
Dual Specificity Kinase DYRK3 Promotes Aggressiveness of Glioblastoma by Altering Mitochondrial Morphology and Function

Abstract
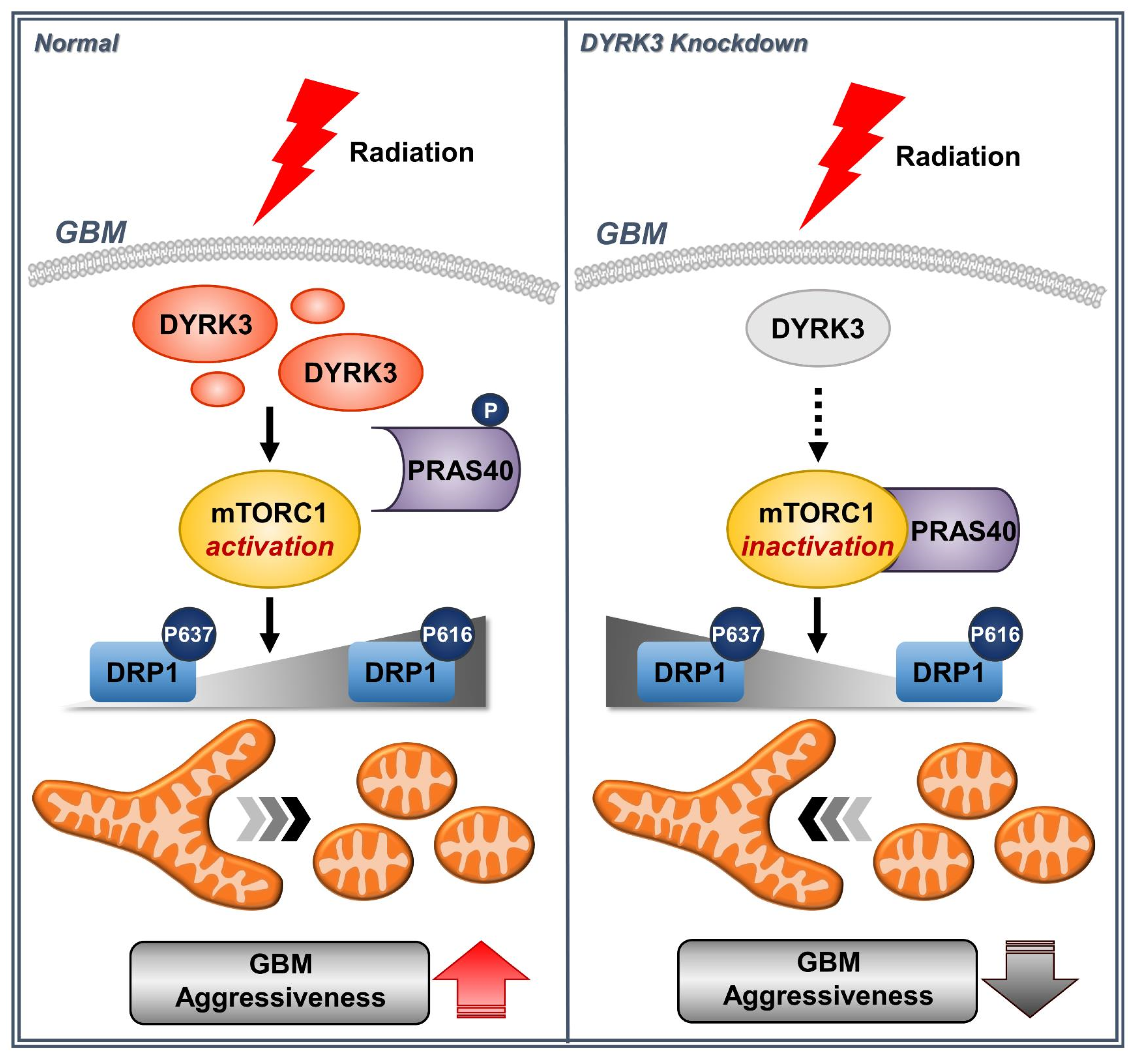
:1. Introduction
2. Results
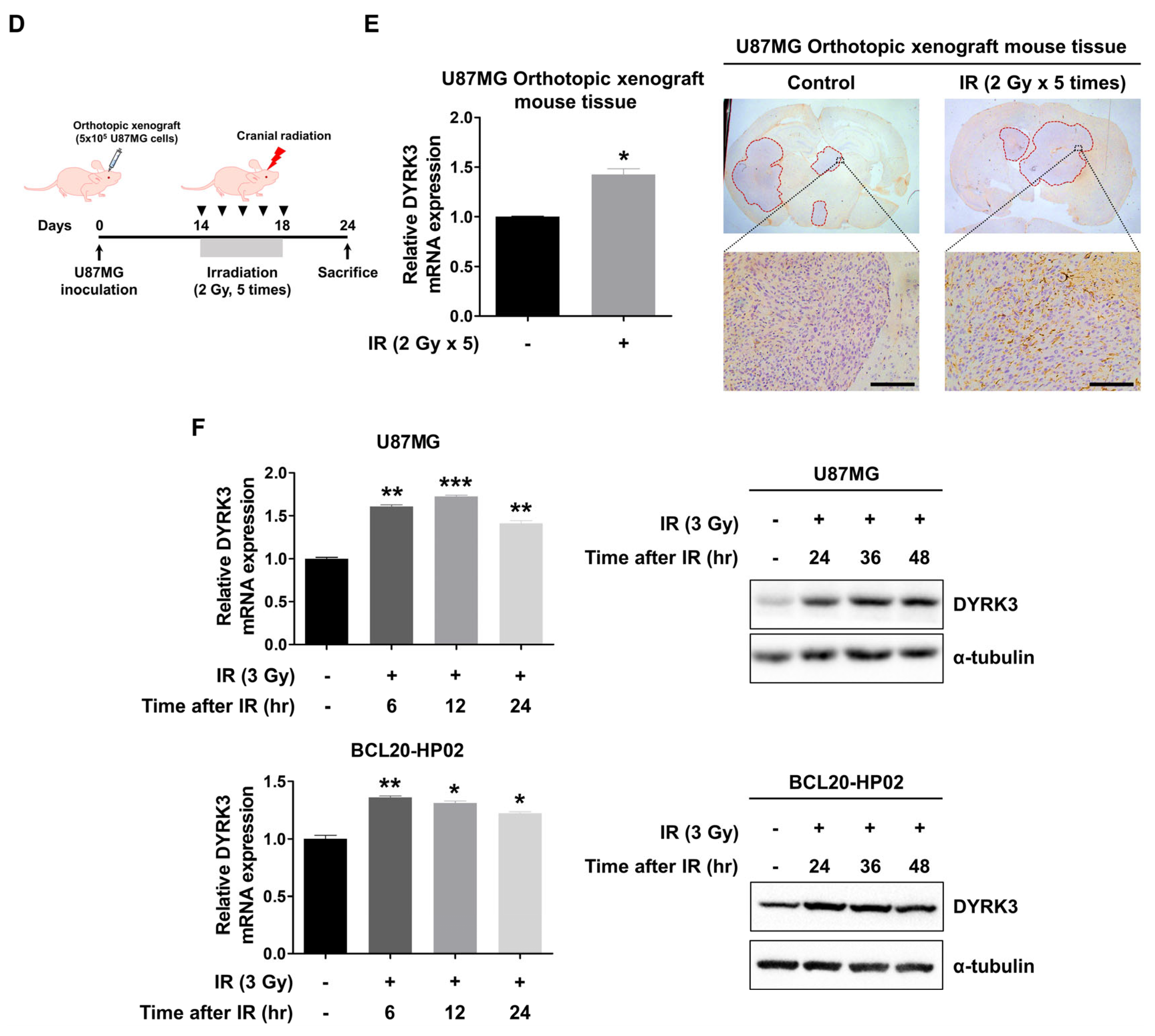
2.1. DYRK3 Is Upregulated by Radiation and Contributes to GBM Progression
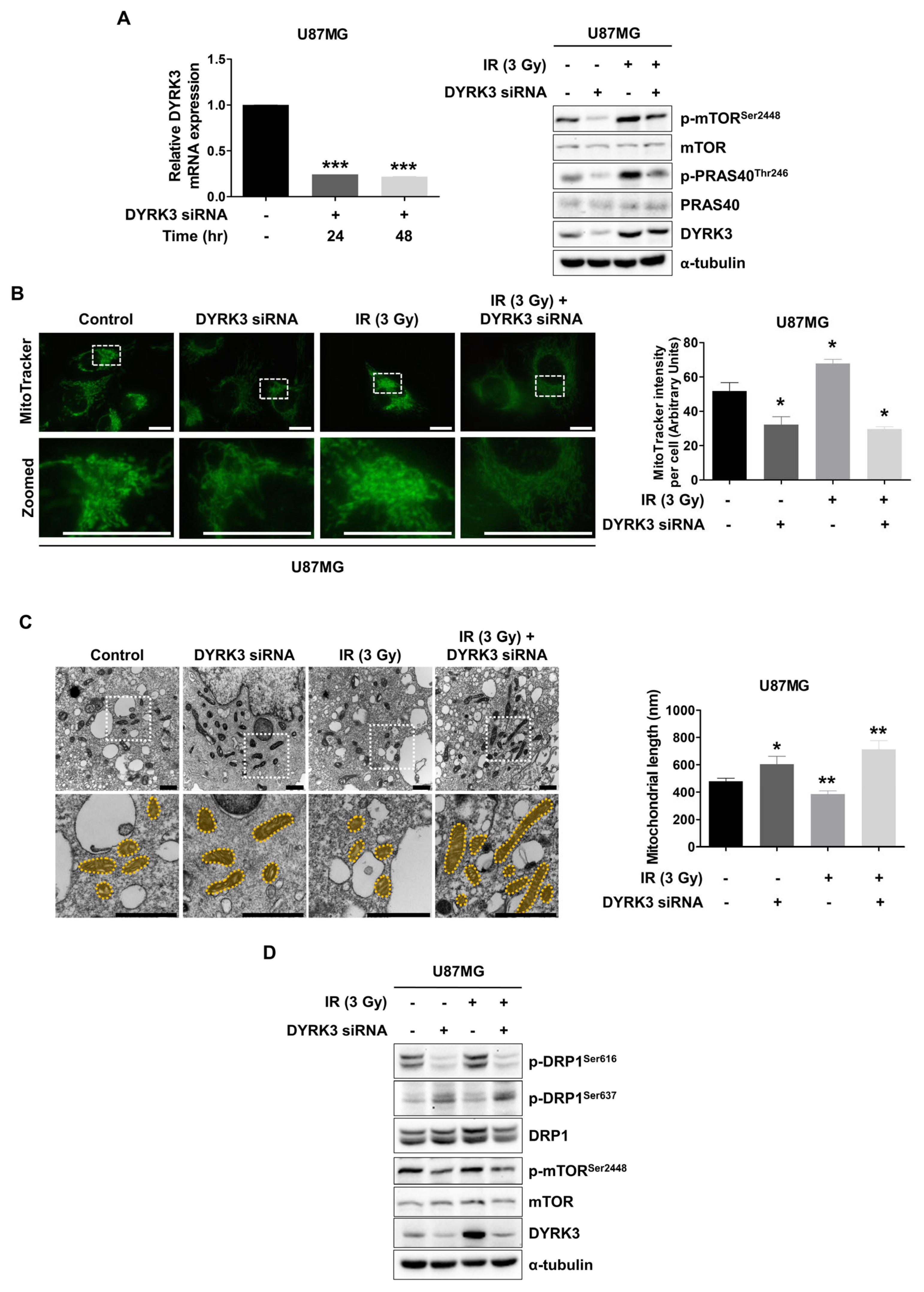
2.2. Radiation-Induced DYRK3 Induces Mitochondrial Fission
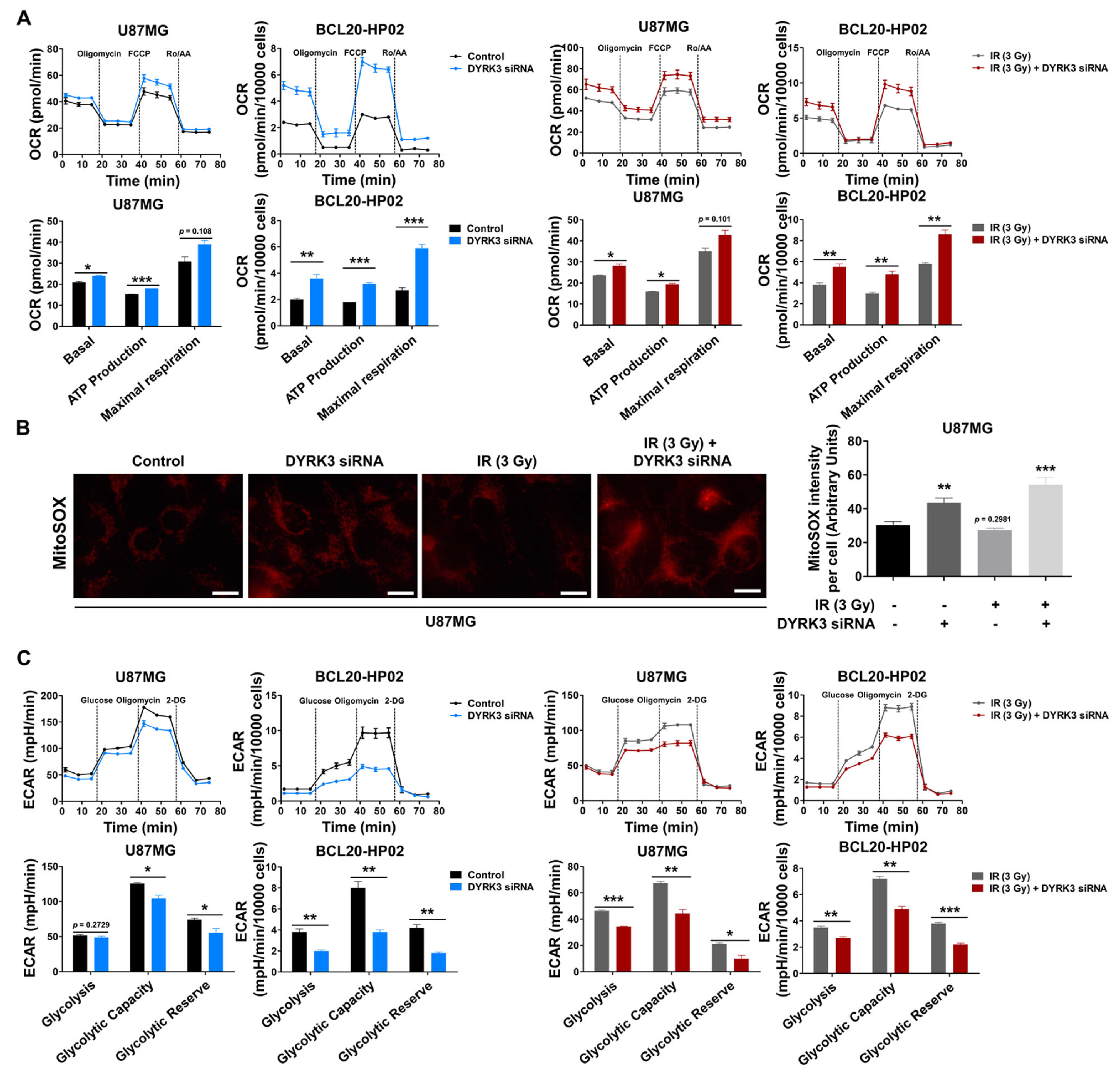
2.3. Radiation-Induced DYRK3 Inhibits OXPHOS and Increases Glycolytic Activity
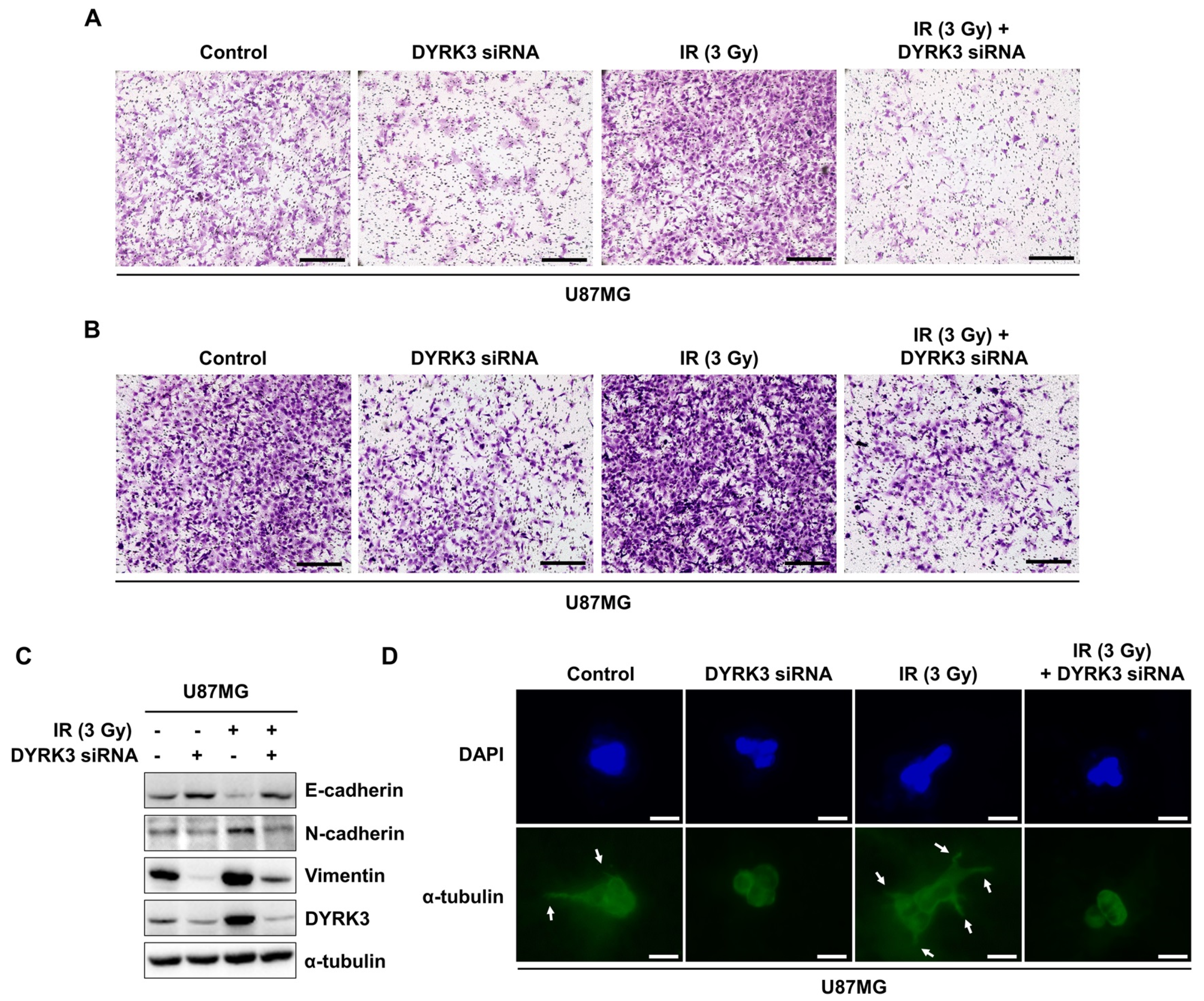
2.4. Radiation-Induced DYRK3 Promotes Migration and Invasion of GBM
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Line, Cell Culture, Irradiation, and Transfection
4.3. Animal Care Protocol and Orthotopic Xenograft Mouse Model
4.4. Total RNA Isolation and qRT-PCR
4.5. Immunohistochemistry (IHC)
4.6. Western Blot Analysis
4.7. MitoTrackerTM Green FM and MitoSOXTM Red Staining
4.8. Transmission Electron Microscopy (TEM)
4.9. Seahorse Analysis
4.10. Migration and Invasion Assay
4.11. Three-Dimensional (3D) Culture
4.12. Database Analysis and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DRP1 | Dynamin related protein 1 |
| DYRK3 | Dual specificity tyrosine-phosphorylation-regulated kinase 3 |
| GBM | Glioblastoma multiforme |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| OXPHOS | Oxidative phosphorylation |
| PRAS40 | Proline-rich AKT1 substrate 1 |
| ROS | Reactive oxygen species |
References
- Delgado-Martín, B.; Medina, M. Ángel Advances in the knowledge of the molecular biology of glioblastoma and its impact in patient diagnosis, stratification, and treatment. Adv. Sci. 2020, 7. [Google Scholar] [CrossRef]
- Jang, M.; Kim, S.S.; Lee, J. Cancer cell metabolism: Implications for therapeutic targets. Exp. Mol. Med. 2013, 45, e45. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarrado-Castellarnau, M.; De Atauri, P.; Cascante, M. Oncogenic regulation of tumor metabolic reprogramming. Oncotarget 2016, 7, 62726–62753. [Google Scholar] [CrossRef] [Green Version]
- Toulany, M.; Rodemann, H.P. Phosphatidylinositol 3-kinase/Akt signaling as a key mediator of tumor cell responsiveness to radiation. Semin. Cancer Biol. 2015, 35, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.W.; Miller, N. Durability of kinase-directed therapies—a network perspective on response and resistance. Mol. Cancer Ther. 2015, 14, 1975–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.C.; Duarte, C.W.; Welaya, K.; Rohrbach, T.D.; Bredel, M.; Yang, E.S.; Choradia, N.V.; Thottassery, J.V.; Gillespie, G.Y.; Bonner, J.A.; et al. Kinomic exploration of temozolomide and radiation resistance in Glioblastoma multiforme xenolines. Radiother. Oncol. 2014, 111, 468–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, S.; Rajapakshe, K.; Zhu, B.; Nikolai, B.C.; Yi, P.; Putluri, N.; Choi, J.M.; Jung, S.Y.; Coarfa, C.; Westbrook, T.F.; et al. Metabolic enzyme PFKFB4 activates transcriptional coactivator SRC-3 to drive breast cancer. Nat. Cell Biol. 2018, 556, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Wippich, F.; Bodenmiller, B.; Trajkovska, M.G.; Wanka, S.; Aebersold, R.; Pelkmans, L. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell 2013, 152, 791–805. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Williams, J.G.; Schug, T.T.; Li, X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J. Biol. Chem. 2010, 285, 13223–13232. [Google Scholar] [CrossRef] [Green Version]
- Ma, F.; Zhu, Y.; Hou, J.; Zhong, M.; Zhuo, H.; Zhong, L.; Ye, Z.; Xie, W.; Liu, Y.; Xiong, Y.; et al. Dual-specificity tyrosine phosphorylation—Regulated kinase 3 loss activates purine metabolism and promotes hepatocellular carcinoma progression. Hepatology 2019, 70, 1785–1803. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [Green Version]
- Slepak, T.I.; Salay, L.D.; Lemmon, V.P.; Bixby, J.L. Dyrk kinases regulate phosphorylation of doublecortin, cytoskeletal organization, and neuronal morphology. Cytoskeleton 2012, 69, 514–527. [Google Scholar] [CrossRef]
- Fogg, V.C.; Lanning, N.J.; MacKeigan, J.P. Mitochondria in cancer: At the crossroads of life and death. Chin. J. Cancer 2011, 30, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, M.; Rodrigues, A.S.D.J.; Pereira, S.L.C.; Perestrelo, T.; Correia, M.; Ramalho-Santos, J. Mitochondrial mechanisms of metabolic reprogramming in proliferating cells. Curr. Med. Chem. 2015, 22, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Ferraz, L.S. Therapeutic potential of targeting mitochondrial dynamics in cancer. Biochem. Pharmacol. 2020, 182, 114282. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Prudent, J.; St-Pierre, J.; Larsson, O.; Topisirovic, I.; Vali, H.; McBride, H.M.; Bergeron, J.J.; Sonenberg, N.; Basu, K.; et al. mTOR controls mitochondrial dynamics and cell survival via MTFP1. Mol. Cell 2017, 67, 922–935.e5. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.-Y.; Chow, K.-C.; Chiao, M.-T.; Yang, Y.-C.; Shen, C.-C. Higher levels of dynamin-related protein 1 are associated with reduced radiation sensitivity of glioblastoma cells. Curr. Neurovascular Res. 2020, 17, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; Du, J.; Ni, W.; Liu, W. Tbx2 confers poor prognosis in glioblastoma and promotes temozolomide resistance with change of mitochondrial dynamics. OncoTargets Ther. 2017, 10, 1059–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, B.; Lee, S.; Kim, H.; Kang, H.; Jeon, J.; Jo, S.; Seong, K.M.; Lee, S.-J.; Youn, H.; Youn, B. Decreased FBP1 expression rewires metabolic processes affecting aggressiveness of glioblastoma. Oncogene 2019, 39, 36–49. [Google Scholar] [CrossRef]
- Sathornsumetee, S.; Reardon, D.A. Targeting multiple kinases in glioblastoma multiforme. Expert Opin. Investig. Drugs 2009, 18, 277–292. [Google Scholar] [CrossRef]
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915. [Google Scholar] [CrossRef]
- Haar, E.V.; Lee, S.-I.; Bandhakavi, S.; Griffin, T.J.; Kim, D.-H. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol. 2007, 9, 316–323. [Google Scholar] [CrossRef]
- Taguchi, T.; Ishihara, N.; Gu, J.; Kondo, A.; Taniguchi, N.; Miyoshi, E.; Jofuku, A.; Oka, T.; Mihara, K.; Nakagawa, T.; et al. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-R.; Blackstone, C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Da-Silva, A.; Valacca, C.; Rios, E.; Pópulo, H.; Soares, P.; Sobrinho-Simões, M.; Scorrano, L.; Máximo, V.; Campello, S. Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS ONE 2015, 10, e0122308. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.-Y.; Zhang, J.-F.; Yang, Z.-J.; Jiang, L.-P.; Wei, Y.-F.; Lai, Q.-N.; Wang, J.-B.; Xin, H.-B.; Han, X.-J. Involvement of Drp1 in hypoxia-induced migration of human glioblastoma U251 cells. Oncol. Rep. 2014, 32, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.S. Mitochondria: A target for cancer therapy. Br. J. Pharmacol. 2006, 147, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Serasinghe, M.N.; Wieder, S.Y.; Chipuk, J.E.; Renault, T.T.; Elkholi, R.; Asciolla, J.J.; Yao, J.L.; Jabado, O.; Hoehn, K.; Kageyama, Y.; et al. Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol. Cell 2015, 57, 521–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Shi, F.; Li, Y.; Li, W.; Yu, X.; Zhao, L.; Zhou, M.; Hu, J.; Luo, X.; Tang, M.; et al. Drp1-dependent remodeling of mitochondrial morphology triggered by EBV-LMP1 increases cisplatin resistance. Signal Transduct. Target. Ther. 2020, 5, 1–12. [Google Scholar] [CrossRef]
- Xie, Q.; Wu, Q.; Horbinski, C.M.; Flavahan, W.A.; Yang, K.; Zhou, W.; Dombrowski, S.M.; Huang, Z.; Fang, X.; Shi, Y.; et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat. Neurosci. 2015, 18, 501–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arron, J.R.; Winslow, M.M.; Polleri, A.; Chang, C.-P.; Wu, H.; Gao, X.; Neilson, J.R.; Chen, L.; Heit, J.J.; Kim, S.K.; et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nat. Cell Biol. 2006, 441, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Y.; Lin, J.-R.; Tsai, F.-C.; Meyer, T. Dosage of Dyrk1a shifts cells within a p21-cyclin D1 signaling map to control the decision to enter the cell cycle. Mol. Cell 2013, 52, 87–100. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Liu, N.; Zang, S.; Liu, H.; Wang, P.; Ji, C.; Sun, X. Tumor suppressor DYRK1A effects on proliferation and chemoresistance of AML cells by downregulating C-Myc. PLoS ONE 2014, 9, e98853. [Google Scholar] [CrossRef]
- Boichuk, S.; Parry, J.A.; Schöffski, P.; Debiec-Rychter, M.; DeCaprio, J.A.; Duensing, A.; Makielski, K.R.; Litovchick, L.; Baron, J.L.; Zewe, J.P.; et al. The DREAM complex mediates GIST cell quiescence and is a novel therapeutic target to enhance imatinib-induced apoptosis. Cancer Res. 2013, 73, 5120–5129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Yang, X.; Yin, P.; Hu, W.; Liao, H.; Miao, Z.; Pan, C.; Li, N. The involvement of FoxO in cell survival and chemosensitivity mediated by Mirk/Dyrk1B in ovarian cancer. Int. J. Oncol. 2011, 40, 1203–1209. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, S.; He, Z.; Sun, F.; Huang, Y.; Ni, Q.; Wang, H.; Wang, Y.; Cheng, C. Dyrk1B overexpression is associated with breast cancer growth and a poor prognosis. Hum. Pathol. 2017, 66, 48–58. [Google Scholar] [CrossRef]
- Taira, N.; Mimoto, R.; Kurata, M.; Yamaguchi, T.; Kitagawa, M.; Miki, Y.; Yoshida, K. DYRK2 priming phosphorylation of c-Jun and c-Myc modulates cell cycle progression in human cancer cells. J. Clin. Investig. 2012, 122, 859–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimoto, R.; Taira, N.; Takahashi, H.; Yamaguchi, T.; Okabe, M.; Uchida, K.; Miki, Y.; Yoshida, K. DYRK2 controls the epithelial–mesenchymal transition in breast cancer by degrading Snail. Cancer Lett. 2013, 339, 214–225. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Mimoto, R.; Yanaihara, N.; Imawari, Y.; Hirooka, S.; Okamoto, A.; Yoshida, K. DYRK2 regulates epithelial-mesenchymal-transition and chemosensitivity through Snail degradation in ovarian serous adenocarcinoma. Tumor Biol. 2015, 36, 5913–5923. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Veeraraghavan, J.; Tan, Y.; Kim, J.-A.; Wang, X.; Loo, S.K.; Lee, S.; Hu, Y.; Wang, X.-S. A novel neoplastic fusion transcript, RAD51AP1-DYRK4, confers sensitivity to the MEK inhibitor trametinib in aggressive breast cancers. Clin. Cancer Res. 2021, 27, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Clerc, P.; Francis, T.C.; Kennedy, N.W.; Picton, L.K.; Kumar, T.; Uppuluri, S.; Miller, A.M.; Itoh, K.; Karbowski, M.; et al. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev. Cell 2017, 40, 583–594.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, W.; Liu, Y.; Yin, H. Mitochondrial dynamics: Biogenesis, fission, fusion, and mitophagy in the regulation of stem cell behaviors. Stem Cells Int. 2019, 2019, 9757201. [Google Scholar] [CrossRef] [Green Version]
- Boni, J.; Rubio-Perez, C.; López-Bigas, N.; Fillat, C.; De La Luna, S. The DYRK family of kinases in cancer: Molecular functions and therapeutic opportunities. Cancers 2020, 12, 2106. [Google Scholar] [CrossRef]
- Wu, L.-W.; Zhang, J.-K.; Rao, M.; Zhang, Z.-Y.; Zhu, H.-J.; Zhang, C. Harmine suppresses the proliferation of pancreatic cancer cells and sensitizes pancreatic cancer to gemcitabine treatment. OncoTargets Ther. 2019, ume 12, 4585–4593. [Google Scholar] [CrossRef] [Green Version]
- Meinguet, C.; Bruyère, C.; Frederick, R.; Mathieu, V.; Vancraeynest, C.; Pochet, L.; Laloy, J.; Mortier, J.; Wolber, G.; Kiss, R.; et al. 3D-QSAR, design, synthesis and characterization of trisubstituted harmine derivatives with in vitro antiproliferative properties. Eur. J. Med. Chem. 2015, 94, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Kim, W.; Lee, S.; Kwon, D.; Chun, J.; Son, B.; Kim, E.; Lee, J.-M.; Youn, H.; Youn, B. TFAP2C promotes lung tumorigenesis and aggressiveness through miR-183- and miR-33a-mediated cell cycle regulation. Oncogene 2016, 36, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.J.; Youn, H.; Seong, K.M.; Yun, Y.J.; Kim, W.; Kim, Y.H.; Lee, J.Y.; Kim, C.S.; Jin, Y.-W.; Youn, B. Psoralidin, a dual inhibitor of COX-2 and 5-LOX, regulates ionizing radiation (IR)-induced pulmonary inflammation. Biochem. Pharmacol. 2011, 82, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Park, G.; Son, B.; Youn, B.; Kang, J.; Lee, S.; Jeon, J.; Kim, J.-H.; Yi, G.-R.; Youn, H.; Moon, C.; et al. LDR-induced miR-30a and miR-30b target the PAI-1 pathway to control adverse effects of NSCLC radiotherapy. Mol. Ther. 2019, 27, 342–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Primer Sequences |
|---|---|
| Human GAPDH | FW 5′- ATGACATCAAGAAGGTGGTG -3′ RV 5′- CATACCAGGAAATGAGCTTG -3′ |
| Human DYRK3 | FW 5′- CCCTCTGCCCGCTTGAC-3′ RV 5′- CCCGTTTCCCTGACACCT T -3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, K.; Lee, S.; Kang, H.; Shin, E.; Kim, H.Y.; Youn, H.; Youn, B. Dual Specificity Kinase DYRK3 Promotes Aggressiveness of Glioblastoma by Altering Mitochondrial Morphology and Function. Int. J. Mol. Sci. 2021, 22, 2982. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062982
Kim K, Lee S, Kang H, Shin E, Kim HY, Youn H, Youn B. Dual Specificity Kinase DYRK3 Promotes Aggressiveness of Glioblastoma by Altering Mitochondrial Morphology and Function. International Journal of Molecular Sciences. 2021; 22(6):2982. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062982
Chicago/Turabian StyleKim, Kyeongmin, Sungmin Lee, Hyunkoo Kang, Eunguk Shin, Hae Yu Kim, HyeSook Youn, and BuHyun Youn. 2021. "Dual Specificity Kinase DYRK3 Promotes Aggressiveness of Glioblastoma by Altering Mitochondrial Morphology and Function" International Journal of Molecular Sciences 22, no. 6: 2982. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22062982