
Pertussis Toxin Inhibits Encephalitogenic T-Cell Infiltration and Promotes a B-Cell-Driven Disease during Th17-EAE



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Pertussis Toxin Reduced Th17-EAE Disease Severity
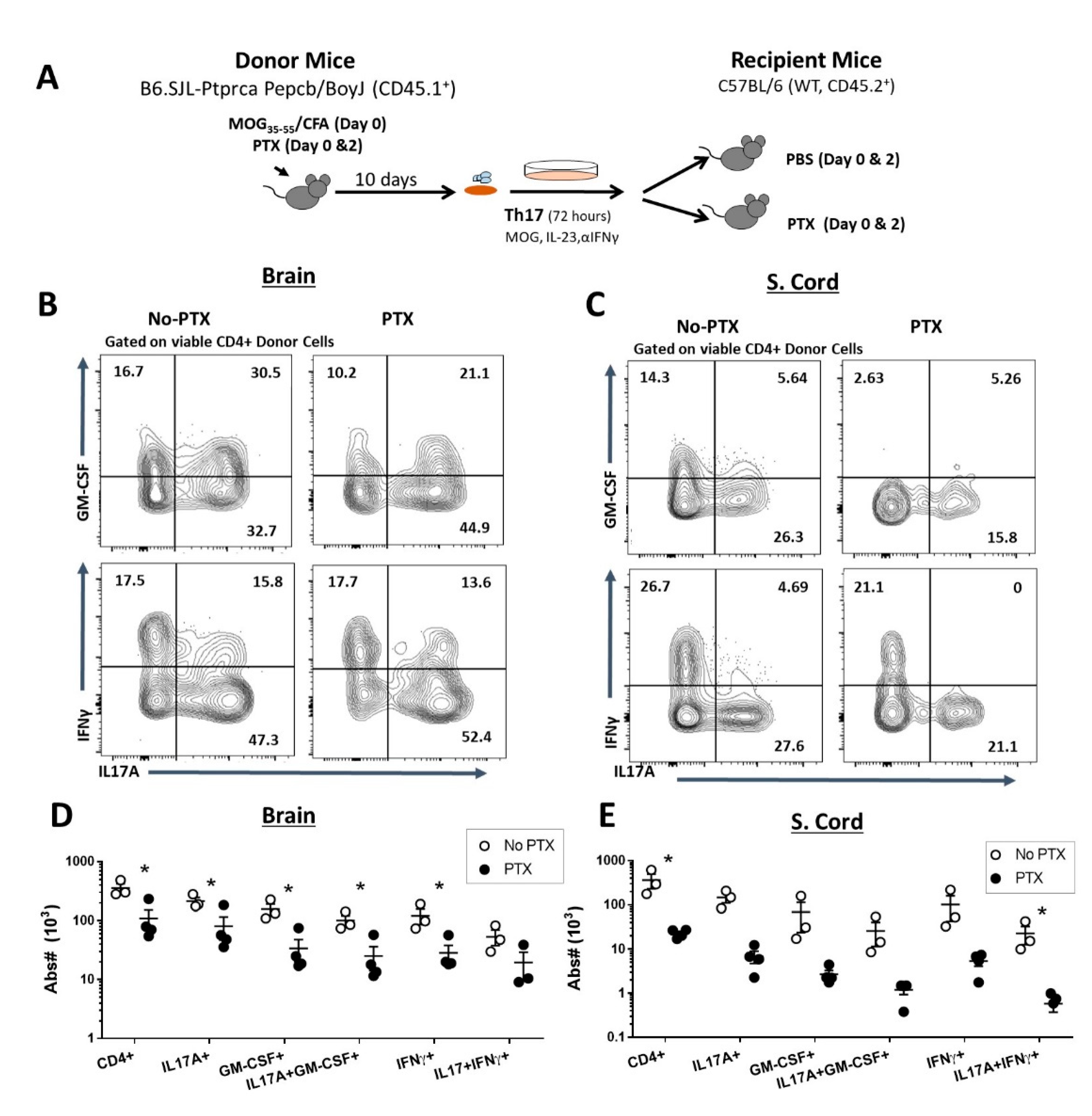
2.2. PTX Reduced Accumulation of Encephalitogenic CD4+ T Cells in the CNS
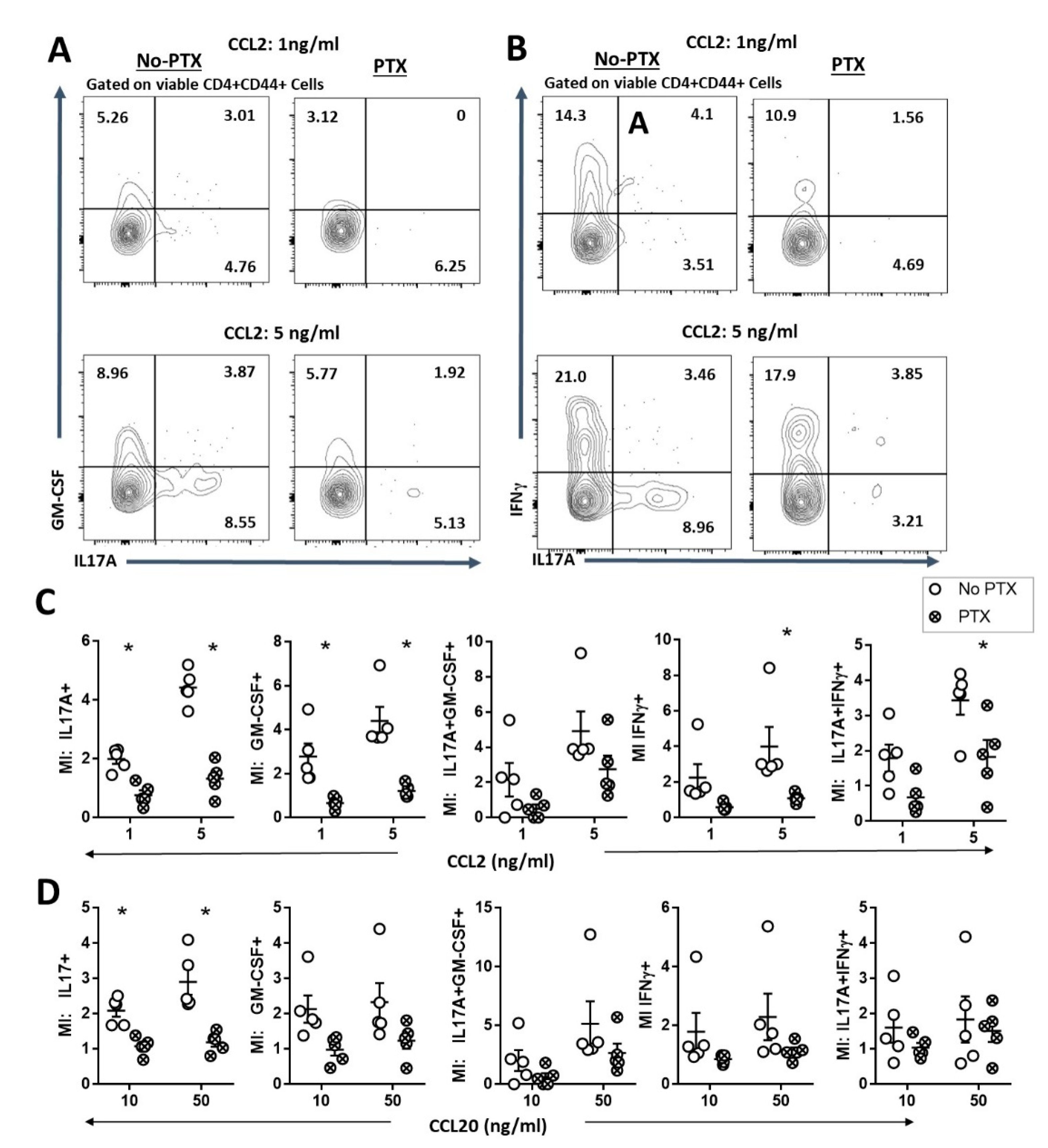
2.3. PTX Impaired Chemokine-Dependent Recruitment of Inflammatory Th17 Cells
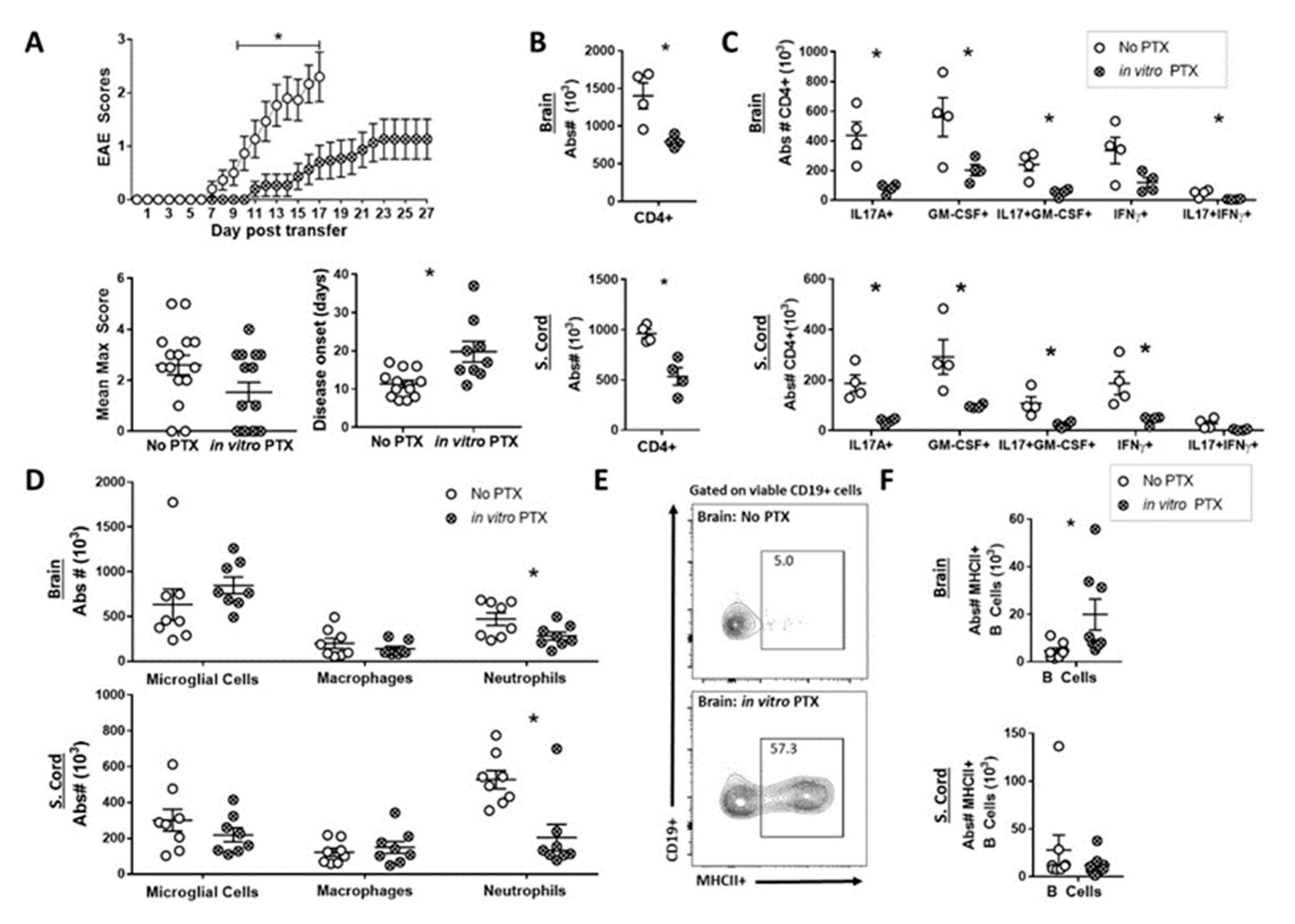
2.4. In Vitro PTX treatment Reduced the Capacity of Th17 Cells to Induce EAE
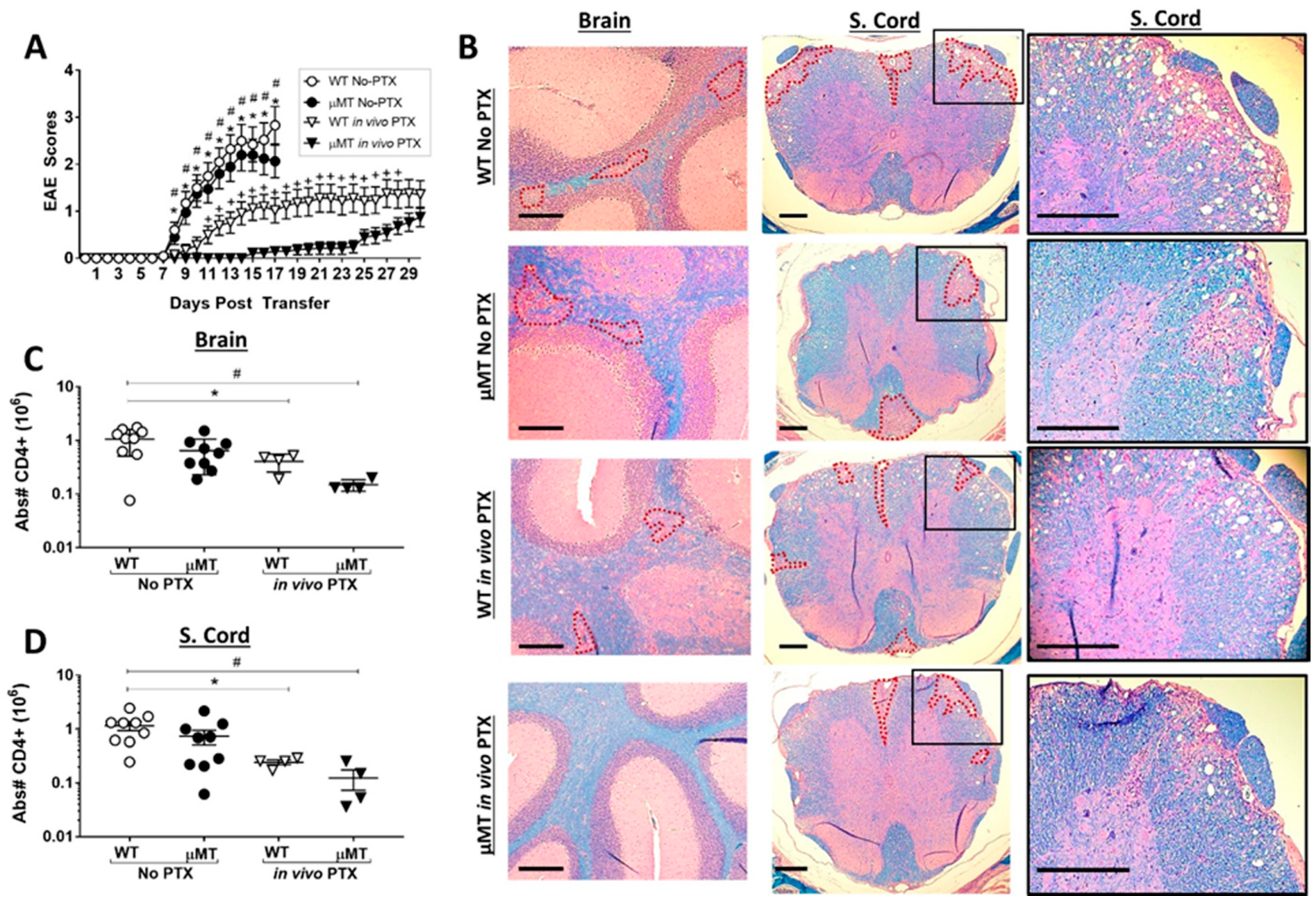
2.5. PTX Treatment Reveals a Pro-Inflammatory Function of B Cells in the Th17-EAE Model
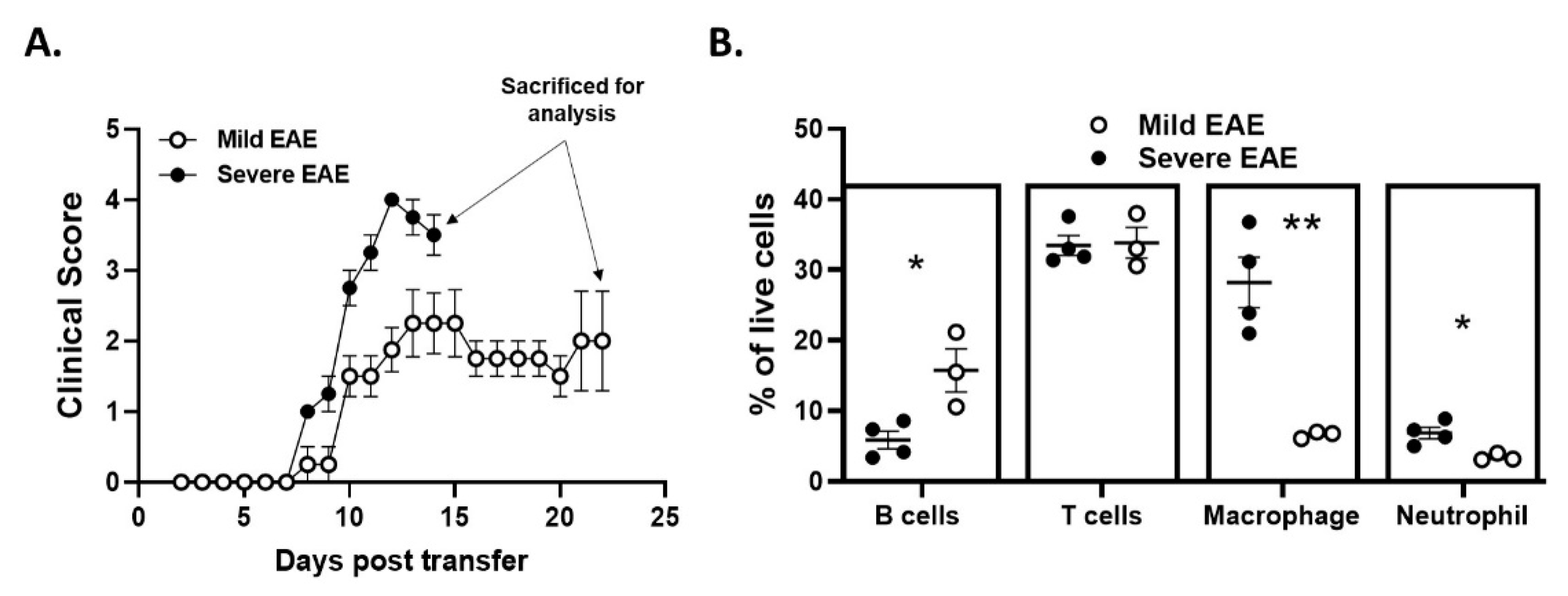
2.6. Disease Duration and/or Severity Affects B-Cell Accumulation in the CNS
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. EAE Induction
4.3. Isolation of CNS-Infiltrating Cells
4.4. Flow Cytometry
4.5. Histology
4.6. Chemotaxis Assay
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bar-Or, A.; Calabresi, P.A.J.; Arnlod, D.; Markowitz, C.; Shafer, S.; Kasper, L.H.; Waubant, E.; Gazda, S.; Fox, R.J.; Panzara, M.; et al. Rituximab in relapsing-remitting multiple sclerosis: A 72-week, open-label, phase I trial. Ann. Neurol. 2008, 63, 395–400. [Google Scholar] [CrossRef]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-cell depletion with Rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [Green Version]
- Kappos, L.; Li, D.; Calabresi, P.A.; O’Connor, P.; Bar-Or, A.; Barkhof, F.; Yin, M.; Leppert, D.; Glanzman, R.; Tinbergen, J.; et al. Ocrelizumab in relapsing-remitting multiple sclerosis: A phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011, 378, 1779–1787. [Google Scholar] [CrossRef]
- Blauth, K.; Soltys, J.; Matschulat, A.; Reiter, C.R.; Ritchie, A.; Baird, N.L.; Bennett, J.L.; Owens, G.P. Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid cause demyelination of spinal cord explants. Acta Neuropathol. 2015, 130, 765–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harp, C.R.P.; Archambault, A.S.; Sim, J.; Ferris, S.T.; Mikesell, R.J.; Koni, P.A.; Shimoda, M.; Linington, C.; Russell, J.H.; Wu, G.F. B Cell Antigen Presentation Is Sufficient To Drive Neuroinflammation in an Animal Model of Multiple Sclerosis. J. Immunol. 2015, 194, 5077–5084. [Google Scholar] [CrossRef] [Green Version]
- Molnarfi, N.; Schulze-Topphoff, U.; Weber, M.S.; Patarroyo, J.C.; Prod’homme, T.; Varrin-Doyer, M.; Shetty, A.; Linington, C.; Slavin, A.J.; Hidalgo, J.; et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J. Exp. Med. 2013, 210, 2921–2937. [Google Scholar] [CrossRef] [PubMed]
- Barr, T.A.; Shen, P.; Brown, S.; Lampropoulou, V.; Roch, T.; Lawrie, S.; Fan, B.; O’Connor, R.A.; Anderton, S.M.; Bar-Or, A.; et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. J. Exp. Med. 2012, 209, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Hartung, H.P.; Freedman, M.S.; Boyko, A.; Mikol, D.; Freudensprung, U.; Plitz, T.; Grp, A.S. ATAMS: A randomised trial of the B-cell-targeting agent atacicept in patients with relapsing multiple sclerosis. Mult. Scler. J. 2011, 17, S40. [Google Scholar]
- Matsushita, T.; Yanaba, K.; Bouaziz, J.D.; Fujimoto, M.; Tedder, T.F. Regulatory B cells inhibit EAE initiation in mice while other B cells promote disease progression. J. Clin. Investig. 2008, 118, 3420–3430. [Google Scholar] [CrossRef] [Green Version]
- Pierson, E.R.; Stromnes, I.M.; Goverman, J.M. B Cells Promote Induction of Experimental Autoimmune Encephalomyelitis by Facilitating Reactivation of T Cells in the Central Nervous System. J. Immunol. 2014, 192, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.L.; Kumar, G.; Agasing, A.; Ko, R.M.; Axtell, R.C. Role of TFH Cells in Promoting T Helper 17-Induced Neuroinflammation. Front. Immunol. 2018, 9, 382. [Google Scholar] [CrossRef] [Green Version]
- Agasing, A.M.; Wu, Q.; Khatri, B.; Borisow, N.; Ruprecht, K.; Brandt, A.U.; Gawde, S.; Kumar, G.; Quinn, J.L.; Ko, R.M.; et al. Transcriptomics and proteomics reveal a cooperation between interferon and T-helper 17 cells in neuromyelitis optica. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Mitsdoerffer, M.; Lee, Y.; Jager, A.; Kim, H.J.; Korn, T.; Kolls, J.K.; Cantor, H.; Bettelli, E.; Kuchroo, V.K. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc. Natl. Acad. Sci. USA 2010, 107, 14292–14297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archambault, A.S.; Carrero, J.A.; Barnett, L.G.; McGee, N.G.; Sim, J.; Wright, J.O.; Raabe, T.; Chen, P.Q.; Ding, H.; Allenspach, E.J.; et al. Cutting Edge: Conditional MHC Class II Expression Reveals a Limited Role for B Cell Antigen Presentation in Primary and Secondary CD4 T Cell Responses. J. Immunol. 2013, 191, 545–550. [Google Scholar] [CrossRef] [Green Version]
- Agasing, A.M.; Gawde, S.; Kumar, G.; Turner, E.; Axtell, R.C. B cell function impacts the efficacy of IFN-beta therapy in EAE. J. Neuroimmunol. 2020, 338, 577106. [Google Scholar] [CrossRef] [PubMed]
- Alfano, M.; Schmidtmayerova, H.; Amella, C.A.; Pushkarsky, T.; Bukrinsky, M. The B-oligomer of pertussis toxin deactivates CC chemokine receptor 5 and blocks entry of M-tropic HIV-1 strains. J. Exp. Med. 1999, 190, 597–605. [Google Scholar] [CrossRef]
- Mangmool, S.; Kurose, H. G(i/o) Protein-Dependent and -Independent Actions of Pertussis Toxin (PTX). Toxins 2011, 3, 884–899. [Google Scholar] [CrossRef] [Green Version]
- Andreasen, C.; Carbonetti, N.H. Pertussis Toxin Inhibits Early Chemokine Production To Delay Neutrophil Recruitment in Response to Bordetella pertussis Respiratory Tract Infection in Mice. Infect. Immun. 2008, 76, 5139–5148. [Google Scholar] [CrossRef] [Green Version]
- Su, S.B.; Silver, P.B.; Zhang, M.F.; Chan, C.C.; Caspi, R.R. Pertussis toxin inhibits induction of tissue-specific autoimmune disease by disrupting G protein-coupled signals. J. Immunol. 2001, 167, 250–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cyster, J.G.; Goodnow, C.C. Pertussis Toxin Inhibits Migration of B-Lymphocyte and T-Lymphocyte into Splenic White Pulp Cords. J. Exp. Med. 1995, 182, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Schneider, O.D.; Weiss, A.A.; Miller, W.E. Pertussis Toxin Signals through the TCR to Initiate Cross-Desensitization of the Chemokine Receptor CXCR4. J. Immunol. 2009, 182, 5730–5739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, M.; Kang, J.; Lee, Y.; Riu, K.; Kim, Y.; Jee, Y.; Matsumoto, Y.; Shin, T. Pertussis toxin-induced hyperacute autoimmune encephalomyelitis in Lewis rats is correlated with increased expression of inducible nitric oxide synthase and tumor necrosis factor alpha. Neurosci. Lett. 2001, 308, 41–44. [Google Scholar] [CrossRef]
- Arimoto, H.; Tanuma, N.; Jee, Y.; Miyazawa, T.; Shima, K.; Matsumoto, Y. Analysis of experimental autoimmune encephalomyelitis induced in F344 rats by pertussis toxin administration. J. Neuroimmunol. 2000, 104, 15–21. [Google Scholar] [CrossRef]
- Kerfoot, S.M.; Long, E.M.; Hickey, M.J.; Andonegui, G.; Lapointe, B.M.; Zanardo, R.C.O.; Bonder, C.; James, W.G.; Robbins, S.M.; Kubes, P. TLR4 contributes to disease-inducing mechanisms resulting in central nervous system autoimmune disease. J. Immunol. 2004, 173, 7070–7077. [Google Scholar] [CrossRef] [PubMed]
- Kugler, S.; Bocker, K.; Heusipp, G.; Greune, L.; Kim, K.S.; Schmidt, M.A. Pertussis toxin transiently affects barrier integrity, organelle organization and transmigration of monocytes in a human brain microvascular endothelial cell barrier model. Cell Microbiol. 2007, 9, 619–632. [Google Scholar] [CrossRef]
- Ronchi, F.; Basso, C.; Preite, S.; Reboldi, A.; Baumjohann, D.; Perlini, L.; Lanzavecchia, A.; Sallusto, F. Experimental priming of encephalitogenic Th1/Th17 cells requires pertussis toxin-driven IL-1 beta production by myeloid cells. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Bradstreet, T.R.; Schwarzkopf, E.A.; Sim, J.; Carrero, J.A.; Chou, C.; Cook, L.E.; Egawa, T.; Taneja, R.; Murphy, T.L.; et al. Bhlhe40 controls cytokine production by Tcells and is essential for pathogenicity in autoimmune neuroinflammation. Nat. Commun. 2014, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumas, A.; Amiable, N.; Vaccari, J.P.D.; Chae, J.J.; Keane, R.W.; Lacroix, S.; Vallieres, L. The Inflammasome Pyrin Contributes to Pertussis Toxin-Induced IL-1 beta Synthesis, Neutrophil Intravascular Crawling and Autoimmune Encephalomyelitis. PLoS Pathog. 2014, 10, e1004150. [Google Scholar] [CrossRef] [Green Version]
- Robbinson, D.; Cockle, S.; Singh, B.; Strejan, G.H. Native, but not genetically inactivated, pertussis toxin protects mice against experimental allergic encephalomyelitis. Cell Immunol. 1996, 168, 165–173. [Google Scholar] [CrossRef]
- Jager, A.; Dardalhon, V.; Sobel, R.A.; Bettelli, E.; Kuchroo, V.K. Th1, Th17, and Th9 Effector Cells Induce Experimental Autoimmune Encephalomyelitis with Different Pathological Phenotypes. J. Immunol. 2009, 183, 7169–7177. [Google Scholar] [CrossRef]
- Codarri, L.; Gyulveszi, G.; Tosevski, V.; Hesske, L.; Fontana, A.; Magnenat, L.; Suter, T.; Becher, B. ROR gamma t drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 2011, 12, 560–567. [Google Scholar] [CrossRef] [PubMed]
- El-Behi, M.; Ciric, B.; Dai, H.; Yan, Y.P.; Cullimore, M.; Safavi, F.; Zhang, G.X.; Dittel, B.N.; Rostami, A. The encephalitogenicity of T(H)17 cells is dependent on IL-1-and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 2011, 12, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Kara, E.E.; McKenzie, D.R.; Bastow, C.R.; Gregor, C.E.; Fenix, K.A.; Ogunniyi, A.D.; Paton, J.C.; Mack, M.; Pombal, D.R.; Seillet, C.; et al. CCR2 defines in vivo development and homing of IL-23-driven GM-CSF-producing Th17 cells. Nat. Commun. 2015, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Wojkowska, D.W.; Szpakowski, P.; Ksiazek-Winiarek, D.; Leszczynski, M.; Glabinski, A. Interactions between Neutrophils, Th17 Cells, and Chemokines during the Initiation of Experimental Model of Multiple Sclerosis. Mediat. Inflamm. 2014, 2014, 590409. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.X.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef]
- Lu, C.M.; Diehl, S.A.; Noubade, R.; Ledoux, J.; Nelson, M.T.; Spach, K.; Zachary, J.F.; Blankenhorn, E.P.; Teuscher, C. Endothelial histamine H-1 receptor signaling reduces blood-brain barrier permeability and susceptibility to autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2010, 107, 18967–18972. [Google Scholar] [CrossRef] [Green Version]
- Racke, M.K.; Hu, W.; Lovett-Racke, A.E. PTX cruiser: Driving autoimmunity via TLR4. Trends Immunol. 2005, 26, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Q.; Hester, S.E.; Kennett, M.J.; Karanikas, A.T.; Bendor, L.; Place, D.E.; Harvill, E.T. Interleukin-1 Receptor Signaling Is Required To Overcome the Effects of Pertussis Toxin and for Efficient Infection- or Vaccination-Induced Immunity against Bordetella pertussis. Infect. Immun. 2011, 79, 527–541. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, A.J.; Regan-Komito, D.; Christou, I.; White, G.E.; McNeill, E.; Kenyon, A.; Taylor, L.; Kapellos, T.S.; Fisher, E.A.; Channon, K.M.; et al. A Real Time Chemotaxis Assay Unveils Unique Migratory Profiles amongst Different Primary Murine Macrophages. PLoS ONE 2013, 8, e58744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQualter, J.L.; Darwiche, R.; Ewing, C.; Onuki, M.; Kay, T.W.; Hamilton, J.A.; Reid, H.H.; Bernard, C.C.A. Granulocyte macrophage colony-stimulating factor: A new putative therapeutic target in multiple sclerosis. J. Exp. Med. 2001, 194, 873–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haak, S.; Croxford, A.L.; Kreymborg, K.; Heppner, F.L.; Pouly, S.; Becher, B.; Waisman, A. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J. Clin. Investig. 2009, 119, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Forde, E.A.; Dogan, R.N.E.; Karpus, W.J. CCR4 contributes to the pathogenesis of experimental autoimmune encephalomyelitis by regulating inflammatory macrophage function. J. Neuroimmunol. 2011, 236, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Rottman, J.B.; Slavin, A.J.; Silva, R.; Weiner, H.L.; Gerard, C.G.; Hancock, W.W. Leukocyte recruitment during onset of experimental allergic encephalomyelitis is CCR1 dependent. Eur. J. Immunol. 2000, 30, 2372–2377. [Google Scholar] [CrossRef]
- Vogel, D.Y.S.; Kooij, G.; Heijnen, P.D.A.M.; Breur, M.; Peferoen, L.A.N.; van der Valk, P.; de Vries, H.E.; Amor, S.; Dijkstra, C.D. GM-CSF promotes migration of human monocytes across the blood brain barrier. Eur. J. Immunol. 2015, 45, 1808–1819. [Google Scholar] [CrossRef] [PubMed]
- Simmons, S.B.; Liggitt, D.; Goverman, J.M. Cytokine-Regulated Neutrophil Recruitment Is Required for Brain but Not Spinal Cord Inflammation during Experimental Autoimmune Encephalomyelitis. J. Immunol. 2014, 193, 555–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reboldi, A.; Coisne, C.; Baumjohann, D.; Benvenuto, F.; Bottinelli, D.; Lira, S.; Uccelli, A.; Lanzavecchia, A.; Engelhardt, B.; Sallusto, F. C-C chemokine receptor 6-regulated entry of T-H-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat. Immunol. 2009, 10, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Pizza, M.; Bartoloni, A.; Prugnola, A.; Silvestri, S.; Rappuoli, R. Subunit-S1 of Pertussis Toxin—Mapping of the Regions Essential for Adp-Ribosyltransferase Activity. Proc. Natl. Acad. Sci. USA 1988, 85, 7521–7525. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.S.; Prod’homme, T.; Patarroyo, J.C.; Molnarfi, N.; Karnezis, T.; Lehmann-Horn, K.; Danilenko, D.M.; Eastham-Anderson, J.; Slavin, A.J.; Linington, C.; et al. B-Cell Activation Influences T-Cell Polarization and Outcome of Anti-CD20 B-Cell Depletion in Central Nervous System Autoimmunity. Ann. Neurol. 2010, 68, 369–383. [Google Scholar] [CrossRef]
- Bar-Or, A.; Fawaz, L.; Fan, B.L.; Darlington, P.J.; Rieger, A.; Ghorayeb, C.; Calabresi, P.A.; Waubant, E.; Hauser, S.L.; Zhang, J.M.; et al. Abnormal B-Cell Cytokine Responses A Trigger of T-Cell Mediated Disease in MS? Ann. Neurol. 2010, 67, 452–461. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maria, Z.; Turner, E.; Agasing, A.; Kumar, G.; Axtell, R.C. Pertussis Toxin Inhibits Encephalitogenic T-Cell Infiltration and Promotes a B-Cell-Driven Disease during Th17-EAE. Int. J. Mol. Sci. 2021, 22, 2924. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062924
Maria Z, Turner E, Agasing A, Kumar G, Axtell RC. Pertussis Toxin Inhibits Encephalitogenic T-Cell Infiltration and Promotes a B-Cell-Driven Disease during Th17-EAE. International Journal of Molecular Sciences. 2021; 22(6):2924. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062924
Chicago/Turabian StyleMaria, Zahra, Emma Turner, Agnieshka Agasing, Gaurav Kumar, and Robert C. Axtell. 2021. "Pertussis Toxin Inhibits Encephalitogenic T-Cell Infiltration and Promotes a B-Cell-Driven Disease during Th17-EAE" International Journal of Molecular Sciences 22, no. 6: 2924. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062924