
Mechanisms by Which Probiotic Bacteria Attenuate the Risk of Hepatocellular Carcinoma



Abstract
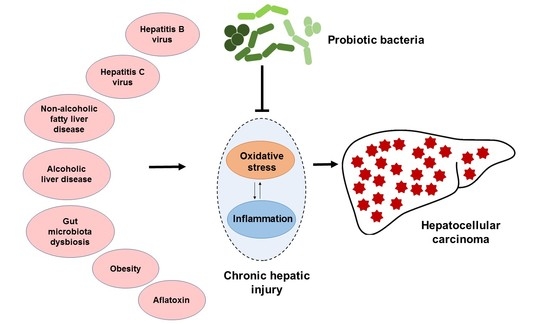
:
1. Introduction
2. Hepatocellular Carcinoma
2.1. HCC Etiology
2.2. Tumor Microenvironment and Molecular Pathogenesis of HCC
3. Association of Gut Microbiota with the Pathogenesis of HCC
4. Probiotic Bacteria-Mediated Mechanisms to Attenuate HCC
5. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [Green Version]
- Castelli, G.; Pelosi, E.; Testa, U. Liver cancer: Molecular characterization, clonal evolution and cancer stem cells. Cancers 2017, 9, 127. [Google Scholar] [CrossRef] [Green Version]
- Ng, C.K.Y.; Piscuoglio, S.; Terracciano, L.M. Molecular classification of hepatocellular carcinoma: The view from metabolic zonation. Hepatology 2017, 66, 1377–1380. [Google Scholar] [CrossRef]
- Ko, K.-L.; Mak, L.-Y.; Cheung, K.-S.; Yuen, M.-F. Hepatocellular carcinoma: Recent advances and emerging medical therapies. F1000Research 2020, 9. [Google Scholar] [CrossRef]
- Gupta, H.; Youn, G.S.; Shin, M.J.; Suk, K.T. Role of gut microbiota in hepatocarcinogenesis. Microorganisms 2019, 7, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, M.L.Y.; El-Nezami, H. Targeting gut microbiota in hepatocellular carcinoma: Probiotics as a novel therapy. Hepatobiliary Surg. Nutr. 2018, 7, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowiak-Kopeć, P.; Śliżewska, K. The effect of probiotics on the production of short-chain fatty acids by human intestinal microbiome. Nutrients 2020, 12, 1107. [Google Scholar] [CrossRef]
- Tajiri, K.; Shimizu, Y. Gut bacteria may control development of hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2017, 6, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Yu, Z.; Tian, F.; Zhao, J.; Zhang, H.; Zhai, Q.; Chen, W. Surface components and metabolites of probiotics for regulation of intestinal epithelial barrier. Microb. Cell Factories 2020, 19, 23. [Google Scholar] [CrossRef]
- Brandi, G.; De Lorenzo, S.; Candela, M.; Pantaleo, M.A.; Bellentani, S.; Tovoli, F.; Saccoccio, G.; Biasco, G. Microbiota, NASH, HCC and the potential role of probiotics. Carcinogenesis 2017, 38, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Abenavoli, L.; Luzza, F.; Mendez-Sanchez, N. Probiotics supplementation in the management of hepatocellular carcinoma. Hepatobiliary Surg. Nutr. 2019, 8, 632–634. [Google Scholar] [CrossRef] [PubMed]
- Reeves, H.L.; Zaki, M.Y.W.; Day, C.P. Hepatocellular carcinoma in obesity, type 2 diabetes, and NAFLD. Dig. Dis. Sci. 2016, 61, 1234–1245. [Google Scholar] [CrossRef]
- Chávez-Tapia, N.C.; González-Rodríguez, L.; Jeong, M.; López-Ramírez, Y.; Barbero-Becerra, V.; Juárez-Hernández, E.; Romero-Flores, J.L.; Arrese, M.; Méndez-Sánchez, N.; Uribe, M. Current evidence on the use of probiotics in liver diseases. J. Funct. Foods 2015, 17, 137–151. [Google Scholar] [CrossRef]
- Markowiak, P.; Śliżewska, K. Effects of probiotics, prebiotics, and synbiotics on human health. Nutrients 2017, 9, 1021. [Google Scholar] [CrossRef]
- Peredo-Lovillo, A.; Romero-Luna, H.E.; Jiménez-Fernández, M. Health promoting microbial metabolites produced by gut microbiota after prebiotics metabolism. Food Res. Int. 2020, 136, 109473. [Google Scholar] [CrossRef] [PubMed]
- Theilmann, M.C.; Goh, Y.J.; Nielsen, K.F.; Klaenhammer, T.R.; Barrangou, R.; Hachem, M.A. Lactobacillus acidophilus metabolizes dietary plant glucosides and externalizes their bioactive phytochemicals. mBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Dhanasekaran, R.; Bandoh, S.; Roberts, L.R. Molecular pathogenesis of hepatocellular carcinoma and impact of therapeutic advances. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Ghouri, Y.A.; Mian, I.; Rowe, J.H. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J. Carcinog. 2017, 16. [Google Scholar] [CrossRef]
- Janevska, D.; Chaloska-Ivanova, V.; Janevski, V. Hepatocellular carcinoma: Risk factors, diagnosis and treatment. Open Access Maced. J. Med. Sci. 2015, 3, 732–736. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.Y.; Friedman, S.L. Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology 2012, 56, 769–775. [Google Scholar] [CrossRef]
- Lee, D.H.; Lee, J.M. Primary malignant tumours in the non-cirrhotic liver. Eur. J. Radiol. 2017, 95, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Sandhu, S.; Lai, J.-P.; Sandhu, D.S. Hepatocellular carcinoma in non-cirrhotic liver: A comprehensive review. World J. Hepatol. 2019, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of hbv-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef]
- Su, I.-J.; Wang, L.H.-C.; Hsieh, W.-C.; Wu, H.-C.; Teng, C.-F.; Tsai, H.-W.; Huang, W. The emerging role of hepatitis b virus pre-s2 deletion mutant proteins in HBV tumorigenesis. J. Biomed. Sci. 2014, 21, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, C.-J.; Lin, Y.-J.; Yen, C.-S.; Tsai, H.-W.; Tsai, T.-F.; Chang, K.-Y.; Huang, W.-C.; Lin, P.-W.; Chiang, C.-W.; Chang, T.-T. Hepatitis B virus x protein upregulates MTOR signaling through IKKβ to increase cell proliferation and VEGF production in hepatocellular carcinoma. PLoS ONE 2012, 7, e41931. [Google Scholar] [CrossRef]
- Pandey, A.K.; Singhi, E.K.; Arroyo, J.P.; Ikizler, T.A.; Gould, E.R.; Brown, J.; Beckman, J.A.; Harrison, D.G.; Moslehi, J. Mechanisms of VEGF (vascular endothelial growth factor) inhibitor–associated hypertension and vascular disease. Hypertension 2018, 71, e1–e8. [Google Scholar] [CrossRef]
- Thio, C.L.; Hawkins, C. 148—Hepatitis B virus and hepatitis delta virus. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed.; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; Elsevier: Philadelphia, PA, USA, 2015; pp. 1815–1839.e7. ISBN 978-1-4557-4801-3. [Google Scholar]
- Jin, Y.; Wu, D.; Yang, W.; Weng, M.; Li, Y.; Wang, X.; Zhang, X.; Jin, X.; Wang, T. Hepatitis B virus X protein induces epithelial-mesenchymal transition of hepatocellular carcinoma cells by regulating long non-coding RNA. Virol. J. 2017, 14, 238. [Google Scholar] [CrossRef] [Green Version]
- De Martel, C.; Maucort-Boulch, D.; Plummer, M.; Franceschi, S. World-wide relative contribution of hepatitis B and C viruses in hepatocellular carcinoma. Hepatol. Baltim. MD 2015, 62, 1190–1200. [Google Scholar] [CrossRef]
- El-Serag, H.B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 2012, 142, 1264–1273.e1. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.L.; Morgan, T.R. The natural history of hepatitis C virus (HCV) infection. Int. J. Med. Sci. 2006, 3, 47–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axley, P.; Ahmed, Z.; Ravi, S.; Singal, A.K. Hepatitis C virus and hepatocellular carcinoma: A narrative review. J. Clin. Transl. Hepatol. 2018, 6, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Wang, Y.; Li, M.; Tong, W.-M.; Jia, J.-D.; Huang, J. Correlation of TP53 mutations with HCV positivity in hepatocarcinogenesis: Identification of a novel TP53 microindel in hepatocellular carcinoma with HCV infection. Oncol. Rep. 2013, 30, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mileo, A.M.; Mattarocci, S.; Matarrese, P.; Anticoli, S.; Abbruzzese, C.; Catone, S.; Sacco, R.; Paggi, M.G.; Ruggieri, A. Hepatitis C virus core protein modulates PRb2/P130 expression in human hepatocellular carcinoma cell lines through promoter methylation. J. Exp. Clin. Cancer Res. 2015, 34, 140. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhao, Y.; Gao, Y.; Hu, W.; Qu, Y.; Lou, N.; Zhu, Y.; Zhang, X.; Yang, H. Hepatitis C virus NS3 protein enhances hepatocellular carcinoma cell invasion by promoting PPM1A ubiquitination and degradation. J. Exp. Clin. Cancer Res. 2017, 36, 42. [Google Scholar] [CrossRef] [Green Version]
- Irshad, M.; Gupta, P.; Irshad, K. Molecular basis of hepatocellular carcinoma induced by hepatitis C virus infection. World J. Hepatol. 2017, 9, 1305–1314. [Google Scholar] [CrossRef] [Green Version]
- Younes, R.; Bugianesi, E. Should we undertake surveillance for hcc in patients with NAFLD? J. Hepatol. 2018, 68, 326–334. [Google Scholar] [CrossRef]
- Chen, K.; Ma, J.; Jia, X.; Ai, W.; Ma, Z.; Pan, Q. Advancing the understanding of NAFLD to hepatocellular carcinoma development: From experimental models to humans. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 117–125. [Google Scholar] [CrossRef]
- Cobbina, E.; Akhlaghi, F. Non-alcoholic fatty liver disease (NAFLD)—pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab. Rev. 2017, 49, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Mehal, W.Z. Sterile inflammation in the liver. Gastroenterology 2012, 143, 1158–1172. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Yi, J.; Newberg, J.Y.; Tien, J.C.; Wu, H.; Finegold, M.J.; Kodama, M.; Wei, Z.; Tamura, T.; Takehara, T.; et al. Molecular Profiling of nonalcoholic fatty liver disease-associated hepatocellular carcinoma using SB transposon mutagenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E10417–E10426. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Ma, H.; Wang, L.; Song, X.; Zhang, J.; Liu, W.; Ge, Y.; Sun, Y.; Yu, X.; Wang, Z.; et al. Tumor suppressor ZHX2 inhibits NAFLD–HCC progression via blocking LPL-mediated lipid uptake. Cell Death Differ. 2020, 27, 1693–1708. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, P.; Sharma, Y.; Maisuradze, L.; Tchaikovskaya, T.; Gupta, S. Ataxia telangiectasia mutated pathway disruption affects hepatic DNA and tissue damage in nonalcoholic fatty liver disease. Exp. Mol. Pathol. 2020, 113, 104369. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- George, V.C.; Ansari, S.A.; Chelakkot, V.S.; Chelakkot, A.L.; Chelakkot, C.; Menon, V.; Ramadan, W.; Ethiraj, K.R.; El-Awady, R.; Mantso, T.; et al. DNA-dependent protein kinase: Epigenetic alterations and the role in genomic stability of cancer. Mutat. Res. Mutat. Res. 2019, 780, 92–105. [Google Scholar] [CrossRef]
- Ganne-Carrié, N.; Nahon, P. Hepatocellular carcinoma in the setting of alcohol-related liver disease. J. Hepatol. 2019, 70, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [Green Version]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef]
- Mizumoto, A.; Ohashi, S.; Hirohashi, K.; Amanuma, Y.; Matsuda, T.; Muto, M. Molecular mechanisms of acetaldehyde-mediated carcinogenesis in squamous epithelium. Int. J. Mol. Sci. 2017, 18, 1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setshedi, M.; Wands, J.R.; de la Monte, S.M. Acetaldehyde adducts in alcoholic liver disease. Oxid. Med. Cell. Longev. 2010, 3, 178–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paget, V.; Lechevrel, M.; Sichel, F. Acetaldehyde-induced mutational pattern in the tumour suppressor gene TP53 analysed by use of a functional assay, the FASAY (functional analysis of separated alleles in yeast). Mutat. Res. Toxicol. Environ. Mutagen. 2008, 652, 12–19. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.-C.; Llovet, J.M. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, N.; Zhou, H.; Fan, H.; Yuan, Y. Single nucleotide polymorphisms and cancer susceptibility. Oncotarget 2017, 8, 110635–110649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donati, B.; Pietrelli, A.; Pingitore, P.; Dongiovanni, P.; Caddeo, A.; Walker, L.; Baselli, G.; Pelusi, S.; Rosso, C.; Vanni, E.; et al. Telomerase reverse transcriptase germline mutations and hepatocellular carcinoma in patients with nonalcoholic fatty liver disease. Cancer Med. 2017, 6, 1930–1940. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Liu, F.; Li, B.; Chen, X.; Ma, Y.; Yan, L.; Wen, T.; Xu, M.; Wang, W.; Yang, J. Polymorphisms of Tumor necrosis factor-alpha and hepatocellular carcinoma risk: A huge systematic review and meta-analysis. Dig. Dis. Sci. 2011, 56, 2227–2236. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.G.; Ho, R.L.K.; Wong, J.; Lee, K.F.; Lai, P.B.S. Single nucleotide polymorphism in the promoter region of human alpha-fetoprotein (AFP) gene and its significance in hepatocellular carcinoma (HCC). Eur. J. Surg. Oncol. 2007, 33, 882–886. [Google Scholar] [CrossRef]
- Junjie, X.; Songyao, J.; Minmin, S.; Yanyan, S.; Baiyong, S.; Xiaxing, D.; Jiabin, J.; Xi, Z.; Hao, C. The Association between toll-like receptor 2 single-nucleotide polymorphisms and hepatocellular carcinoma susceptibility. BMC Cancer 2012, 12, 57. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, L.; Xiang, X.; Wang, H.; Cai, W.; Xie, J.; Han, Y.; Bao, S.; Xie, Q. Three common functional polymorphisms in microRNA encoding genes in the susceptibility to hepatocellular carcinoma: A systematic review and meta-analysis. Gene 2013, 527, 584–593. [Google Scholar] [CrossRef]
- Hirankarn, N.; Kimkong, I.; Kummee, P.; Tangkijvanich, P.; Poovorawan, Y. Interleukin-1β gene polymorphism associated with hepatocellular carcinoma in hepatitis B virus infection. World J. Gastroenterol. 2006, 12, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-M.; Xie, H.-Y.; Zhou, L.; Yang, Z.; Zhang, F.; Zheng, S.-S. A Single nucleotide polymorphism in the vascular endothelial growth factor gene is associated with recurrence of hepatocellular carcinoma after transplantation. Arch. Med. Res. 2009, 40, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, Y.; Long, X.-D.; Wang, W.; Jiao, H.-K.; Mei, Z.; Yin, Q.-Q.; Ma, L.-N.; Zhou, A.-W.; Wang, L.-S.; et al. Cbx4 governs HIF-1α to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell 2014, 25, 118–131. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.-D.; Ma, Y.-S.; Fang, Y.; Liu, L.-L.; Fu, D.; Shen, X.-Z. Role of the microenvironment in hepatocellular carcinoma development and progression. Cancer Treat. Rev. 2012, 38, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Klaas, M.; Kangur, T.; Viil, J.; Mäemets-Allas, K.; Minajeva, A.; Vadi, K.; Antsov, M.; Lapidus, N.; Järvekülg, M.; Jaks, V. The alterations in the extracellular matrix composition guide the repair of damaged liver tissue. Sci. Rep. 2016, 6, 27398. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.G. Cellular sources of extracellular matrix in hepatic fibrosis. Clin. Liver Dis. 2008, 12, 759–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez–Gea, V.; Toffanin, S.; Friedman, S.L.; Llovet, J.M. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013, 144, 512–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, A.X.; Duda, D.G.; Sahani, D.V.; Jain, R.K. HCC and angiogenesis: Possible targets and future directions. Nat. Rev. Clin. Oncol. 2011, 8, 292–301. [Google Scholar] [CrossRef]
- Shammas, M.A. Telomeres, lifestyle, cancer, and aging. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 28–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaiteerakij, R.; Roberts, L.R. Telomerase mutation: A genetic risk factor for cirrhosis. Hepatology 2011, 53, 1430–1432. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166–2176. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, D.; Saitta, C.; Giosa, D.; Casuscelli di Tocco, F.; Musolino, C.; Caminiti, G.; Chines, V.; Franzè, M.S.; Alibrandi, A.; Navarra, G.; et al. Frequency of somatic mutations in TERT promoter, TP53 and CTNNB1 genes in patients with hepatocellular carcinoma from Southern Italy. Oncol. Lett. 2020, 19, 2368–2374. [Google Scholar] [CrossRef]
- Schulze, K.; Nault, J.-C.; Villanueva, A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. J. Hepatol. 2016, 65, 1031–1042. [Google Scholar] [CrossRef] [Green Version]
- Mancarella, S.; Krol, S.; Crovace, A.; Leporatti, S.; Dituri, F.; Frusciante, M.; Giannelli, G. Validation of hepatocellular carcinoma experimental models for TGF-β promoting tumor progression. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabregat, I.; Caballero-Díaz, D. Transforming growth factor-β-induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Hin Tang, J.J.; Hao Thng, D.K.; Lim, J.J.; Toh, T.B. JAK/STAT signaling in hepatocellular carcinoma. Hepatic Oncol. 2020, 7. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Lim, H.Y.; Shi, S.; Lee, J.; Deng, S.; Xie, T.; Zhu, Z.; Wang, Y.; Pocalyko, D.; Yang, W.J.; et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology 2013, 58, 706–717. [Google Scholar] [CrossRef]
- Zhang, P.; Torres, K.; Liu, X.; Liu, C.; Pollock, R.E. An overview of chromatin-regulating proteins in cells. Curr. Protein Pept. Sci. 2016, 17, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Song, X.; Cao, D.; Cui, K.; Wang, J.; Utpatel, K.; Shang, R.; Wang, H.; Che, L.; Evert, M.; et al. Oncogene-dependent function of BRG1 in hepatocarcinogenesis. Cell Death Dis. 2020, 11, 91. [Google Scholar] [CrossRef]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef]
- Milosevic, I.; Vujovic, A.; Barac, A.; Djelic, M.; Korac, M.; Radovanovic Spurnic, A.; Gmizic, I.; Stevanovic, O.; Djordjevic, V.; Lekic, N.; et al. Gut-liver axis, gut microbiota, and its modulation in the management of liver diseases: A review of the literature. Int. J. Mol. Sci. 2019, 20, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Li, F.; Zhuang, Y.; Xu, J.; Wang, J.; Mao, X.; Zhang, Y.; Liu, X. Alteration in gut microbiota associated with hepatitis B and non-hepatitis virus related hepatocellular carcinoma. Gut Pathog. 2019, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Ohira, H.; Tsutsui, W.; Fujioka, Y. Are short chain fatty acids in gut microbiota defensive players for inflammation and atherosclerosis? J. Atheroscler. Thromb. 2017, 24, 660–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapidot, Y.; Amir, A.; Nosenko, R.; Uzan-Yulzari, A.; Veitsman, E.; Cohen-Ezra, O.; Davidov, Y.; Weiss, P.; Bradichevski, T.; Segev, S.; et al. Alterations in the gut microbiome in the progression of cirrhosis to hepatocellular carcinoma. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Yiu, J.H.C.; Dorweiler, B.; Woo, C.W. Interaction between gut microbiota and toll-like receptor: From immunity to metabolism. J. Mol. Med. 2017, 95, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Petrick, J.L.; Thistle, J.E.; Pinto, L.A.; Kemp, T.J.; Tran, H.Q.; Gewirtz, A.T.; Waterboer, T.; Fedirko, V.; Jenab, M.; et al. Bacterial translocation and risk of liver cancer in a finnish cohort. Cancer Epidemiol. Biomark. Prev. 2019, 28, 807–813. [Google Scholar] [CrossRef]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut microbiota promotes obesity-associated liver cancer through PGE2-mediated suppression of antitumor immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Takashina, Y.; Watanabe, M.; Nagamine, R.; Saito, Y.; Kamada, N.; Saito, H. Bile acid metabolism regulated by the gut microbiota promotes non-alcoholic steatohepatitis-associated hepatocellular carcinoma in mice. Oncotarget 2018, 9, 9925–9939. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Sung, C.Y.J.; Lee, N.; Ni, Y.; Pihlajamäki, J.; Panagiotou, G.; El-Nezami, H. Probiotics modulated gut microbiota suppresses hepatocellular carcinoma growth in mice. Proc. Natl. Acad. Sci. USA 2016, 113, E1306–E1315. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.K.; Kang, J.Y.; Shin, H.S.; Park, I.H.; Ha, N.J. Antiviral activity of Bifidobacterium adolescentis SPM0212 against hepatitis B virus. Arch. Pharm. Res. 2013, 36, 1525–1532. [Google Scholar] [CrossRef]
- El-Adawi, H.; Nour, I.; Fattouh, F.; El-Deeb, N. Investigation of the antiviral bioactivity of Lactobacillus bulgaricus 761N extracellular extract against hepatitis C virus (HCV). Int. J. Pharmacol. 2015, 11, 19–26. [Google Scholar] [CrossRef]
- Rupasinghe, H.P.V.; Parmar, I.; Neir, S.V. Biotransformation of cranberry proanthocyanidins to probiotic metabolites by Lactobacillus rhamnosus enhances their anticancer activity in HepG2 cells in vitro. Oxid. Med. Cell. Longev. 2019, 2019, 4750795. [Google Scholar] [CrossRef] [Green Version]
- Elshaer, A.M.; El-kharashi, O.A.; Hamam, G.G.; Nabih, E.S.; Magdy, Y.M.; Abd El Samad, A.A. Involvement of TLR4/ CXCL9/ PREX-2 pathway in the development of hepatocellular carcinoma (HCC) and the promising role of early administration of Lactobacillus plantarum in Wistar rats. Tissue Cell 2019, 60, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Mihailović, M.; Živković, M.; Jovanović, J.A.; Tolinački, M.; Sinadinović, M.; Rajić, J.; Uskoković, A.; Dinić, S.; Grdović, N.; Golić, N.; et al. Oral administration of probiotic Lactobacillus paraplantarum BGCG11 attenuates diabetes-induced liver and kidney damage in rats. J. Funct. Foods 2017, 38, 427–437. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Q.; Hesketh, J.; Huang, D.; Gan, F.; Hao, S.; Tang, S.; Guo, Y.; Huang, K. Protective effects of selenium-glutathione-enriched probiotics on CCl4-induced liver fibrosis. J. Nutr. Biochem. 2018, 58, 138–149. [Google Scholar] [CrossRef]
- Xue, L.; He, J.; Gao, N.; Lu, X.; Li, M.; Wu, X.; Liu, Z.; Jin, Y.; Liu, J.; Xu, J.; et al. Probiotics may delay the progression of nonalcoholic fatty liver disease by restoring the gut microbiota structure and improving intestinal endotoxemia. Sci. Rep. 2017, 7, 45176. [Google Scholar] [CrossRef] [PubMed]
- Heydari, Z.; Rahaie, M.; Alizadeh, A.M. Different anti-inflammatory effects of Lactobacillus acidophilus and Bifidobactrum bifidioum in hepatocellular carcinoma cancer mouse through impact on microRNAs and their target genes. J. Nutr. Intermed. Metab. 2019, 16, 100096. [Google Scholar] [CrossRef]
- Nduti, N.; McMillan, A.; Seney, S.; Sumarah, M.; Njeru, P.; Mwaniki, M.; Reid, G. Investigating probiotic yoghurt to reduce an aflatoxin B1 biomarker among school children in Eastern Kenya: Preliminary study. Int. Dairy J. 2016, 63, 124–129. [Google Scholar] [CrossRef]
- Oo, K.M.; Lwin, A.A.; Kyam, Y.Y.; Tun, W.M.; Fukada, K.; Goshima, A.; Shimada, T.; Okada, S. Safety and long-term effect of the probiotic FK-23 in patients with hepatitis C virus infection. Biosci. Microbiota Food Health 2016, 35, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Nanis, G.A.; Mohamed, L.S.; Hassan, E.; Maii, M.N. Lactobacillus acidophilus and Bifidobacteria Spp having antibacterial and antiviral effects on chronic HCV infection. Afr. J. Microbiol. Res. 2019, 13, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Nabavi, S.; Rafraf, M.; Somi, M.H.; Homayouni-Rad, A.; Asghari-Jafarabadi, M. Effects of probiotic yogurt consumption on metabolic factors in individuals with nonalcoholic fatty liver disease. J. Dairy Sci. 2014, 97, 7386–7393. [Google Scholar] [CrossRef]
- Ahn, S.B.; Jun, D.W.; Kang, B.-K.; Lim, J.H.; Lim, S.; Chung, M.-J. Randomized, double-blind, placebo-controlled study of a multispecies probiotic mixture in nonalcoholic fatty liver disease. Sci. Rep. 2019, 9, 5688. [Google Scholar] [CrossRef] [Green Version]
- Duseja, A.; Acharya, S.K.; Mehta, M.; Chhabra, S.; Shalimar; Rana, S.; Das, A.; Dattagupta, S.; Dhiman, R.K.; Chawla, Y.K.; et al. High potency multistrain probiotic improves liver histology in non-alcoholic fatty liver disease (NAFLD): A randomised, double-blind, proof of concept study. BMJ Open Gastroenterol. 2019, 6, e000315. [Google Scholar] [CrossRef] [Green Version]
- Mofidi, F.; Poustchi, H.; Yari, Z.; Nourinayyer, B.; Merat, S.; Sharafkhah, M.; Malekzadeh, R.; Hekmatdoost, A. Synbiotic supplementation in lean patients with non-alcoholic fatty liver disease: A pilot, randomised, double-blind, placebo-controlled, clinical trial. Br. J. Nutr. 2017, 117, 662–668. [Google Scholar] [CrossRef] [Green Version]
- Miro-Blanch, J.; Yanes, O. Epigenetic regulation at the interplay between gut microbiota and host metabolism. Front. Genet. 2019, 10. [Google Scholar] [CrossRef]
- Thilakarathna, W.P.D.W.; Rupasinghe, H.P.V. Microbial metabolites of proanthocyanidins reduce chemical carcinogen-induced DNA damage in human lung epithelial and fetal hepatic cells in vitro. Food Chem. Toxicol. 2019, 125, 479–493. [Google Scholar] [CrossRef]
- Thilakarathna, W.P.D.W.; Langille, M.G.I.; Rupasinghe, H.P.V. Polyphenol-based prebiotics and synbiotics: Potential for cancer chemoprevention. Curr. Opin. Food Sci. 2018, 20, 51–57. [Google Scholar] [CrossRef]




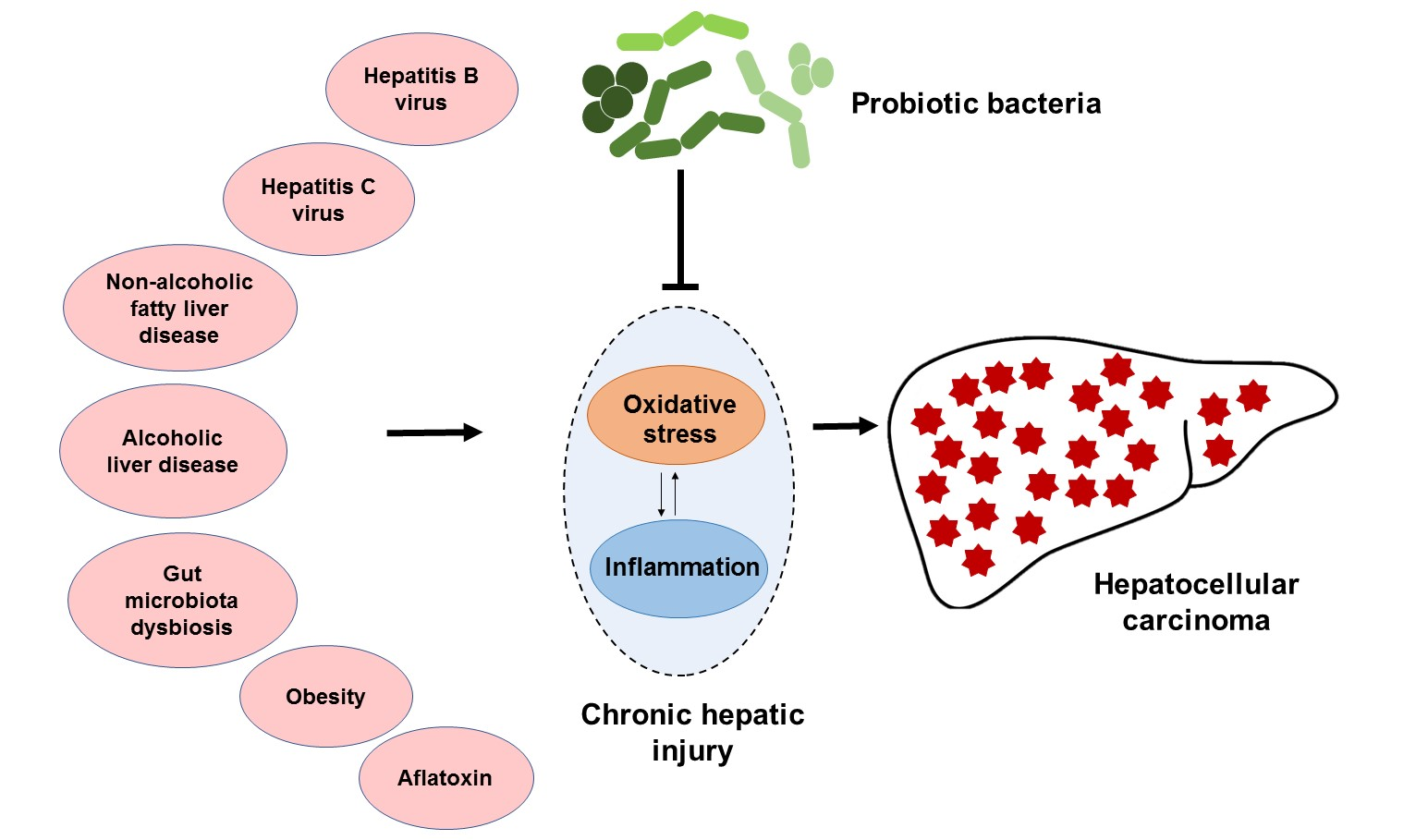{kind=link}
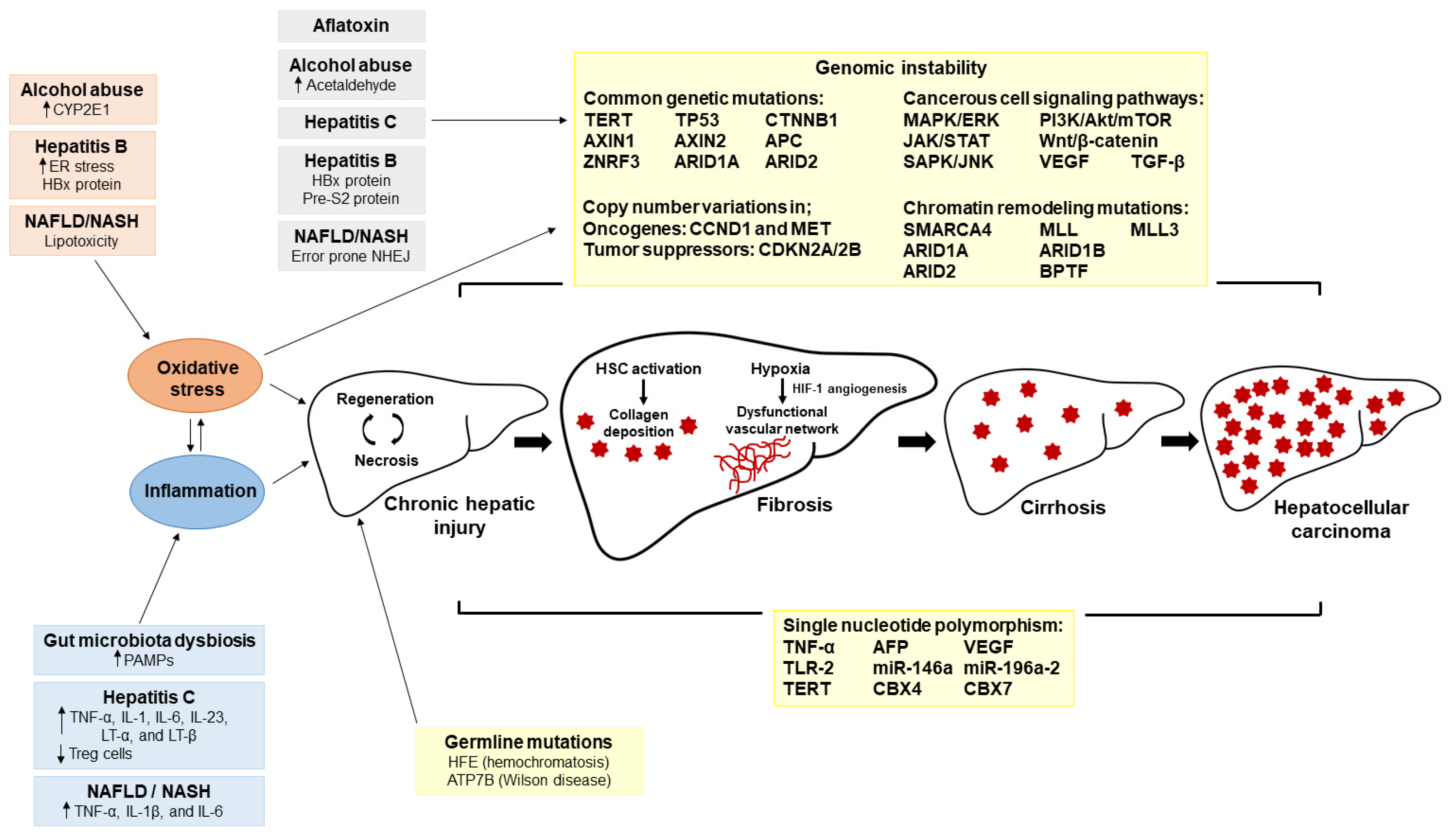{kind=link}
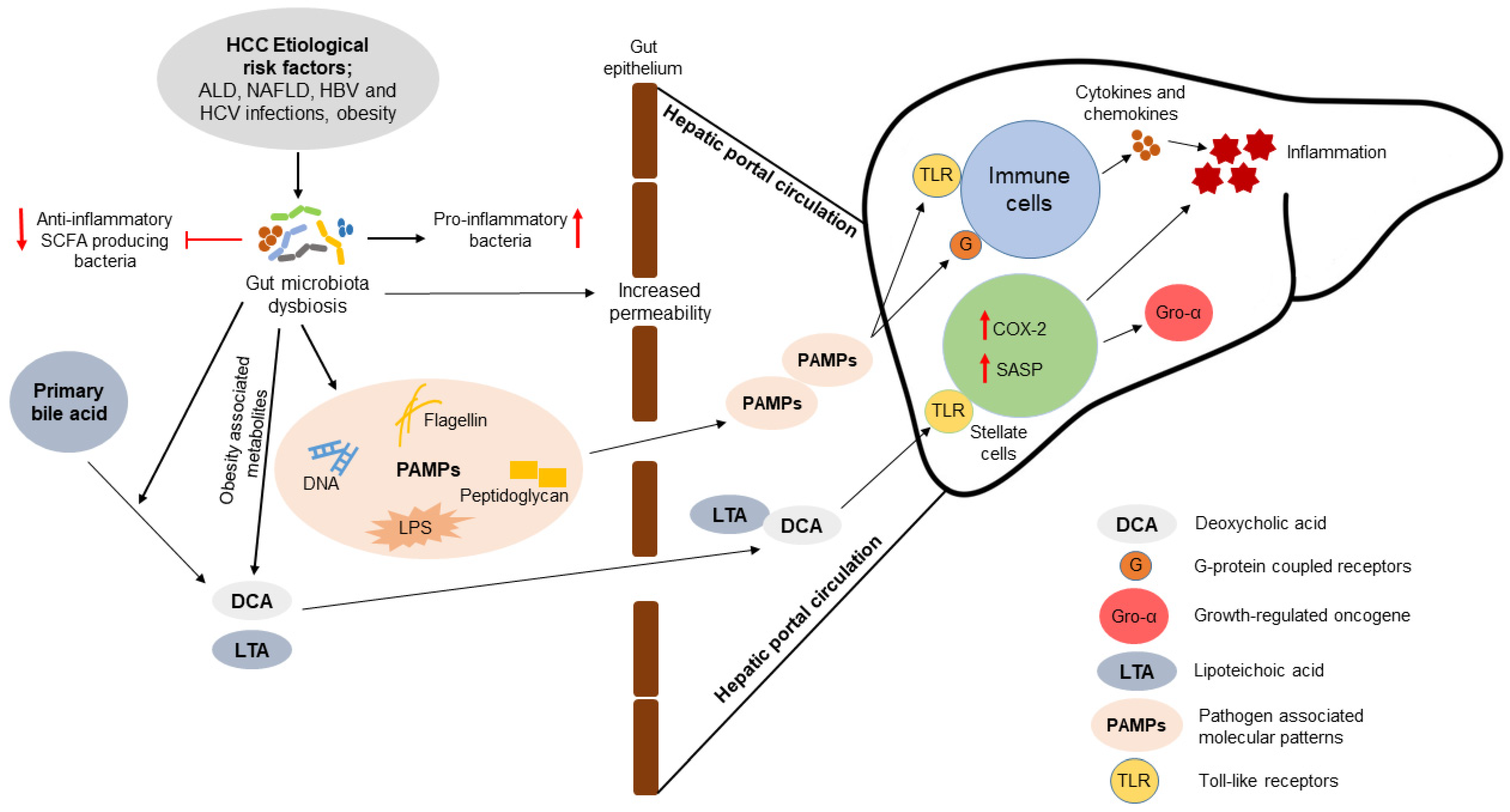{kind=link}
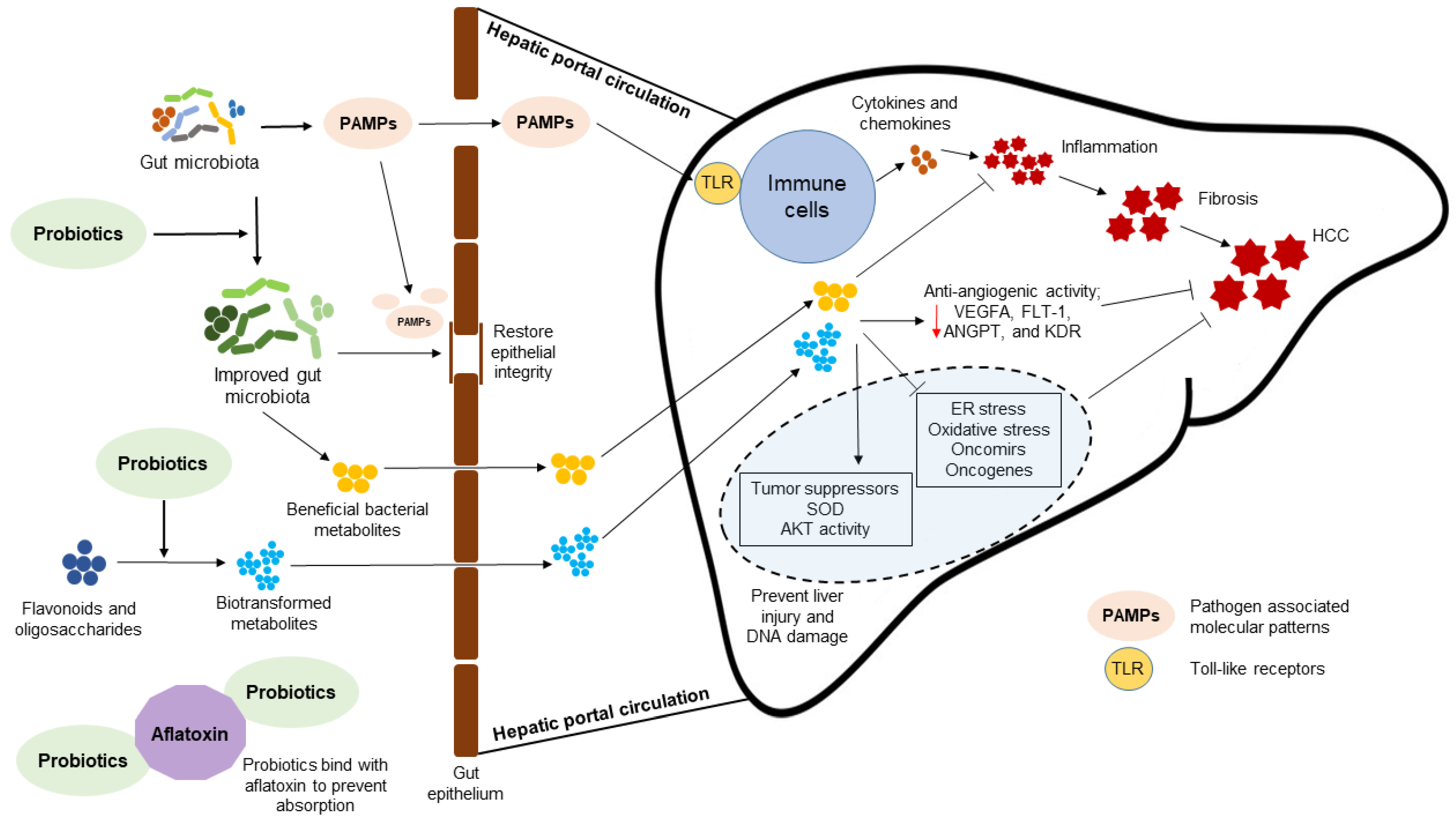{kind=link}
| Probiotic Bacteria | Experimental Model | Findings | Reference |
|---|---|---|---|
| In vitro studies | |||
| (1) B. adolescentis SPM0212 | HepG2.2.15 cells were incubated with the probiotic cell extract and HBV for 24 h. | The probiotic cell extract inhibits the replication of HBV virus by activation of MxA protein through upregulation of STAT1. | [90] |
| (2) L. bulgaricus 761N | HepG2 cells were incubated with a extracellular extract of probiotic bacteria and HCV for 96 h. | Treatment with culture media extract of probiotic bacteria significantly reduced the HCV viral load and HepG2 cell death. | [91] |
| (3) Cranberry proanthocyanidin extract biotransformed by L. rhamnosus | HepG2 cells were incubated with biotransformed proanthocyanidins (10–500 µg/mL) up to 48 h. | Biotransformed proanthocyanidins inhibit the proliferation of HepG2 cells by depleting mitochondria. The effective concentration of biotransformed proanthocyanidins is significantly low compared to the non-biotransformed material. | [92] |
| In vivo studies | |||
| (4) Prohep, a novel probiotic mixture of L. rhamnosus, E. coli Nissle 1917, and heat inactivated VSL#3 (1:1:1) | Male C57BL6/N mice (5–6 weeks) were fed with the probiotic mixture ad libitum and subcutaneously injected with murine hepatoma cells Hepa1-6. | Supplementation with the probiotic mixture modulated GM to suppress the tumor growth by downregulating inflammatory cytokine IL-17 and upregulating the expression of anti-inflammatory cytokines. Probiotic supplementation also downregulated the expression of angiogenic growth factors and receptors. | [89] |
| (5) L. plantarum EMCC-1039 | Wistar rats were supplemented with 1.2 × 109 cfu/mL of probiotic bacteria daily and liver cirrhosis was induced by administration of thioacetamide (200 mg/kg b.w. intraperitoneal) three times a week. | Probiotic supplementation attenuated thioacetamide-induced cirrhosis in rat livers by suppressing the expression of TLR4, CXCL9, and PREX-2. | [93] |
| (6) L. paraplantarum BGCG11 | Albino Wistar rats were intraperitoneally injected with streptozotocin (40 mg/kg b.w./day) for 5 days and supplemented with probiotic bacteria (1 × 108 cfu/day) for 4 weeks. | Probiotic supplementation reduced hepatic DNA damage by restoring the SOD activity in diabetic rats. Probiotics also reduced hepatic inflammation and liver fibrosis by restoring Akt signaling and preventing the degradation of pro-caspase 3. | [94] |
| (7) A novel probiotic mixture of Saccharomyces cerevisiae and L. acidophilus enriched with selenium and glutathione | Liver fibrosis in male Wistar rats was induced by intraperitoneal injection of CCl4 (2 mL/kg) twice a week for seven weeks. Rats were then supplemented with probiotic mixture (1 g/kg b.w./day) for seven weeks. (Daily intake of, selenium, 38.4 µg/kg b.w.; glutathione 34.1 mg/kg b.w.; S, cerevisiae, 1 × 1010 cfu; L. acidophilus, 1 × 1010 cfu) | Probiotic bacteria together with selenium and glutathione synergistically reduced liver damage and fibrosis. The probiotic mixture inhibited CCl4-induced oxidative stress, ER stress and inflammation by the activation of SIRT1. | [95] |
| (8) A probiotic mixture of B. nfantis, L. acidophilus, and Bacillus cereus | Male SPF SD rats on a HSHF diet were supplemented with the probiotic mixture for 12 weeks. (0.5 × 106 cfu/day of B. infantis and L. acidophilus, and 0.5 × 105 cfu/day Bacillus cereus) | Supplementation with probiotic bacteria ameliorated the loss of GM richness, colonization resistance and gut barrier function in rats fed with HSHF diet. This in turn reduced serum LPS levels and activation of TLR4-mediated immune response. | [96] |
| (9) L. acidophilus and B. bifidum | Male Balb/c mice (6 weeks) were supplemented with the two probiotic bacteria separately (1 × 109 cfu/day) for five months. Ten days into the probiotic supplementation, mice were subcutaneously injected with the carcinogen azoxymethane (15 mg/kg b.w.) weekly for three weeks to induce colon cancer. | Probiotic supplementation downregulated the expression of oncomirs (miR-155 and miR-221) and oncogenes (Bcl-w and KRAS) in liver tissue. Moreover, probiotic supplementation upregulated the expression of tumor suppressor miR-122 and gene PU.1. | [97] |
| Clinical studies | |||
| (10) A yoghurt with S. thermophilus, L. rhamnosus GR-1, and Weissella cibaria NN20 | Children of 6–10 years old were provided 200 mL of the probiotic yogurt daily for 14 days. | Supplementation with the yogurt containing probiotic bacteria significantly reduced the urine availability of aflatoxin metabolites. | [98] |
| (11) Heat-treated Enterococcus faecalis FK-23 | Long term supplementation (2700 mg/day up to 36 months) of HCV positive subjects with the heat-treated probiotic bacteria. | Heat-treated probiotic bacteria significantly reduced the serum levels of ALT and AST. | [99] |
| (12) A mixture of L. acidophilus and Bifidobacteria spp. probiotic bacteria. | Chronic HCV patients were fed the probiotic mixture (1 × 109 cfu/day) daily for one month and subjected to pegylated IFN-α and ribavirin treatment weekly for 12 weeks. | Administration of probiotic bacteria increased the response rate to pegylated IFN-α and ribavirin treatment by 25%. | [100] |
| (13) A yogurt with L. acidophilus La5 and B. lactis Bb12 | Adult NAFLD patients (23–63 years old) were fed 300 g of the probiotic yogurt for 8 weeks. (4.42 × 106 cfu/g yogurt of L. acidophilus La5 and 3.85 × 106 cfu/g yogurt of B. lactis Bb12) | Supplementation with probiotics ameliorated the NAFLD risk factors. Serum levels of ALT, AST, and total cholesterol is significantly reduced in the NAFLD patients supplemented with probiotic bacteria. | [101] |
| (14) A Probiotic mixture of L. acidophilus CBT LA1, L. rhamnosus CBT LR5, L. paracasei CBT LPC5, Pediococcus. pentosaceus CBT SL4, B. lactis CBT BL3, and B. breve CBT BR3 | Obese NAFLD patients were supplemented with the probiotic mixture (1 × 109 cfu/day) for 12 weeks. | Supplementation with the probiotic mixture significantly reduced the body weight, total body fat, total cholesterol and intra hepatic fat fraction of obese NAFLD patients. Probiotics administration also reduced the TNF-α expression in the NAFLD patients. | [102] |
| (15) A multi-strain probiotic mixture of L. paracasei DSM 24733, L. plantarum DSM 24730, L. acidophilus DSM 24735 and L. delbrueckii subsp. bulgaricus DSM 24734, B. longum DSM 24736, B. infantis DSM 24737, B. breve DSM 24732, and S. thermophilus DSM 24731 | NAFLD patients complying with exercise and dietary recommendations were fed a multi-strain probiotic mixture (675 × 109 cfu/day) for 12 months. | Probiotic supplementation improved the liver histology in NAFLD patients by reducing hepatocyte ballooning and hepatic fibrosis. Probiotic bacteria also reduced hepatic lobular inflammation and levels of ALT, adipocytokines, leptin and endotoxins. | [103] |
| (16) A synbiotic of L. casei, L. rhamnosus, S. thermophilus, B. breve, L. acidophilus, B. longum, and L. bulgaricus together with FOS | Non-obese NAFLD patients were supplemented with the synbiotic (0.4 × 109 cfu of probiotics/day and 250 mg of FOS/day) daily for 28 weeks. | Supplementation with the synbiotic significantly reduced haptic steatosis and fibrosis in the non-obese NAFLD patients. Synbiotic administration also reduced the levels of liver damage marker and inflammatory mediators. | [104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thilakarathna, W.P.D.W.; Rupasinghe, H.P.V.; Ridgway, N.D. Mechanisms by Which Probiotic Bacteria Attenuate the Risk of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 2606. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052606
Thilakarathna WPDW, Rupasinghe HPV, Ridgway ND. Mechanisms by Which Probiotic Bacteria Attenuate the Risk of Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2021; 22(5):2606. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052606
Chicago/Turabian StyleThilakarathna, Wasitha P.D. Wass, H.P. Vasantha Rupasinghe, and Neale D. Ridgway. 2021. "Mechanisms by Which Probiotic Bacteria Attenuate the Risk of Hepatocellular Carcinoma" International Journal of Molecular Sciences 22, no. 5: 2606. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22052606