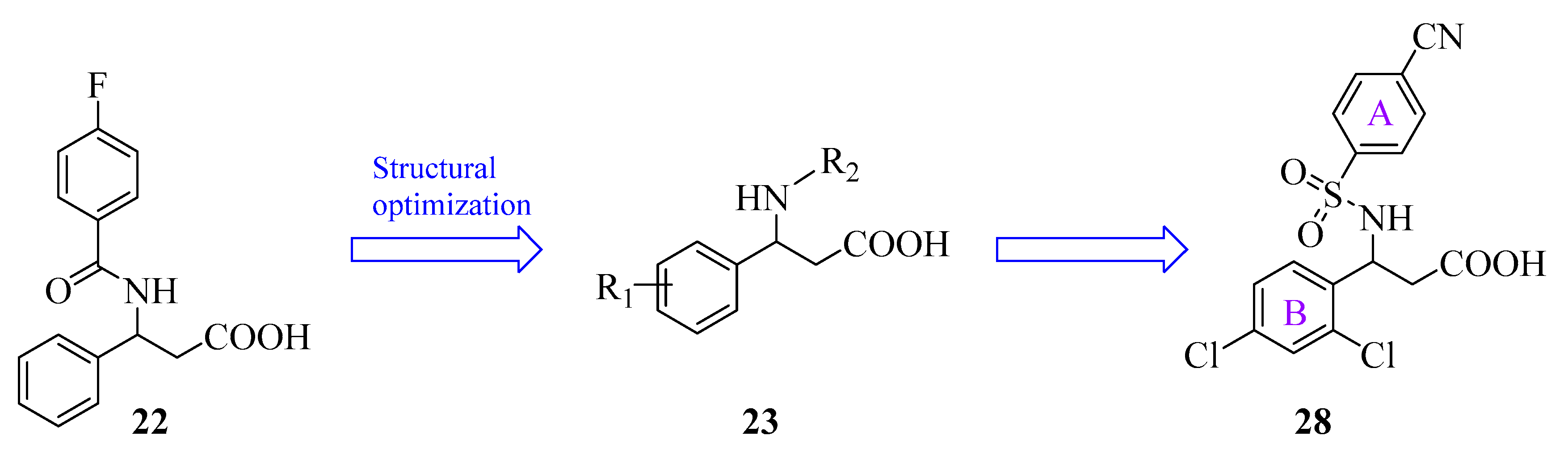
| 22 | compound 9 | CADD | Recent | [94] |
| 28 | compound 21l | CADD | Recent | [94] |
| 29 | fluoxetine | DRR | Recent | [95] |
| 30 | rottlerin | NPDD | NA | [96,97,98,99,100,101,102,103,104,105,106] |
| 31 | thymoquinone | NPDD | Recent | [107,108,109,110,111,112,113,114,115] |
| 32 | 6-hydroxystaurosporinone | NPDD | NA | [116,117] |
| 33 | myriaporone 3/4 | NPDD | NA | [118,119] |
| 34 | leptosin M | NPDD | NA | [120] |
| 35 | calyxin Y | NPDD | Recent | [121] |
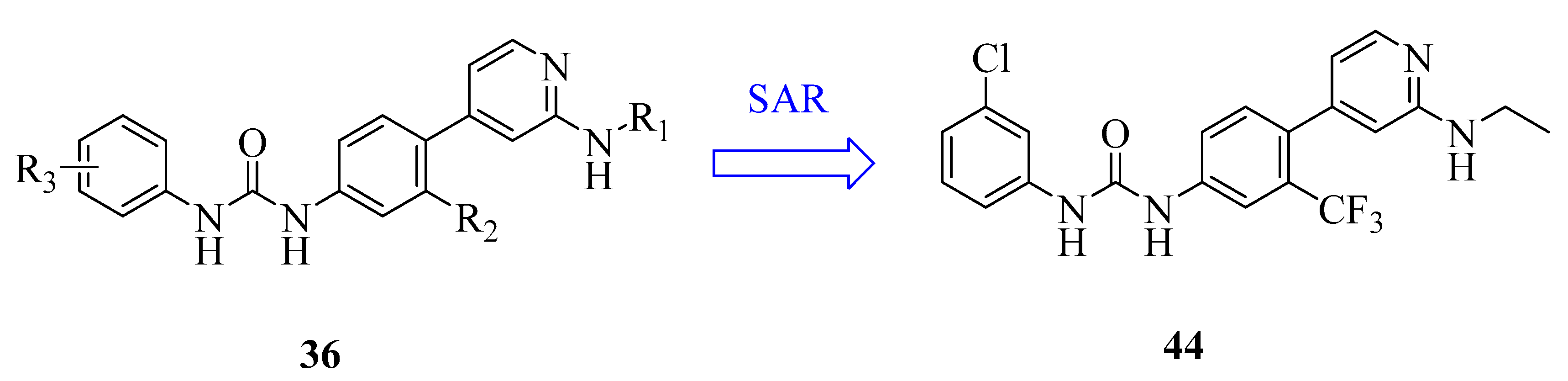
| 44 | compound 18i | MTDD | Recent | [122] |
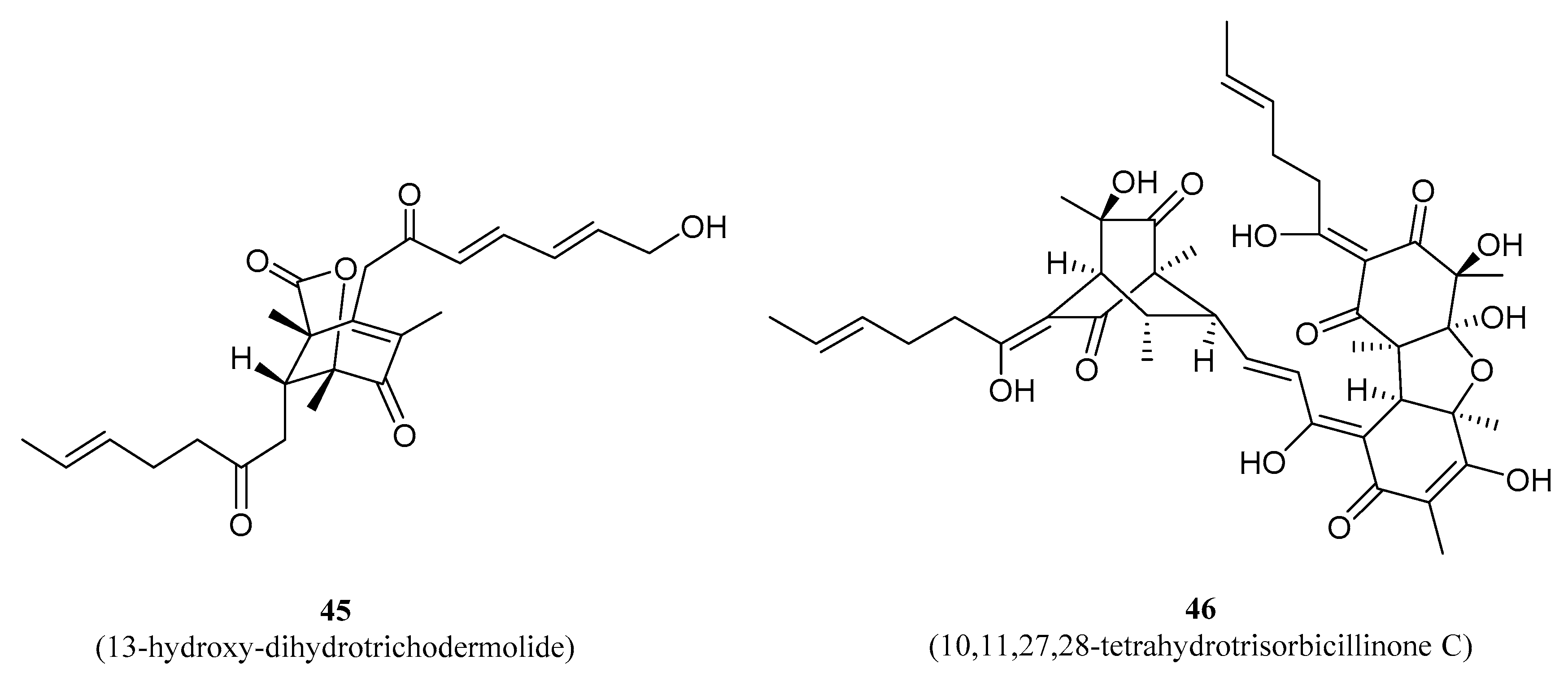
| 45 | 13-hydroxy-dihydro- trichodermolide | NPDD | Recent | [123] |
| 46 | 10,11,27,28-tetrahydro- trisorbicillinone C | NPDD | Recent | [123] |
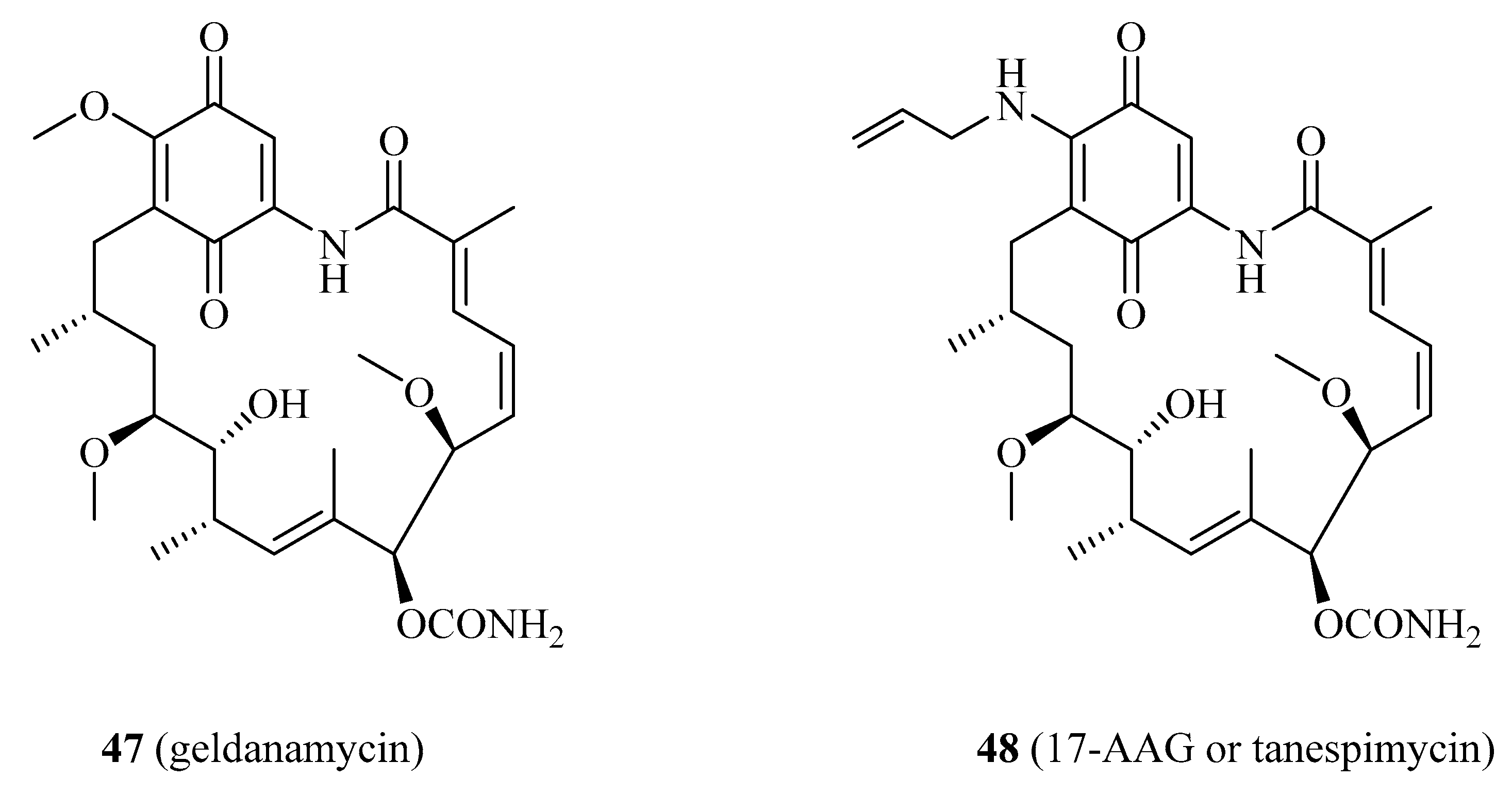
| 47 | geldanamycin | NPDD | Recent | [124,125] |
| 48 | 17-AAG | NPDD | Recent | [124,126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, B.; Zou, J.; Zhang, Q.; Wang, Z.; Wang, N.; He, S.; Zhao, Y.; Naman, C.B. Progress in the Development of Eukaryotic Elongation Factor 2 Kinase (eEF2K) Natural Product and Synthetic Small Molecule Inhibitors for Cancer Chemotherapy. Int. J. Mol. Sci. 2021, 22, 2408. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052408
Zhang B, Zou J, Zhang Q, Wang Z, Wang N, He S, Zhao Y, Naman CB. Progress in the Development of Eukaryotic Elongation Factor 2 Kinase (eEF2K) Natural Product and Synthetic Small Molecule Inhibitors for Cancer Chemotherapy. International Journal of Molecular Sciences. 2021; 22(5):2408. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052408
Chicago/Turabian StyleZhang, Bin, Jiamei Zou, Qiting Zhang, Ze Wang, Ning Wang, Shan He, Yufen Zhao, and C. Benjamin Naman. 2021. "Progress in the Development of Eukaryotic Elongation Factor 2 Kinase (eEF2K) Natural Product and Synthetic Small Molecule Inhibitors for Cancer Chemotherapy" International Journal of Molecular Sciences 22, no. 5: 2408. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052408