
Distinct miRNA Signatures and Networks Discern Fetal from Adult Erythroid Differentiation and Primary from Immortalized Erythroid Cells

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
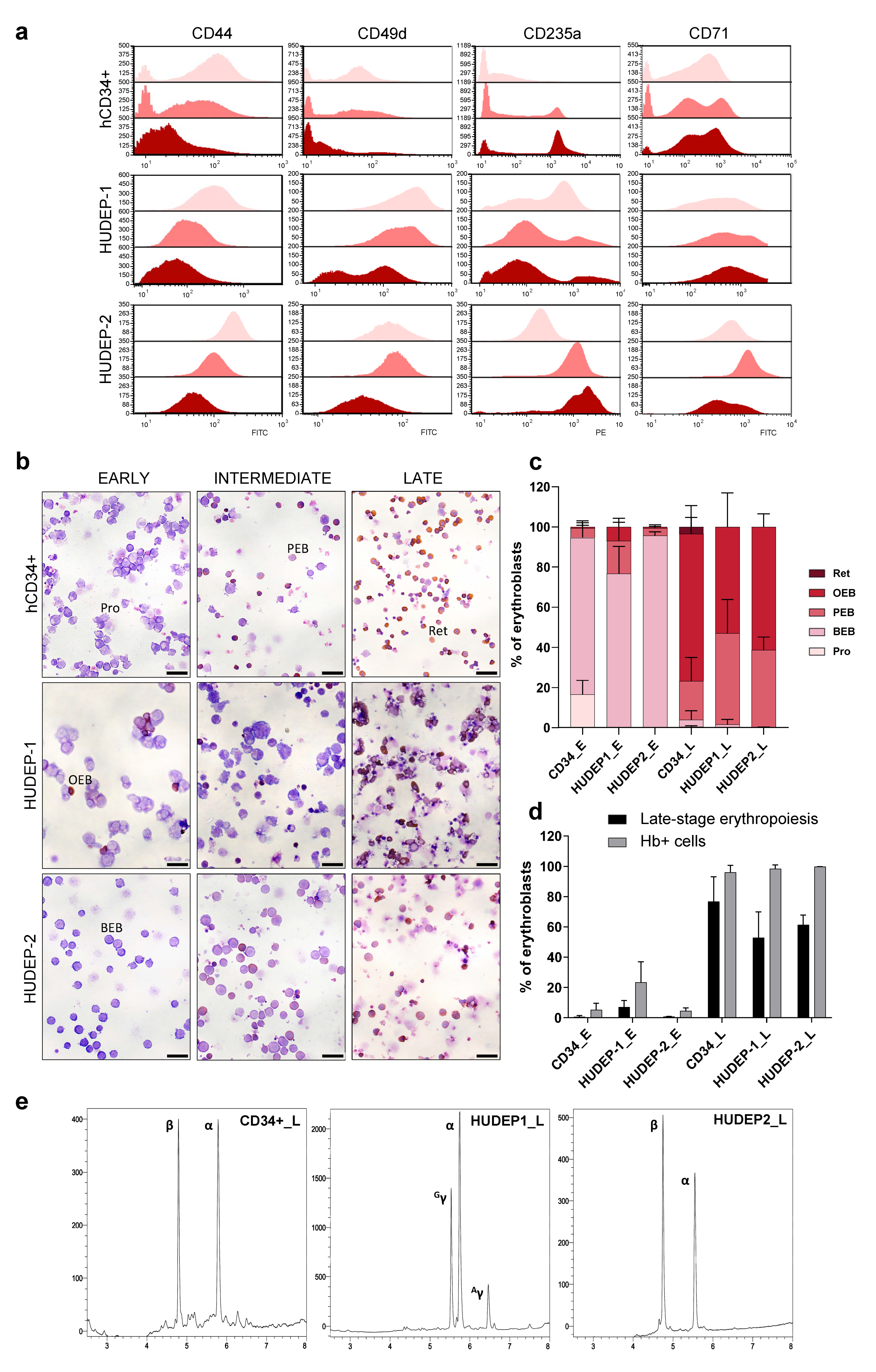
2.1. Flow Cytometry-Based Cell-Surface Protein Analysis Confirms Consistent Erythroid Differentiation in Cultures
2.2. Morphological Changes Characterize Erythroid Differentiation in Cultures
2.3. Reversed-Phase High-Performance Liquid Chromatography (RP-HPLC) Analysis Verifies Adult and Fetal Globin Expression Profiles of Differentiated Cells
2.4. Small-RNA Profiling during Erythroid Differentiation Detects Novel and Known miRNAs and Delineates Cell Types and Stages for HUDEP and Primary hCD34+ Cells
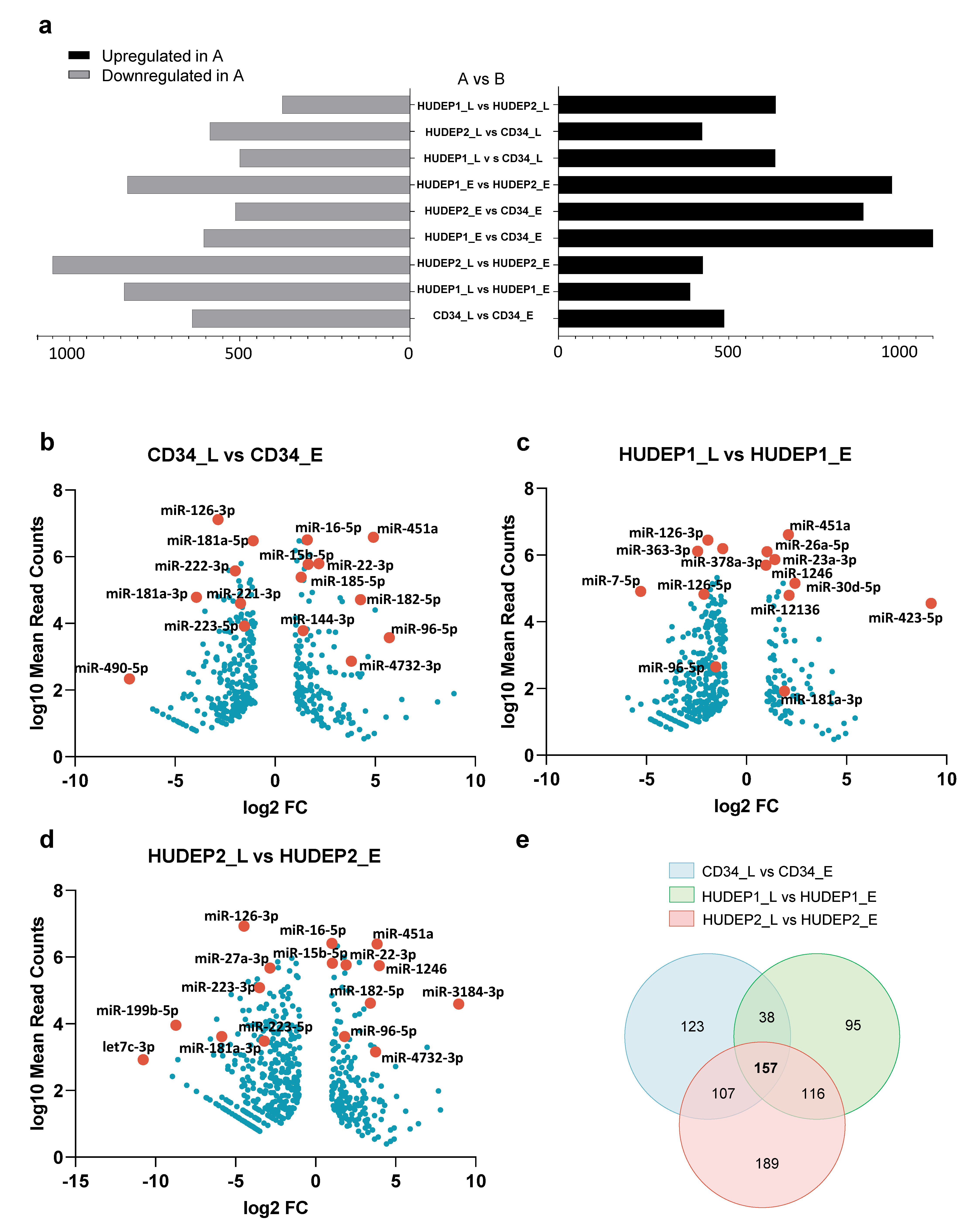
2.5. Temporal miRNA Expression Profiling Shows Dynamic Regulation of Erythroid Differentiation
2.6. Characterisation of the miRNA Transcriptome Highlights Differences between Primary hCD34+ Cells and HUDEP Cell Lines
2.7. Distinct HUDEP-1 and HUDEP-2 miRNA Patterns Reveal Signatures Associated with Hemoglobin Switching
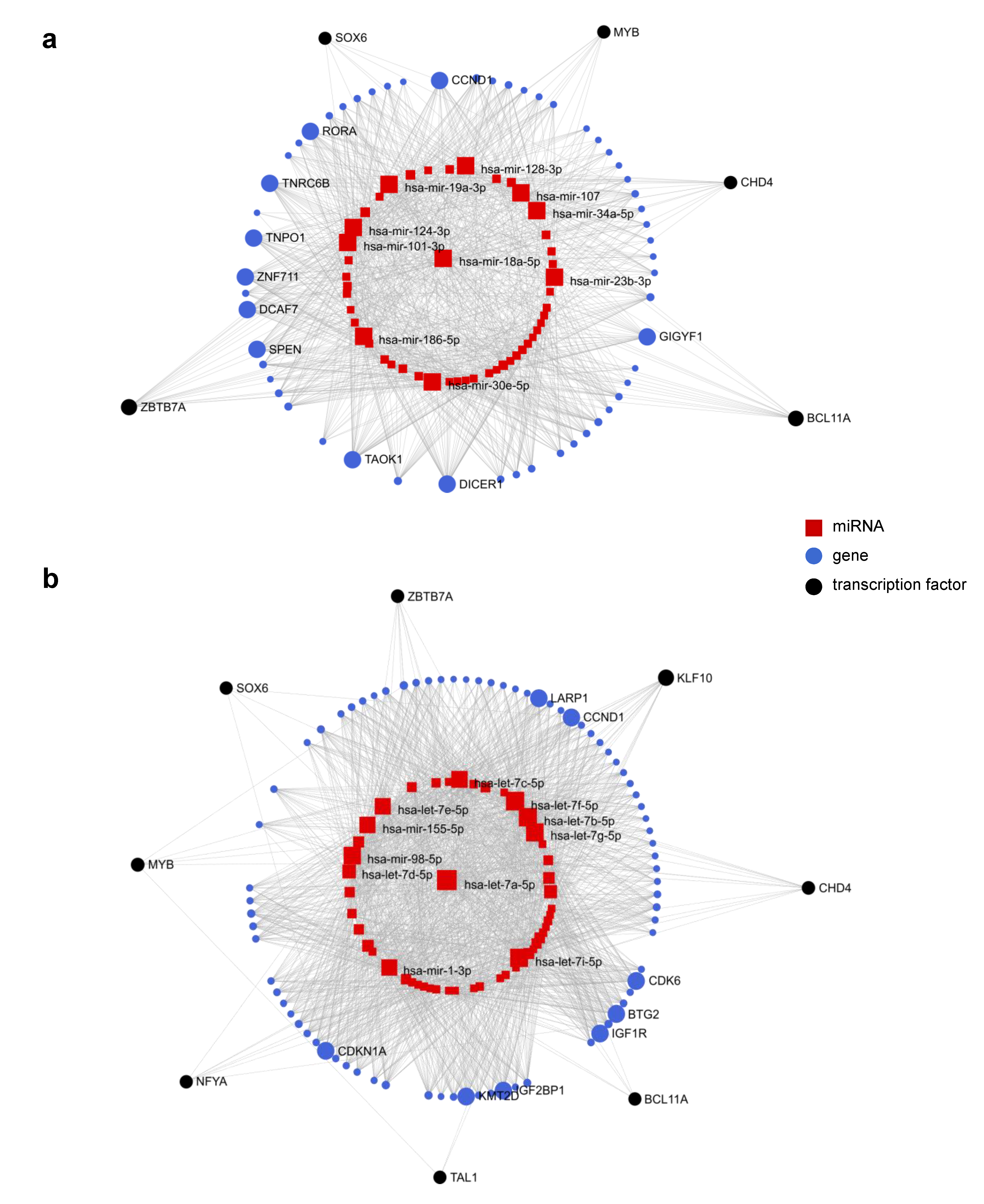
2.8. Evidence Suggests Cross Talk between miRNAs and Long Non-Coding RNAs (lncRNAs)
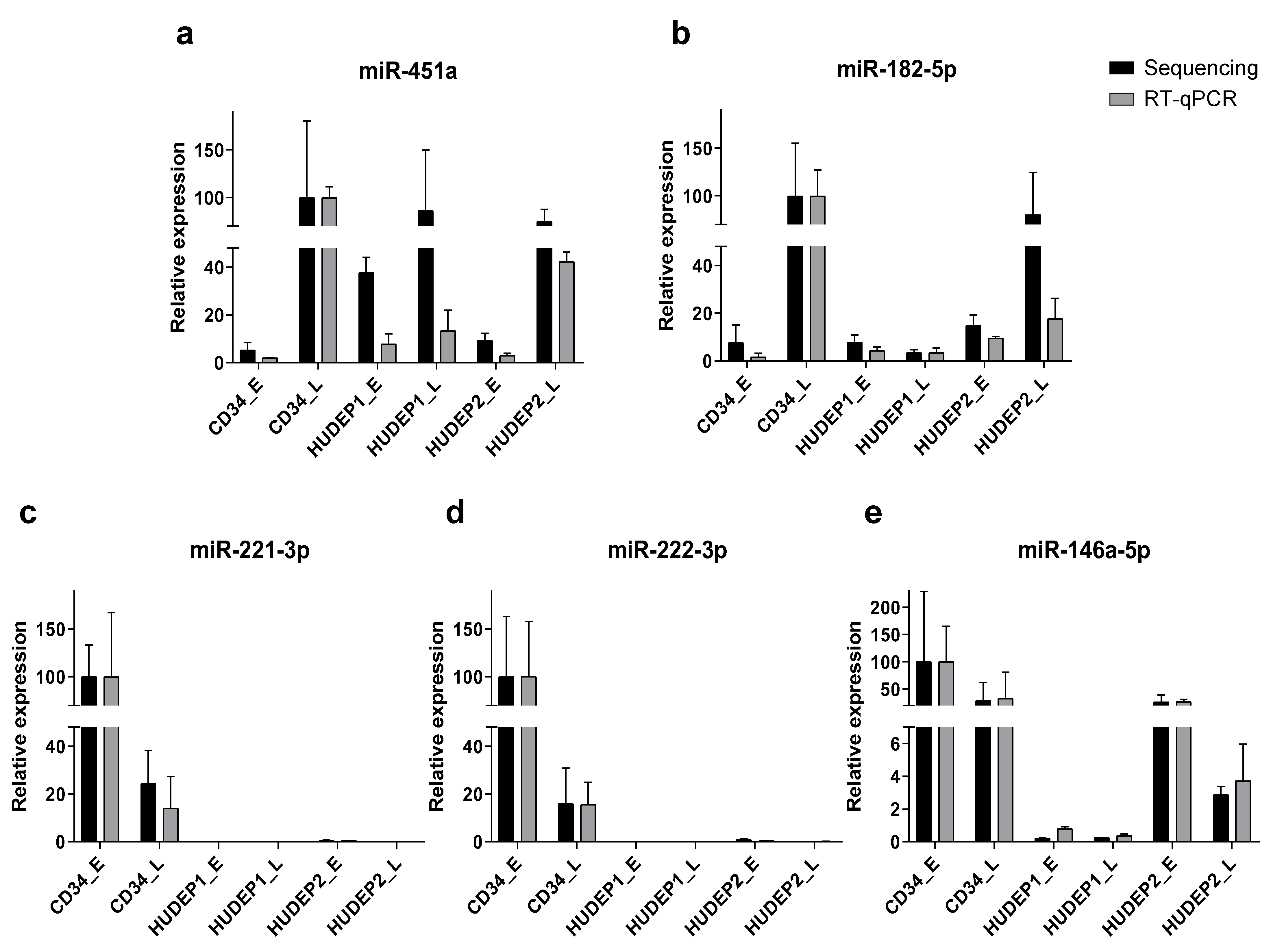
2.9. Reverse-Transcription Quantitative PCR (RT-qPCR) Validates Small RNA Sequencing Results
3. Discussion
4. Materials and Methods
4.1. Culture of Human Umbilical Cord Blood-Derived Erythroid Progenitor (HUDEP) Cells
4.2. Isolation and Culture of PB-Derived hCD34+ HSPCs
4.3. Cytocentrifugation and Microscopy
4.4. Flow Cytometry and Analysis
4.5. Globin Chain Analysis by RP-HPLC
4.6. DNA Nanoball (DNB) Small RNA Sequencing and Analysis
4.7. Reverse-Transcription Quantitative PCR (RT-qPCR)
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BB | beading buffer |
| BM | bone marrow |
| ceRNA | competing endogenous RNA |
| DE | differentially expressed |
| DNB | DNA nanoball |
| FC | fold change |
| GO | gene ontology |
| GRN | gene regulatory network |
| h | human |
| HbA | adult hemoglobin |
| HbF | fetal hemoglobin |
| hCD34+ | human peripheral-blood-derived CD34+ stem/progenitor |
| HPV16 | human papillomavirus 16 |
| HSC | hematopoietic stem cell |
| HSPC | hematopoietic stem and progenitor cell |
| HUDEP | human umbilical cord blood-derived erythroid progenitor |
| lncRNA | long non-coding RNA |
| miRNA | microRNA |
| ncRNA | non-coding RNA |
| PB | peripheral blood |
| PBS | phosphate-buffered saline |
| PC | principal component |
| piRNA | piwi-interacting RNA |
| pre-miRNA | precursor miRNA |
| pri-miRNA | primary miRNA |
| RISC | RNA-induced silencing complex |
| RP-HPLC | reversed-phase high-performance liquid chromatography |
| rRNA | ribosomal RNA |
| RT-qPCR | reverse-transcription quantitative PCR |
| siRNA | small interfering RNA |
| sncRNA | small non-coding RNA |
| snoRNA | small nucleolar RNA |
| snRNA | small nuclear RNA |
| TF | transcription factor |
| TPM | transcripts per million |
| tRNA | transfer RNA |
References
- Orkin, S.H.; Zon, L.I. Hematopoiesis: An Evolving Paradigm for Stem Cell Biology. Cell 2008, 132, 631–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramson, S.; Miller, R.G.; Phillips, R.A. The identification in adult bone marrow of pluripotent and restricted stem cells of the myeloid and lymphoid systems*. J. Exp. Med. 1977, 145, 1567–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, A.J.; McCulloch, E.A.; Till, J.E. Cytological demonstration of the clonal nature of spleen colonies derived from transplanted mouse marrow cells. Nature 1963, 197, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Scala, S.; Aiuti, A. In vivo dynamics of human hematopoietic stem cells: Novel concepts and future directions. Blood Adv. 2019, 3, 1916–1924. [Google Scholar] [CrossRef] [Green Version]
- Goode, D.K.; Obier, N.; Vijayabaskar, M.S.; Lie-A-Ling, M.; Lilly, A.J.; Hannah, R.; Lichtinger, M.; Batta, K.; Florkowska, M.; Patel, R.; et al. Dynamic Gene Regulatory Networks Drive Hematopoietic Specification and Differentiation. Dev. Cell 2016, 36, 572–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, F.; Huang, W.; Guo, G. Studying hematopoiesis using single-cell technologies. J. Hematol. Oncol. 2017, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, Y.; Liu, Y.; Ji, P. Understanding terminal erythropoiesis: An update on chromatin condensation, enucleation, and reticulocyte maturation. Blood Rev. 2020. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Vasudevan, S. Posttranscriptional Upregulation by MicroRNAs. Wiley Interdiscip. Rev. RNA 2012, 3, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Stevens, K. When microRNAs activate translation. Nat. Methods 2008, 5, 122–123. [Google Scholar] [CrossRef]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in control of gene expression: An overview of nuclear functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [Green Version]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, S.; Imai-Sumida, M.; Tanaka, Y.; Dahiya, R. Interaction and cross-talk between non-coding RNAs. Cell. Mol. Life Sci. 2018, 75, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Felli, N.; Fontana, L.; Pelosi, E.; Botta, R.; Bonci, D.; Facchiano, F.; Liuzzi, F.; Lulli, V.; Morsilli, O.; Santoro, S.; et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc. Natl. Acad. Sci. USA 2005, 102, 18081–18086. [Google Scholar] [CrossRef] [Green Version]
- Masaki, S.; Ohtsuka, R.; Abe, Y.; Muta, K.; Umemura, T. Expression patterns of microRNAs 155 and 451 during normal human erythropoiesis. Biochem. Biophys. Res. Commun. 2007, 364, 509–514. [Google Scholar] [CrossRef]
- Zhan, M.; Miller, C.P.; Papayannopoulou, T.; Stamatoyannopoulos, G.; Song, C.Z. MicroRNA expression dynamics during murine and human erythroid differentiation. Exp. Hematol. 2007, 35, 1015–1025. [Google Scholar] [CrossRef] [Green Version]
- Dore, L.C.; Amigo, J.D.; Dos Santos, C.O.; Zhang, Z.; Gai, X.; Tobias, J.W.; Yu, D.; Klein, A.M.; Dorman, C.; Wu, W.; et al. A GATA-1-regulated microRNA locus essential for erythropoiesis. Proc. Natl. Acad. Sci. USA 2008, 105, 3333–3338. [Google Scholar] [CrossRef] [Green Version]
- Felli, N.; Pedini, F.; Romania, P.; Biffoni, M.; Morsilli, O.; Castelli, G.; Santoro, S.; Chicarella, S.; Sorrentino, A.; Peschle, C.; et al. MicroRNA 223-dependent expression of LMO2 regulates normal erythropoiesis. Haematologica 2009, 94, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Byon, J.C.H.; Papayannopoulou, T. MicroRNAs: Allies or Foes in erythropoiesis? J. Cell. Physiol. 2012, 227, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Thein, S.L. Genetic modifiers of the β-haemoglobinopathies. Proc. Br. J. Haematol. 2008, 141, 357–366. [Google Scholar] [CrossRef]
- Stamatoyannopoulos, G. Control of globin gene expression during development and erythroid differentiation. Exp. Hematol. 2005, 33, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Menzel, S.; Thein, S.L. Genetic Modifiers of Fetal Haemoglobin in Sickle Cell Disease. Mol. Diagnosis Ther. 2019, 23, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Vinjamur, D.S.; Bauer, D.E.; Orkin, S.H. Recent progress in understanding and manipulating haemoglobin switching for the haemoglobinopathies. Br. J. Haematol. 2018, 180, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Saki, N.; Abroun, S.; Soleimani, M.; Kavianpour, M.; Shahjahani, M.; Mohammadi-Asl, J.; Hajizamani, S. MicroRNA expression in β-Thalassemia and sickle cell disease: A role in the induction of fetal hemoglobin. Cell J. 2016, 17, 583–592. [Google Scholar] [PubMed]
- Lulli, V.; Romania, P.; Morsilli, O.; Cianciulli, P.; Gabbianelli, M.; Testa, U.; Giuliani, A.; Marziali, G. MicroRNA-486-3p Regulates γ-Globin Expression in Human Erythroid Cells by Directly Modulating BCL11A. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzouzi, I.; Moest, H.; Winkler, J.; Fauchère, J.-C.C.; Gerber, A.P.; Wollscheid, B.; Stoffel, M.; Schmugge, M.; Speer, O. Microrna-96 directly inhibits γ-Globin expression in human erythropoiesis. PLoS ONE 2011, 6, e22838. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.T.; Huang, Y.N.; Palanisamy, K.; Chang, S.S.; Wang, I.K.; Wu, K.H.; Chen, P.; Peng, C.T.; Li, C.Y. Reciprocal regulation of γ-globin expression by exo-miRNAs: Relevance to γ-globin silencing in β-thalassemia major. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Akiyama, Y.; Yuasa, Y. Multiple-to-Multiple Relationships between MicroRNAs and Target Genes in Gastric Cancer. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Huang, S.; Ding, J.; Zhao, Y.; Liang, L.; Liu, T.; Zhan, R.; He, X. Multiple microRNAs modulate p21Cip1/Waf1 expression by directly targeting its 3′ untranslated region. Oncogene 2010, 29, 2302–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, M.E. Targeting of mRNAs by multiple miRNAs: The next step. Oncogene 2010, 29, 2161–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Shaikho, E.M.; Perez, J.; Wilson, C.A.; Liu, L.Y.; White, M.R.; Farrell, J.J.; Chui, D.H.K.; Sebastiani, P.; Steinberg, M.H. BCL2L1 is associated with γ-globin gene expression. Blood Adv. 2019, 3, 2995–3001. [Google Scholar] [CrossRef]
- Daniels, D.E.; Downes, D.J.; Ferrer-Vicens, I.; Ferguson, D.C.J.; Singleton, B.K.; Wilson, M.C.; Trakarnsanga, K.; Kurita, R.; Nakamura, Y.; Anstee, D.J.; et al. Comparing the two leading erythroid lines BEL-A and HUDEP-2. Haematologica 2020, 105, E389–E394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demirci, S.; Leonard, A.; Haro-Mora, J.J.; Uchida, N.; Tisdale, J.F. CRISPR/Cas9 for Sickle Cell Disease: Applications, Future Possibilities, and Challenges. Adv. Exp. Med. Biol. 2019, 1144, 37–52. [Google Scholar] [CrossRef]
- Vinjamur, D.S.; Bauer, D.E. Growing and Genetically Manipulating Human Umbilical Cord Blood-Derived Erythroid Progenitor (HUDEP) Cell Lines; Humana Press: New York, NY, USA, 2018; pp. 275–284. [Google Scholar]
- Wienert, B.; Martyn, G.E.; Kurita, R.; Nakamura, Y.; Quinlan, K.G.R.R.; Crossley, M. KLF1 drives the expression of fetal hemoglobin in British HPFH. Blood 2017, 130, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Norton, L.J.; Funnell, A.P.W.; Burdach, J.; Wienert, B.; Kurita, R.; Nakamura, Y.; Philipsen, S.; Pearson, R.C.M.; Quinlan, K.G.R.; Crossley, M. KLF1 directly activates expression of the novel fetal globin repressor ZBTB7A/LRF in erythroid cells. Blood Adv. 2017, 1, 685–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurita, R.; Suda, N.; Sudo, K.; Miharada, K.; Hiroyama, T.; Miyoshi, H.; Tani, K.; Nakamura, Y. Establishment of Immortalized Human Erythroid Progenitor Cell Lines Able to Produce Enucleated Red Blood Cells. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [Green Version]
- Kosaka, N.; Sugiura, K.; Yamamoto, Y.; Yoshioka, Y.; Miyazaki, H.; Komatsu, N.; Ochiya, T.; Kato, T. Identification of erythropoietin-induced microRNAs in haematopoietic cells during erythroid differentiation. Br. J. Haematol. 2008, 142, 293–300. [Google Scholar] [CrossRef]
- Choong, M.L.; Yang, H.H.; McNiece, I. MicroRNA expression profiling during human cord blood-derived CD34 cell erythropoiesis. Exp. Hematol. 2007, 35, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Eshghi, S.; Vogelezang, M.G.; Hynes, R.O.; Griffith, L.G.; Lodish, H.F. α4β1 integrin and erythropoietin mediate temporally distinct steps in erythropoiesis: Integrins in red cell development. J. Cell Biol. 2007, 177, 871–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Liu, J.; Heck, S.; Chasis, J.A.; An, X.; Mohandas, N. Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc. Natl. Acad. Sci. USA 2009, 106, 17413–17418. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Liu, J.; Xue, F.; Halverson, G.; Reid, M.; Guo, A.; Chen, L.; Raza, A.; Galili, N.; Jaffray, J.; et al. Isolation and functional characterization of human erythroblasts at distinct stages: Implications for understanding of normal and disordered erythropoiesis in vivo. Blood 2013, 121, 3246–3253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, L.C.; von Willebrand, E.; Jokinen, M.; Karhi, K.K.; Gahmberg, C.G. Glycophorin A as an erythroid marker in normal and malignant hematopoiesis. Haematol. Blood Transfus. 1981, 26, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Wang, X.; Maeda, M.; Canver, M.C.; Sher, F.; Funnell, A.P.W.; Fisher, C.; Suciu, M.; Martyn, G.E.; Norton, L.J.; et al. Gene regulation: Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science 2016, 351, 285–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq: An R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2009, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.A.; Addya, S.; Vadigepalli, R.; Banini, B.; Delgrosso, K.; Huang, H.; Surrey, S. Transcriptional regulatory network analysis of developing human erythroid progenitors reveals patterns of coregulation and potential transcriptional regulators. Physiol. Genom. 2006, 28, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Merryweather-Clarke, A.T.; Atzberger, A.; Soneji, S.; Gray, N.; Clark, K.; Waugh, C.; McGowan, S.J.; Taylor, S.; Nandi, A.K.; Wood, W.G.; et al. Global gene expression analysis of human erythroid progenitors. Blood 2011, 117. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, H.; Chang, K.H.; Qu, H.; Zhang, Z.; Xiong, Q.; Qi, H.; Cui, P.; Lin, Q.; Ruan, X.; et al. Transcriptome dynamics during human erythroid differentiation and development. Genomics 2013, 102, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruchova, H.; Yoon, D.; Agarwal, A.M.; Mendell, J.; Josef, T. The Regulated Expression of miRNAs in Normal and Polycythemia Vera Erythropoiesis. Transfusion 2009, 35, 1657–1667. [Google Scholar] [CrossRef]
- Doss, J.F.; Corcoran, D.L.; Jima, D.D.; Telen, M.J.; Dave, S.S.; Chi, J.-T. A comprehensive joint analysis of the long and short RNA transcriptomes of human erythrocytes. BMC Genom. 2015, 16, 952. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, A.A.; Fasano, J.D.; Morilla, S.M.; Venkatesan, S.; Kupfer, G.; Hattangadi, S.M. MiR-181a regulates erythroid enucleation via the regulation of Xpo7 expression. Haematologica 2018, 103, e341–e344. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Maruyama, K.; Miyata, K.; Yoshimura, A. Proliferation and erythroid differentiation through the cytoplasmic domain of the erythropoietin receptor. J. Biol. Chem. 1994, 269, 5976–5980. [Google Scholar] [CrossRef]
- Haas, N.; Riedt, T.; Labbaf, Z.; Baßler, K.; Gergis, D.; Fröhlich, H.; Gütgemann, I.; Janzen, V.; Schorle, H. Kit transduced signals counteract erythroid maturation by MAPK-dependent modulation of erythropoietin signaling and apoptosis induction in mouse fetal liver. Cell Death Differ. 2015, 22, 790–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Shi, X.; Sheng, K.; Han, G.; Li, W.; Zhao, Q.; Jiang, B.; Feng, J.; Li, J.; Gu, Y. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia. Mol. Med. Rep. 2019, 19, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Lum, A.M.; Wang, B.B.; Li, L.; Channa, N.; Bartha, G.; Wabl, M. Retroviral activation of the mir-106a microRNA cistron in T lymphoma. Retrovirology 2007, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harden, M.E. Dysregulation of Host CellularmicroRNA Expression by the HumanPapillomavirus E6 and E7 Oncoproteins. Ph.D. Thesis, Harvard University, Cambridge, MA, USA, 2017. [Google Scholar]
- Ghorai, A.; Ghosh, U. miRNA gene counts in chromosomes vary widely in a species and biogenesis of miRNA largely depends on transcription or post-transcriptional processing of coding genes. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, W.; Wu, S.; Wu, Y.; Liu, T.; Zhao, X.; Li, Y. MicroRNA-196a/-196b regulate the progression of hepatocellular carcinoma through modulating the JAK/STAT pathway via targeting SOCS2. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Tan, X.; Liu, P.; Yang, Y.; Huang, Y.; Liu, X.; Meng, X.; Yu, B.; Wu, Y.; Jin, H. Role of exosomal microRNA-125b-5p in conferring the metastatic phenotype among pancreatic cancer cells with different potential of metastasis. Life Sci. 2020, 255. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Xi, J.; Li, Y.; Xie, X.; Shi, J.; Zhang, Z.; Li, Y.; Fang, F.; Wang, S.; Yue, W.; et al. Global transcriptome analysis for identification of interactions between coding and noncoding RNAs during human erythroid differentiation. Front. Med. 2016, 10, 297–310. [Google Scholar] [CrossRef]
- Xu, J.; Wan, X.; Chen, X.; Fang, Y.; Cheng, X.; Xie, X.; Lu, W. MiR-2861 acts as a tumor suppressor via targeting EGFR/AKT2/CCND1 pathway in cervical cancer induced by human papillomavirus virus 16 E6. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, Y.; Zhao, F.; Wang, H.; Cai, W.; Gao, J.; Li, Y.; Cai, S. MiR-34a-5p promotes the multi-drug resistance of osteosarcoma by targeting the CD117 gene. Oncotarget 2016, 7, 28420–28434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessard, S.; Beaudoin, M.; Orkin, S.H.; Bauer, D.E.; Lettre, G. 14q32 and let-7 microRNAs regulate transcriptional networks in fetal and adult human erythroblasts. Hum. Mol. Genet. 2018, 27, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Noh, S.-J.J.; Miller, S.H.; Lee, Y.T.; Goh, S.-H.H.; Marincola, F.M.; Stroncek, D.F.; Reed, C.; Wang, E.; Miller, J.L. Let-7 microRNAs are developmentally regulated in circulating human erythroid cells. J. Transl. Med. 2009, 7, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Wang, B.; Jiang, F.; Wang, D.; Liu, H.; Yan, Y.; Dong, H.; Wang, F.; Gong, B.; Zhu, Y.; et al. A Feedback Loop Consisting of MicroRNA 23a/27a and the β-Like Globin Suppressors KLF3 and SP1 Regulates Globin Gene Expression. Mol. Cell. Biol. 2013, 33, 3994–4007. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Liu, D.; Zhang, X.; Li, Z.; Ye, Y.; Liu, Q.; Shen, J.; Chen, Z.; Huang, H.; Liang, Y.; et al. miR-326 regulates HbF synthesis by targeting EKLF in human erythroid cells. Exp. Hematol. 2018, 63, 33–40.e2. [Google Scholar] [CrossRef]
- Ward, C.M.; Li, B.; Pace, B.S. Original Research: Stable expression of miR-34a mediates fetal hemoglobin induction in K562 cells. Exp. Biol. Med. 2016, 241, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mnika, K.; Mazandu, G.K.; Jonas, M.; Pule, G.D.; Chimusa, E.R.; Hanchard, N.A.; Wonkam, A. Hydroxyurea-induced miRNA expression in sickle cell disease patients in Africa. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Dambal, S.; Shah, M.; Mihelich, B.; Nonn, L. The microRNA-183 cluster: The family that plays together stays together. Nucleic Acids Res. 2015, 43, 7173–7188. [Google Scholar] [CrossRef] [PubMed]
- Shaham, L.; Binder, V.; Gefen, N.; Borkhardt, A.; Izraeli, S. MiR-125 in normal and malignant hematopoiesis. Leukemia 2012, 26, 2011–2018. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Rosner, M.R. MicroRNA-regulated feed forward loop network. Cell Cycle 2009, 8, 2477–2478. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Siklenka, K.; Arora, S.K.; Ribeiro, P.; Kimmins, S.; Xia, J. miRNet—Dissecting miRNA-target interactions and functional associations through network-based visual analysis. Nucleic Acids Res. 2016, 44, W135–W141. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Yamamoto, M.; Engel, J.D. Fetal Globin Gene Repressors as Drug Targets for Molecular Therapies to Treat the β-Globinopathies. Mol. Cell. Biol. 2014, 34, 3560–3569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borg, J.; Phylactides, M.; Bartsakoulia, M.; Tafrali, C.; Lederer, C.; Felice, A.E.; Papachatzopoulou, A.; Kourakli, A.; Stavrou, E.F.; Christou, S.; et al. KLF10 gene expression is associated with high fetal hemoglobin levels and with response to hydroxyurea treatment in β-hemoglobinopathy patients. Pharmacogenomics 2012, 13, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- Amaya, M.; Desai, M.; Gnanapragasam, M.N.; Wang, S.Z.; Zu Zhu, S.; Williams, D.C.; Ginder, G.D. Mi2β-mediated silencing of the fetal γ-globin gene in adult erythroid cells. Blood 2013, 121, 3493–3501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrocini, G.C. de S.; Venancio, L.P.R.; Bonini-Domingos, C.R. Screening of transcription factors involved in fetal hemoglobin regulation using phylogenetic footprinting. Evol. Bioinform. 2015, 11, 239–244. [Google Scholar] [CrossRef] [Green Version]
- Martyn, G.E.; Wienert, B.; Yang, L.; Shah, M.; Norton, L.J.; Burdach, J.; Kurita, R.; Nakamura, Y.; Pearson, R.C.M.; Funnell, A.P.W.; et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat. Genet. 2018, 50, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Morrison, T.A.; Wilcox, I.; Luo, H.Y.; Farrell, J.J.; Kurita, R.; Nakamura, Y.; Murphy, G.J.; Cui, S.; Steinberg, M.H.; Chui, D.H.K. A long noncoding RNA from the HBS1L-MYB intergenic region on chr6q23 regulates human fetal hemoglobin expression. Blood Cells Mol. Dis. 2018. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [Green Version]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef]
- Hornstein, E.; Shomron, N. Canalization of development by micrornas. Nat. Genet. 2006, 38, S20. [Google Scholar] [CrossRef] [PubMed]
- Basak, A.; Munschauer, M.; Lareau, C.A.; Montbleau, K.E.; Ulirsch, J.C.; Hartigan, C.R.; Schenone, M.; Lian, J.; Wang, Y.; Huang, Y.; et al. Control of human hemoglobin switching by LIN28B-mediated regulation of BCL11A translation. Nat. Genet. 2020, 52, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Douglass, E.; Dunn, N.; Good, B.; Harris, N.L.; Lewis, S.E.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; et al. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; DIgles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018, 46, D661–D667. [Google Scholar] [CrossRef] [PubMed]
- Pace, B.S.; Liu, L.; Li, B.; Makala, L.H. Cell signaling pathways involved in drug-mediated fetal hemoglobin induction: Strategies to treat sickle cell disease. Exp. Biol. Med. 2015, 240, 1050–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnan, V.; Pace, B.S. Regulation of γ-globin gene expression involves signaling through the p38 MAPK/CREB1 pathway. Blood Cells Mol. Dis. 2011, 47, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Paikari, A.; Zhang, Y.; Chang, A.; Goyal, A.; Rampersaud, E.; Flanagan, J.M.; Weiss, M.J.; Sheehan, V.A. Regulation of Fetal Hemoglobin through the Insulin Signaling Pathway. Blood 2019, 134, 811. [Google Scholar] [CrossRef]
- Tang, D.C.; Zhu, J.; Liu, W.; Chin, K.; Sun, J.; Chen, L.; Hanover, J.A.; Rodgers, G.P. The hydroxyurea-induced small GTP-binding protein SAR modulates γ-globin gene expression in human erythroid cells. Blood 2005, 106, 3256–3263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahim, F.; Allahmoradi, H.; Salari, F.; Shahjahani, M.; Fard, A.D.; Hosseini, S.A.; Mousakhani, H. Evaluation of signaling pathways involved in γ-globin gene induction using fetal hemoglobin inducer drugs. Int. J. Hematol. Stem Cell Res. 2013, 7, 40–45. [Google Scholar]
- Wilusz, J.E.; Sunwoo, H.; Spector, D.L. Long noncoding RNAs: Functional surprises from the RNA world. Genes Dev. 2009, 23, 1494–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Shi, L. Long non-coding RNAs during normal erythropoiesis. Blood Sci. 2019, 1, 137–140. [Google Scholar] [CrossRef]
- Kulczyńska, K.; Siatecka, M. A regulatory function of long non-coding RNAs in red blood cell development. Acta Biochim. Pol. 2016, 63, 675–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Dominguez, J.R.; Hu, W.; Yuan, B.; Shi, J.; Park, S.S.; Gromatzky, A.A.; Van Oudenaarden, A.; Lodish, H.F. Global discovery of erythroid long noncoding RNAs reveals novel regulators of red cell maturation. Blood 2014, 123, 570–581. [Google Scholar] [CrossRef] [Green Version]
- Ivaldi, M.S.; Diaz, L.F.; Chakalova, L.; Lee, J.; Krivega, I.; Dean, A. Fetal γ-globin genes are regulated by the BGLT3 long noncoding RNA locus. Blood 2018, 132, 1963–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeggari, A.; Marks, D.S.; Larsson, E. miRcode: A map of putative microrna target sites in the long non-coding transcriptome. Bioinformatics 2012, 28, 2062–2063. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Jia, S.; Chen, P.; He, Y. Construction and analysis of a long non-coding RNA (lncRNA)-associated ceRNA network in β-thalassemia and hereditary persistence of fetal hemoglobin. Med. Sci. Monit. 2019, 25, 7079–7086. [Google Scholar] [CrossRef]
- Tsang, J.; Zhu, J.; van Oudenaarden, A. MicroRNA-mediated Feedback and Feedforward Loops are Recurrent Network Motifs in Mammals. Mol. Cell 2007, 26, 753–767. [Google Scholar] [CrossRef] [Green Version]
- Qu, W.; Shi, S.; Sun, L.; Zhang, F.; Zhang, S.; Mu, S.; Zhao, Y.; Liu, B.; Cao, X. Construction of a microRNA-associated feed-forward loop network that identifies regulators of cardiac hypertrophy and acute myocardial infarction. Int. J. Mol. Med. 2018, 42, 2062–2070. [Google Scholar] [CrossRef] [Green Version]
- Moreno, A.M.; Fu, X.; Zhu, J.; Katrekar, D.; Shih, Y.-R.V.; Marlett, J.; Cabotaje, J.; Tat, J.; Naughton, J.; Lisowski, L.; et al. In situ gene therapy via AAV-CRISPR-Cas9 mediated targeted gene regulation. Mol. Ther. 2018, 26, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cora’, D.; Caselle, M. The Epithelial-Mesenchymal Transition, as Hacked by a microRNA Combinatorial Code. Cell Syst. 2018, 7, 3–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.H.; Abdelmohsen, K.; Gorospe, M. Functional interactions among microRNAs and long noncoding RNAs. Semin. Cell Dev. Biol. 2014, 34, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Lopes-Ramos, C.M.; Paulson, J.N.; Chen, C.Y.; Kuijjer, M.L.; Fagny, M.; Platig, J.; Sonawane, A.R.; DeMeo, D.L.; Quackenbush, J.; Glass, K. Regulatory network changes between cell lines and their tissues of origin. BMC Genom. 2017, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehlmann, T.; Reinheimer, S.; Geng, C.; Su, X.; Drmanac, S.; Alexeev, A.; Zhang, C.; Backes, C.; Ludwig, N.; Hart, M.; et al. cPAS-based sequencing on the BGISEQ-500 to explore small non-coding RNAs. Clin. Epigenet. 2016, 8, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosenza, L.C.; Breda, L.; Breveglieri, G.; Zuccato, C.; Finotti, A.; Lampronti, I.; Borgatti, M.; Chiavilli, F.; Gamberini, M.R.; Satta, S.; et al. A validated cellular biobank for β-thalassemia. J. Transl. Med. 2016, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loucari, C.C.; Patsali, P.; van Dijk, T.B.; Stephanou, C.; Papasavva, P.; Zanti, M.; Kurita, R.; Nakamura, Y.; Christou, S.; Sitarou, M.; et al. Rapid and Sensitive Assessment of Globin Chains for Gene and Cell Therapy of Hemoglobinopathies. Hum. Gene Ther. Methods 2018, 29, 60–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths-Jones, S.; Saini, H.K.; Van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths-Jones, S.; Bateman, A.; Marshall, M.; Khanna, A.; Eddy, S.R. Rfam: An RNA family database. Nucleic Acids Res. 2003, 31, 439–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [Green Version]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef]
- Sai lakshmi, S.; Agrawal, S. piRNABank: A web resource on classified and clustered Piwi-interacting RNAs. Nucleic Acids Res. 2008, 36. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Lai, J.; Lehman, M.L.; Nelson, C.C. MiRDeep*: An integrated application tool for miRNA identification from RNA sequencing data. Nucleic Acids Res. 2013. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liang, C.; Liu, J.; Xiao, H.; Huang, S.; Xu, J.; Li, F. Prediction of piRNAs using transposon interaction and a support vector machine. BMC Bioinform. 2014, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Kolde, R. Pheatmap v1.0.12. Available online: https://www.rdocumentation.org/packages/pheatmap (accessed on 1 May 2020).
- Rehmsmeier, M.; Steffen, P.; Höchsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2004, 10, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human microRNA targets. PLoS Biol. 2004, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhang, Y.; Cui, H.; Liu, J.; Wu, Y.; Cheng, Y.; Xu, H.; Huang, X.; Li, S.; Zhou, A.; et al. WEGO 2.0: A web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef] [PubMed]
- Lechman, E.R.; Gentner, B.; van Galen, P.; Giustacchini, A.; Saini, M.; Boccalatte, F.E.; Hiramatsu, H.; Restuccia, U.; Bachi, A.; Voisin, V.; et al. Attenuation of miR-126 activity expands HSC in vivo without exhaustion. Cell Stem Cell 2012, 11, 799–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Y.; Wu, Y.; Huang, J.; Li, Q.; Kang, K.; Qu, J.; Li, F.; Gou, D. Identification of reference genes for circulating microRNA analysis in colorectal cancer. Sci. Rep. 2016, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Bai, Z.; Han, W.; Zhang, J.; Meng, H.; Bi, J.; Ma, X.; Han, S.; Zhang, Z. Identification of suitable reference genes for qPCR analysis of serum microRNA in gastric cancer patients. Dig. Dis. Sci. 2012, 57, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Ragni, E.; De Luca, P.; Marmotti, A.; de Girolamo, L. miR-26a-5p is a Stable Reference Gene for miRNA Studies in Chondrocytes from Developing Human Cartilage. Cells 2019, 8, 631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathways | p-Value | Adjusted p-Value | Odds Ratio | Combined Score |
|---|---|---|---|---|
| Focal Adhesion WP306 | 3.195 × 10−13 | 1.508 × 10−10 | 1.53 | 43.95 |
| Focal Adhesion-PI3K-Akt-mTOR-signaling pathway WP3932 | 4.242 × 10−13 | 1.001 × 10−10 | 1.42 | 40.59 |
| Integrin-mediated Cell Adhesion WP185 | 3.947 × 10−10 | 6.210 × 10−8 | 1.63 | 35.32 |
| EGF/EGFR Signaling Pathway WP437 | 5.413 × 10−9 | 4.258 × 10−7 | 1.47 | 27.94 |
| Epithelial to mesenchymal transition in colorectal cancer WP4239 | 6.715 × 10−9 | 4.528 × 10−7 | 1.47 | 27.65 |
| Ras Signaling WP4223 | 5.153 × 10−9 | 4.864 × 10−7 | 1.44 | 27.47 |
| Insulin Signaling WP481 | 1.184 × 10−8 | 6.986 × 10−7 | 1.46 | 26.65 |
| MAPK Signaling Pathway WP382 | 4.312 × 10−9 | 5.088 × 10−7 | 1.38 | 26.62 |
| ErbB Signaling Pathway WP673 | 4.975 × 10−8 | 2.348 × 10−6 | 1.58 | 26.59 |
| VEGFA-VEGFR2 Signaling Pathway WP3888 | 2.530 × 10−8 | 1.327 × 10−6 | 1.37 | 23.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papasavva, P.L.; Papaioannou, N.Y.; Patsali, P.; Kurita, R.; Nakamura, Y.; Sitarou, M.; Christou, S.; Kleanthous, M.; Lederer, C.W. Distinct miRNA Signatures and Networks Discern Fetal from Adult Erythroid Differentiation and Primary from Immortalized Erythroid Cells. Int. J. Mol. Sci. 2021, 22, 3626. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073626
Papasavva PL, Papaioannou NY, Patsali P, Kurita R, Nakamura Y, Sitarou M, Christou S, Kleanthous M, Lederer CW. Distinct miRNA Signatures and Networks Discern Fetal from Adult Erythroid Differentiation and Primary from Immortalized Erythroid Cells. International Journal of Molecular Sciences. 2021; 22(7):3626. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073626
Chicago/Turabian StylePapasavva, Panayiota L., Nikoletta Y. Papaioannou, Petros Patsali, Ryo Kurita, Yukio Nakamura, Maria Sitarou, Soteroulla Christou, Marina Kleanthous, and Carsten W. Lederer. 2021. "Distinct miRNA Signatures and Networks Discern Fetal from Adult Erythroid Differentiation and Primary from Immortalized Erythroid Cells" International Journal of Molecular Sciences 22, no. 7: 3626. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073626