
Endogenous Opioid Peptides and Alternatively Spliced Mu Opioid Receptor Seven Transmembrane Carboxyl-Terminal Variants

Abstract
:1. Introduction
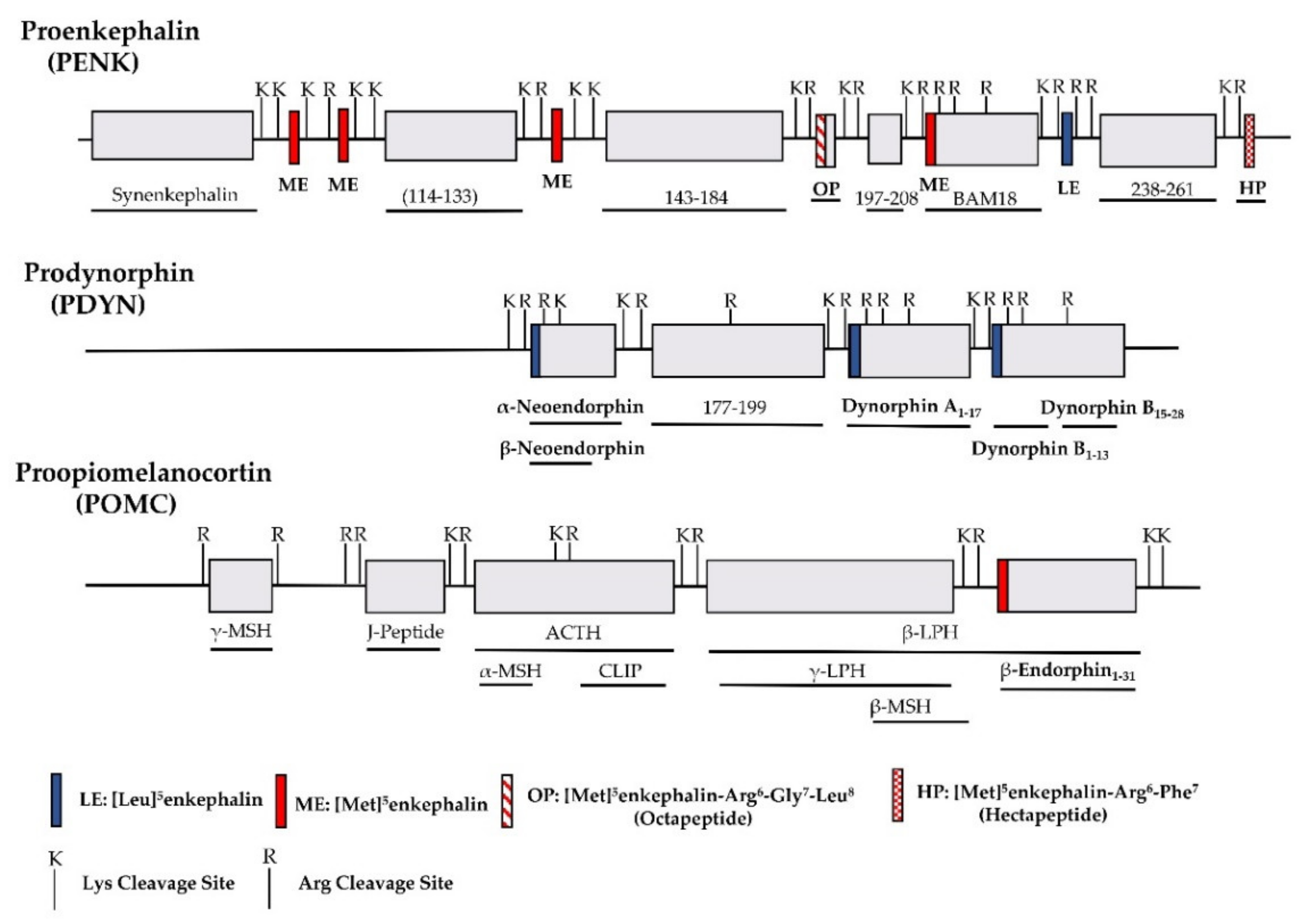
2. The Opioid Receptors and Endogenous Opioid Peptides
3. Alternative Splicing of Mu-Opioid Receptor Gene, OPRM1
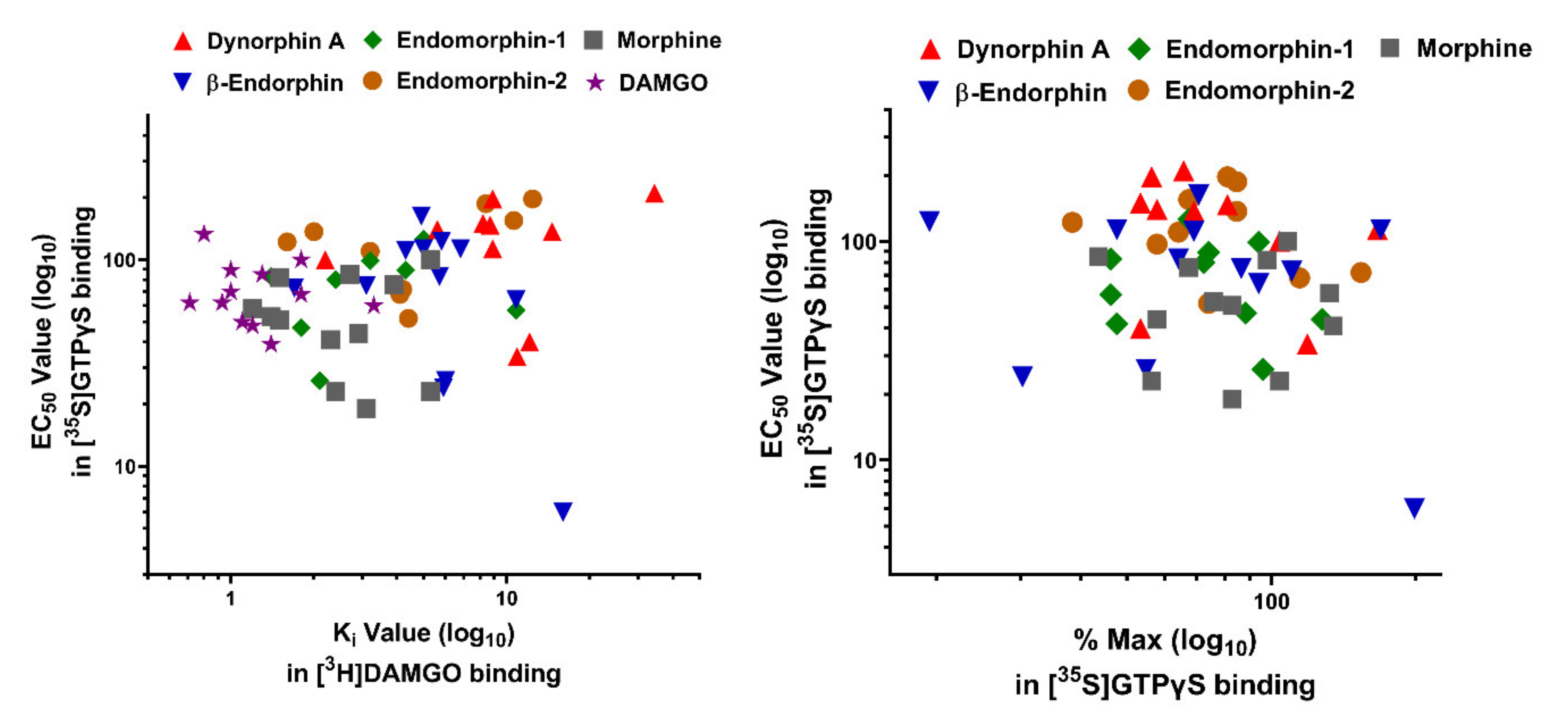
4. Binding Affinities of Endogenous Opioid Peptides in the Full-Length 7TM C-terminal Splice Variants
5. G Protein Coupling Induced by Endogenous Opioid Peptides in the Full-Length 7TM C-terminal Splice Variants
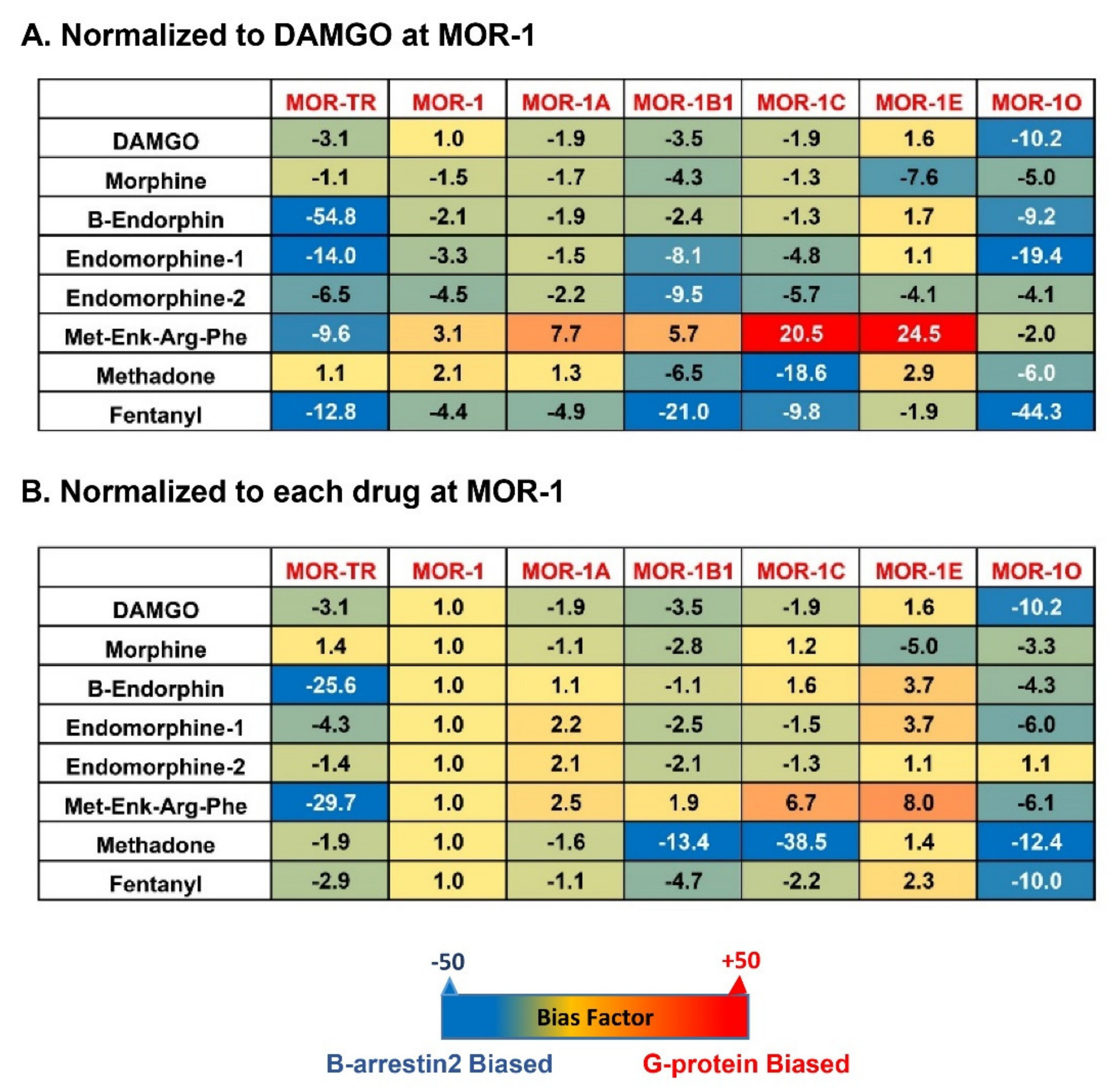
6. Biased Signaling of Endogenous Opioid Peptides in the Full-Length 7TM C-Terminal Splice Variants
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cox, B.M.; Opheim, K.E.; Teschemacher, H.; Goldstein, A. A peptide-like substance from pituitary that acts like morphine. 2. Purification and properties. Life Sci. 1975, 16, 1777–1782. [Google Scholar] [CrossRef]
- Goldstein, A. Opioid peptides (endorphins) in pituitary and brain. Science 1976, 193, 1081–1086. [Google Scholar] [CrossRef]
- Goldstein, A.; Tachibana, S.; Lowney, L.I.; Hunkapiller, M.; Hood, L. Dynorphin-(1-13), an extraordinarily potent opioid peptide. Proc. Natl. Acad. Sci. USA 1979, 76, 6666–6670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birdsall, N.J.M.; Hulme, E.C. C-fragment of lipotropin has a high affinity for brain opiate receptors. Nature 1976, 260, 793–795. [Google Scholar] [PubMed]
- Pert, C.B.; Pasternak, G.W.; Snyder, S.H. Opiate agonists and antagonists discriminated by receptor binding in brain. Science 1973, 182, 1359–1361. [Google Scholar] [CrossRef] [PubMed]
- Terenius, L. Stereospecific interaction between narcotic analgesics and a synaptic plasma membrane fraction of rat cerebral cortex. Acta Pharmacol. Toxicol. 1973, 32, 856. [Google Scholar] [CrossRef]
- Simon, E.J.; Hiller, J.M.; Edelman, I. Stereospecific binding of the potent narcotic analgesice [3H]etorphine to rat-brain homogenate. Proc. Natl. Acad. Sci. USA 1973, 70, 1947–1949. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.J.; Keith, D.E., Jr.; Morrison, H.; Magendzo, K.; Edwards, R.H. Cloning of a delta opioid receptor by functional expression. Science 1992, 258, 1952–1955. [Google Scholar] [CrossRef]
- Kieffer, B.L.; Befort, K.; Gaveriaux-Ruff, C.; Hirth, C.G. The d-opioid receptor: Isolation of a cDNA by expression cloning and pharmacological characterization. Proc. Natl. Acad. Sci. USA 1992, 89, 12048–12052. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Mestek, A.; Liu, J.; Hurley, J.A.; Yu, L. Molecular cloning and functional expression of a m-opioid receptor from rat brain. Mol. Pharmacol. 1993, 44, 8–12. [Google Scholar] [CrossRef]
- Eppler, C.M.; Hulmes, J.D.; Wang, J.-B.; Johnson, B.; Corbett, M.; Luthin, D.R.; Uhl, G.R.; Linden, J. Purification and partial amino acid sequence of a m opioid receptor from rat brain. J. Biol. Chem. 1993, 268, 26447–26451. [Google Scholar] [CrossRef]
- Thompson, R.C.; Mansour, A.; Akil, H.; Watson, S.J. Cloning and pharmacological characterization of a rat m opioid receptor. Neuron 1993, 11, 903–913. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.B.; Imai, Y.; Eppler, C.M.; Gregor, P.; Spivak, C.E.; Uhl, G.R. μ opiate receptor: cDNA cloning and expression. Proc. Natl. Acad. Sci. USA 1993, 90, 10230–10234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Mestek, A.; Liu, J.; Yu, L. Molecular cloning of a rat kappa opioid receptor reveals sequence similarities to the m and d opioid receptors. Biochem. J. 1993, 295, 625–628. [Google Scholar] [CrossRef]
- Li, S.; Zhu, J.; Chen, C.; Chen, Y.-W.; Deriel, J.K.; Ashby, B.; Liu-Chen, L.-Y. Molecular cloning and expression of a rat kappa opioid receptor. Biochem. J. 1993, 295, 629–633. [Google Scholar] [CrossRef]
- Meng, F.; Xie, G.-X.; Thompson, R.C.; Mansour, A.; Goldstein, A.; Watson, S.J.; Akil, H. Cloning and pharmacological characterization of a rat kappa opioid receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 9954–9958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.X. Diversity and complexity of the mu opioid receptor gene: Alternative pre-mRNA splicing and promoters. DNA Cell Biol. 2005, 24, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W.; Pan, Y.X. Mu opioids and their receptors: Evolution of a concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef] [Green Version]
- Pasternak, G.W.; Childers, S.R.; Pan, Y.X. Emerging insights into mu opioid pharmacology. Handb. Exp. Pharmacol. 2020, 258, 89–125. [Google Scholar] [CrossRef]
- Piltonen, M.; Parisien, M.; Gregoire, S.; Chabot-Dore, A.J.; Jafarnejad, S.M.; Berube, P.; Djambazian, H.; Sladek, R.; Geneau, G.; Willett, P.; et al. Alternative splicing of the delta-opioid receptor gene suggests existence of new functional isoforms. Mol. Neurobiol. 2019, 56, 2855–2869. [Google Scholar] [CrossRef]
- Gavériaux-Ruff, C.; Peluso, J.; Befort, K.; Simonin, F.; Zilliox, C.; Kieffer, B.L. Detection of opioid receptor mRNA by RT-PCR reveals alternative splicing for the d- and kappa-opioid receptors. Mol. Brain Res. 1997, 48, 298–304. [Google Scholar] [CrossRef]
- Belkowski, S.M.; Zhu, J.M.; Liu-Chen, L.Y.; Eisenstein, T.K.; Adler, M.W.; Rogers, T.J. Sequence of kappa-opioid receptor cDNA in the R1.1 thymoma cell line. J. Neuroimmunol. 1995, 62, 113–117. [Google Scholar] [CrossRef]
- Alicea, C.; Belkowski, S.M.; Sliker, J.K.; Zhu, J.M.; Liu-Chen, L.Y.; Eisenstein, T.K.; Adler, M.W.; Rogers, T.J. Characterization of kappa-opioid receptor transcripts expressed by T cells and macrophages. J. Neuroimmunol. 1998, 91, 55–62. [Google Scholar] [CrossRef]
- Beckett, A.H.; Casy, A.F. Synthetic analgesics: Sterochemical considerations. J. Pharm. Pharmacol. 1954, 6, 986–1001. [Google Scholar] [CrossRef] [PubMed]
- Beckett, A.H.; Casy, A.F. Analgesics and their antagonists: Biochemical aspects and structure-activity relationships. Prog. Med. Chem. 1965, 4, 171–218. [Google Scholar]
- Portoghese, P.S. Stereochemical factors and receptor interactions associated with narcotic analgesics. J. Pharmacol. Sci. 1966, 55, 865–887. [Google Scholar] [CrossRef]
- Janssen, P.A.H., Jr.; Schider, O.; Besendorf, L.; Pellmont, B. Diphenulpropylamines, morphinans. In Synthetic Analgesics Part I; Pergamon Press: New York, NY, USA, 1960. [Google Scholar]
- Janssen, P.A.H., Jr.; Schider, O.; Besendorf, L.; Pellmont, B. Diphenulpropylamine, morphinans. In Synthetic Analgesics Part II; Pergamon: New York, NY, USA, 1966. [Google Scholar]
- Jacobson, A.E.; Morrison, E.L.; Sargent, L.J. Analgesics. In Medicinal Chemistry (Part II); Burger, A., Ed.; Wiley Interscience: New York, NY, USA, 1970. [Google Scholar]
- Martin, W.R. Opioid antagonists. Pharmacol. Rev. 1967, 19, 463–521. [Google Scholar] [PubMed]
- Martin, W.R.; Eades, C.G.; Thompson, J.A.; Huppler, R.E.; Gilbert, P.E. The effects of morphine and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532. [Google Scholar] [PubMed]
- Hughes, J.; Smith, T.W.; Kosterlitz, H.W.; Fothergill, L.A.; Morgan, B.A.; Morris, H.R. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature 1975, 258, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W.; Goodman, R.; Snyder, S.H. An endogenous morphine like factor in mammalian brain. Life Sci. 1975, 16, 1765–1769. [Google Scholar] [CrossRef]
- Terenius, L.; Wahlstrom, A. Search for an endogenous ligand for the opiate receptor. Acta Physiol. Scand. 1975, 94, 74–81. [Google Scholar] [CrossRef]
- Pert, C.B.; Snyder, S.H. Properties of opiate-receptor binding in rat brain. Proc. Natl. Acad. Sci. USA 1973, 70, 2243–2247. [Google Scholar] [CrossRef] [Green Version]
- Pasternak, G.W.; Snyder, S.H. Opiate receptor binding: Enzymatic treatments and discrimination between agonists and antagonists. Mol. Pharmacol. 1975, 11, 735–744. [Google Scholar] [PubMed]
- Wilson, H.A.; Pasternak, G.W.; Snyder, S.H. Differentiation of opiate agonist and antagonist receptor binding by protein-modifying reagants. Nature 1975, 256, 448–450. [Google Scholar] [CrossRef]
- Snyder, S.H.; Maurty, S. Opiate Receptor Mechanisms; MIT Press: Boston, MA, USA, 1975. [Google Scholar]
- Li, C.H.; Chung, D.; Doneen, B.A. Isolation, characterization and opiate activity of beta-endorphin from human pituitary glands. Biochem. Biophys. Res. Commun. 1976, 72, 1542–1547. [Google Scholar] [CrossRef]
- Berezniuk, I.F.; John, L.D. Endogenous opioids. In The Opiate Receptors; Pasternak, G.W., Ed.; Springer: New York, NY, USA, 2011; pp. 93–120. [Google Scholar]
- Zhang, S.; Tong, Y.; Tian, M.; Dehaven, R.N.; Cortesburgos, L.; Mansson, E.; Simonin, F.; Kieffer, B.; Yu, L. Dynorphin A as a potential endogenous ligand for four members of the opioid receptor gene family. J. Pharmacol. Exp. Ther. 1998, 286, 136–141. [Google Scholar]
- Fricker, L.D.; Margolis, E.B.; Gomes, I.; Devi, L.A. Five decades of research on opioid peptides: current knowledge and unanswered questions. Mol. Pharmacol. 2020, 98, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Zadina, J.E.; Hackler, L.; Ge, L.J.; Kastin, A.J. A potent and selective endogenous agonist for the m-opiate receptor. Nature 1997, 386, 499–502. [Google Scholar] [CrossRef]
- Fichna, J.; Janecka, A.; Costentin, J.; Do Rego, J.C. The endomorphin system and its evolving neurophysiological role. Pharmacol. Rev. 2007, 59, 88–123. [Google Scholar] [CrossRef]
- Emery, M.A.; Akil, H. Endogenous opioids at the intersection of opioid addiction, pain, and depression: The search for a precision medicine approach. Annu. Rev. Neurosci. 2020, 43, 355–374. [Google Scholar] [CrossRef] [Green Version]
- Bodnar, R.J. Endogenous opiates and behavior: 2018. Peptides 2020, 132, 170348. [Google Scholar] [CrossRef] [PubMed]
- Gianoulakis, C. Endogenous opioids and addiction to alcohol and other drugs of abuse. Curr. Top. Med. Chem. 2009, 9, 999–1015. [Google Scholar] [CrossRef] [PubMed]
- Trigo, J.M.; Martin-Garcia, E.; Berrendero, F.; Robledo, P.; Maldonado, R. The endogenous opioid system: A common substrate in drug addiction. Drug Alcohol Depend. 2010, 108, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Pecina, M.; Karp, J.F.; Mathew, S.; Todtenkopf, M.S.; Ehrich, E.W.; Zubieta, J.K. Endogenous opioid system dysregulation in depression: Implications for new therapeutic approaches. Mol. Psychiatry 2019, 24, 576–587. [Google Scholar] [CrossRef]
- Charbogne, P.; Kieffer, B.L.; Befort, K. 15 years of genetic approaches in vivo for addiction research: Opioid receptor and peptide gene knockout in mouse models of drug abuse. Neuropharmacology 2014, 76 Pt B, 204–217. [Google Scholar] [CrossRef] [Green Version]
- Drolet, G.; Dumont, E.C.; Gosselin, I.; Kinkead, R.; Laforest, S.; Trottier, J.F. Role of endogenous opioid system in the regulation of the stress response. Prog. Neuropsychopharmacol. Biol. Psychiatry 2001, 25, 729–741. [Google Scholar] [CrossRef]
- Zimmer, A.; Valjent, E.; Konig, M.; Zimmer, A.M.; Robledo, P.; Hahn, H.; Valverde, O.; Maldonado, R. Absence of delta-9-tetrahydrocannabinol dysphoric effects in dynorphin-deficient mice. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 9499–9505. [Google Scholar] [CrossRef]
- Konig, M.; Zimmer, A.M.; Steiner, H.; Holmes, P.V.; Crawley, J.N.; Brownstein, M.J.; Zimmer, A. Pain responses, anxiety and aggression in mice deficient in pre-proenkephalin. Nature 1996, 383, 535–538. [Google Scholar] [CrossRef]
- Yaswen, L.; Diehl, N.; Brennan, M.B.; Hochgeschwender, U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat. Med. 1999, 5, 1066–1070. [Google Scholar] [CrossRef]
- Rubinstein, M.; Mogil, J.S.; Japon, M.; Chan, E.C.; Allen, R.G.; Low, M.J. Absence of opioid stress-induced analgesia in mice lacking beta-endorphin by site-directed mutagenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 3995–4000. [Google Scholar] [CrossRef] [Green Version]
- Appleyard, S.M.; Hayward, M.; Young, J.I.; Butler, A.A.; Cone, R.D.; Rubinstein, M.; Low, M.J. A role for the endogenous opioid beta-endorphin in energy homeostasis. Endocrinology 2003, 144, 1753–1760. [Google Scholar] [CrossRef] [Green Version]
- Mogil, J.S.; Kest, B.; Sadowski, B.; Belknap, J.K. Differential genetic mediation of sensitivity to morphine in genetic models of opiate antinociception: Influence of nociceptive assay. J. Pharmacol. Exp. Ther. 1996, 276, 532–544. [Google Scholar]
- Reith, M.E.A.; Sershen, H.; Vadasz, C.; Lajtha, A. Strain differences in opiate receptors in mouse brain. Eur. J. Pharmacol. 1981, 74, 377–380. [Google Scholar] [CrossRef]
- Baron, A.; Shuster, L.; Elefterhiou, B.E.; Bailey, D.W. Opiate receptors in mice: Genetic differences. Life Sci. 1975, 17, 633–640. [Google Scholar] [CrossRef]
- Pick, C.G.; Nejat, R.; Pasternak, G.W. Independent expression of two pharmacologically distinct supraspinal mu analgesic systems in genetically different mouse strains. J. Pharmacol. Exp. Ther. 1993, 2265, 166–171. [Google Scholar]
- Chang, A.; Emmel, D.W.; Rossi, G.C.; Pasternak, G.W. Methadone analgesia in morphine-insensitive CXBK mice. Eur. J. Pharmacol. 1998, 351, 189–191. [Google Scholar] [CrossRef]
- Pasternak, G.W.; Childers, S.R.; Snyder, S.H. Naloxazone, long-acting opiate antagonist: Effects in intact animals and on opiate receptor binding in vitro. J. Pharmacol. Exp. Ther. 1980, 214, 455–462. [Google Scholar] [PubMed]
- Pasternak, G.W.; Childers, S.R.; Snyder, S.H. Opiate analgesia: Evidence for mediation by a subpopulation of opiate receptors. Science 1980, 208, 514–516. [Google Scholar] [CrossRef]
- Hazum, E.; Chang, K.J.; Cuatrescasas, P.; Pasternak, G.W. Naloxazone irreversibility inhibits the high affinity binding of [124I]D-ala2-D-leu5-enkephalin. Life Sci. 1981, 29, 843–851. [Google Scholar]
- Hahn, E.F.; Carroll-Buatti, M.; Pasternak, G.W. Irreversible opiate agonists and antagonists: The 14-hydroxydihydromorphinone azines. J. Neurosci. 1982, 2, 572–576. [Google Scholar] [CrossRef] [Green Version]
- Pasternak, G.W. Multiple morphine and enkephalin receptors: Biochemical and pharmacological aspects. In Stress-Induced Analgesia; Kelly, D., Ed.; New York Academy of Sciences: New York, NY, USA, 1986; pp. 130–139. [Google Scholar]
- Pasternak, G.W. Incomplete cross tolerance and multiple mu opioid peptide receptors. Trends Pharmacol. Sci. 2001, 22, 67–70. [Google Scholar] [CrossRef]
- Christensen, C.B.; Jorgensen, L.N. Morphine-6-glucuronide has high affinity for the opioid receptor. Pharmacol. Toxicol. 1987, 60, 75–76. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, G.W.; Bodnar, R.J.; Clark, J.A.; Inturrisi, C.E. Morphine-6-glucuronide, a potent mu agonist. Life Sci. 1987, 41, 2845–2849. [Google Scholar] [CrossRef]
- Paul, D.; Standifer, K.M.; Inturrisi, C.E.; Pasternak, G.W. Pharmacological characterization of morphine-6b-glucuronide, a very potent morphine metabolite. J. Pharmacol. Exp. Ther. 1989, 251, 477–483. [Google Scholar]
- Schuller, A.G.P.; King, M.A.; Zhang, J.W.; Bolan, E.; Pan, Y.X.; Morgan, D.J.; Chang, A.; Czick, M.E.; Unterwald, E.M.; Pasternak, G.W.; et al. Retention of heroin and morphine-6 beta-glucuronide analgesia in a new line of mice lacking exon 1 of MOR-1. Nat. Neurosci. 1999, 2, 151–156. [Google Scholar] [CrossRef]
- Zhou, X.E.; He, Y.; de Waal, P.W.; Gao, X.; Kang, Y.; Van Eps, N.; Yin, Y.; Pal, K.; Goswami, D.; White, T.A.; et al. Identification of phosphorylation codes for arrestin recruitment by g protein-coupled receptors. Cell 2017, 170, 457–469.e413. [Google Scholar] [CrossRef] [Green Version]
- Bolan, E.A.; Pan, Y.X.; Pasternak, G.W. Functional analysis of MOR-1 splice variants of the mouse mu opioid receptor gene Oprm. Synapse 2004, 51, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, D.A.; Pan, L.; Xu, J.; Yu, R.; Xu, M.M.; Pasternak, G.W.; Pan, Y.X. Identification of three new alternatively spliced variants of the rat mu opioid receptor gene: Dissociation of affinity and efficacy. J. Neurochem. 2004, 91, 881–890. [Google Scholar] [CrossRef]
- Pan, Y.X.; Xu, J.; Bolan, E.; Moskowitz, H.S.; Xu, M.; Pasternak, G.W. Identification of four novel exon 5 splice variants of the mouse mu-opioid receptor gene: Functional consequences of C-terminal splicing. Mol. Pharmacol. 2005, 68, 866–875. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Xu, J.; Yu, R.; Xu, M.M.; Pan, Y.X.; Pasternak, G.W. Identification and characterization of six new alternatively spliced variants of the human mu opioid receptor gene, Oprm. Neuroscience 2005, 133, 209–220. [Google Scholar] [CrossRef]
- Xu, J.; Xu, M.; Bolan, E.; Gilbert, A.K.; Pasternak, G.W.; Pan, Y.X. Isolating and characterizing three alternatively spliced mu opioid receptor variants: mMOR-1A, mMOR-1O, and mMOR-1P. Synapse 2014, 68, 144–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.X.; Xu, J.; Bolan, E.; Chang, A.; Mahurter, L.; Rossi, G.; Pasternak, G.W. Isolation and expression of a novel alternatively spliced mu opioid receptor isoform, MOR-1F. FEBS Letters 2000, 466, 337–340. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.X.; Xu, J.; Mahurter, L.; Xu, M.; Gilbert, A.K.; Pasternak, G.W. Identification and characterization of two new human mu opioid receptor splice variants, hMOR-1O and hMOR-1X. Biochem. Biophys. Res. Commun. 2003, 301, 1057–1061. [Google Scholar] [CrossRef]
- Xu, J.; Lu, Z.; Narayan, A.; Le Rouzic, V.P.; Xu, M.; Hunkele, A.; Brown, T.G.; Hoefer, W.F.; Rossi, G.C.; Rice, R.C.; et al. Alternatively spliced mu opioid receptor C termini impact the diverse actions of morphine. J. Clin. Investig. 2017, 127, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Narayan, A.; Hunkele, A.; Xu, J.; Bassoni, D.L.; Pasternak, G.W.; Pan, Y.X. Mu opioids induce biased signaling at the full-length seven transmembrane C-terminal splice variants of the mu opioid receptor gene, Oprm1. Cell Mol. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Koch, T.; Schulz, S.; Schroder, H.; Wolf, R.; Raulf, E.; Hollt, V. Carboxyl-terminal splicing of the rat mu opioid receptor modulates agonist-mediated internalization and receptor resensitization. J. Biol. Chem. 1998, 273, 13652–13657. [Google Scholar] [CrossRef] [Green Version]
- Abbadie, C.; Pasternak, G.W. Differential in vivo internalization of MOR-1 and MOR-1C by morphine. Neuroreport 2001, 12, 3069–3072. [Google Scholar] [CrossRef]
- Tanowitz, M.; Hislop, J.N.; von Zastrow, M. Alternative splicing determines the post-endocytic sorting fate of G-protein-coupled receptors. J. Biol. Chem. 2008, 283, 35614–35621. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Faskowitz, A.J.; Rossi, G.C.; Xu, M.; Lu, Z.; Pan, Y.X.; Pasternak, G.W. Stabilization of morphine tolerance with long-term dosing: Association with selective upregulation of mu-opioid receptor splice variant mRNAs. Proc. Natl. Acad. Sci. USA 2015, 112, 279–284. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lu, Z.; Xu, M.; Rossi, G.C.; Kest, B.; Waxman, A.R.; Pasternak, G.W.; Pan, Y.X. Differential expressions of the alternatively spliced variant mRNAs of the micro opioid receptor gene, OPRM1, in brain regions of four inbred mouse strains. PLoS ONE 2014, 9, e111267. [Google Scholar] [CrossRef] [Green Version]
- Abbadie, C.; Pan, Y.X.; Pasternak, G.W. Differential distribution in rat brain of mu opioid receptor carboxy terminal splice variants MOR-1C-like and MOR-1-like immunoreactivity: Evidence for region-specific processing. J. Comp. Neurol. 2000, 419, 244–256. [Google Scholar] [CrossRef]
- Abbadie, C.; Pan, Y.-X.; Drake, C.T.; Pasternak, G.W. Comparative immunhistochemical distributions of carboxy terminus epitopes from the mu opioid receptor splice variants MOR-1D, MOR-1 and MOR-1C in the mouse and rat central nervous systems. Neuroscience 2000, 100, 141–153. [Google Scholar] [CrossRef]
- Brown, T.G.; Xu, J.; Hurd, Y.L.; Pan, Y.X. Dysregulated expression of the alternatively spliced variant mRNAs of the mu opioid receptor gene, OPRM1, in the medial prefrontal cortex of male human heroin abusers and heroin self-administering male rats. J. Neurosci. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dever, S.M.; Xu, R.; Fitting, S.; Knapp, P.E.; Hauser, K.F. Differential expression and HIV-1 regulation of mu-opioid receptor splice variants across human central nervous system cell types. J. Neurovirol. 2012, 18, 181–190. [Google Scholar] [CrossRef]
- Dever, S.M.; Costin, B.N.; Xu, R.; El-Hage, N.; Balinang, J.; Samoshkin, A.; O’Brien, M.A.; McRae, M.; Diatchenko, L.; Knapp, P.E.; et al. Differential expression of the alternatively spliced OPRM1 isoform mu-opioid receptor-1K in HIV-infected individuals. AIDS 2014, 28, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.Y.; Liu, Z.C.; Sun, Y.G.; Ross, M.; Kim, S.; Tsai, F.F.; Li, Q.F.; Jeffry, J.; Kim, J.Y.; Loh, H.H.; et al. Unidirectional cross-activation of GRPR by MOR1D uncouples itch and analgesia induced by opioids. Cell 2011, 147, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.Y.; Ginosar, Y.; Yazdi, J.; Hincker, A.; Chen, Z.F. Cross-talk between human spinal cord mu-opioid receptor 1y isoform and gastrin-releasing peptide receptor mediates opioid-induced scratching behavior. Anesthesiology 2019, 131, 381–391. [Google Scholar] [CrossRef]
- Pan, Y.X.; Xu, J.; Xu, M.; Rossi, G.C.; Matulonis, J.E.; Pasternak, G.W. Involvement of exon 11-associated variants of the mu opioid receptor MOR-1 in heroin, but not morphine, actions. Proc. Natl. Acad. Sci. USA 2009, 106, 4917–4922. [Google Scholar] [CrossRef] [Green Version]
- Grinnell, S.G.; Ansonoff, M.; Marrone, G.F.; Lu, Z.; Narayan, A.; Xu, J.; Rossi, G.; Majumdar, S.; Pan, Y.X.; Bassoni, D.L.; et al. Mediation of buprenorphine analgesia by a combination of traditional and truncated mu opioid receptor splice variants. Synapse 2016, 70, 395–407. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, S.; Grinnell, S.; Le, R.V.; Burgman, M.; Polikar, L.; Ansonoff, M.; Pintar, J.; Pan, Y.X.; Pasternak, G.W. Truncated G protein-coupled mu opioid receptor MOR-1 splice variants are targets for highly potent opioid analgesics lacking side effects. Proc. Natl. Acad. Sci. USA 2011, 108, 19778–19783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieskopf, J.S.; Pan, Y.X.; Marcovitz, J.; Tuttle, A.H.; Majumdar, S.; Pidakala, J.; Pasternak, G.W.; Mogil, J.S. Broad-spectrum analgesic efficacy of IBNtxA is mediated by exon 11-associated splice variants of the mu-opioid receptor gene. Pain 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Xu, M.; Brown, T.; Rossi, G.C.; Hurd, Y.L.; Inturrisi, C.E.; Pasternak, G.W.; Pan, Y.X. Stabilization of the mu-opioid receptor by truncated single transmembrane splice variants through a chaperone-like action. J. Biol. Chem. 2013, 288, 21211–21227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Xu, J.; Pan, Y.X. A truncated six transmembrane splice variant MOR-1g enhances expression of the full-length seven transmembrane mu-opioid receptor through heterodimerization. Mol. Pharmacol. 2020, 98, 518–527. [Google Scholar] [CrossRef]
- Pan, Y.X.; Xu, J.; Bolan, E.; Abbadie, C.; Chang, A.; Zuckerman, A.; Rossi, G.; Pasternak, G.W. Identification and characterization of three new alternatively spliced mu-opioid receptor isoforms. Mol. Pharmacol. 1999, 56, 396–403. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the mu-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.J.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into mu-opioid receptor activation. Nature 2015, 524, 315. [Google Scholar] [CrossRef] [Green Version]
- Sim, L.J.; Selley, D.E.; Childers, S.R. In vitro autoradiography of receptor-activated G proteins in rat brain by agonist-stimulated guanylyl 5′-[gamma-[35S]thio]triphosphate binding. Proc. Natl. Acad. Sci. USA 1995, 92, 7242–7246. [Google Scholar] [CrossRef] [Green Version]
- Childers, S.R. Opioid receptor-coupled second messenger systems. Life Sci. 1991, 48, 1991–2003. [Google Scholar] [CrossRef]
- Abbadie, C.; Gultekin, S.H.; Pasternak, G.W. Immunohistochemical localization of the carboxy terminus of the novel mu opioid receptor splice variant MOR-1C within the human spinal cord. Neuroreport 2000, 11, 1953–1957. [Google Scholar] [CrossRef]
- Smith, J.S.; Lefkowitz, R.J.; Rajagopal, S. Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. [Google Scholar] [CrossRef]
- Raehal, K.M.; Schmid, C.L.; Groer, C.E.; Bohn, L.M. Functional selectivity at the mu-opioid receptor: Implications for understanding opioid analgesia and tolerance. Pharmacol. Rev. 2011, 63, 1001–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, C.L.; Kennedy, N.M.; Ross, N.C.; Lovell, K.M.; Yue, Z.Z.; Morgenweck, J.; Cameron, M.D.; Bannister, T.D.; Bohn, L.M. Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 2017, 171, 1165. [Google Scholar] [CrossRef] [Green Version]
- Grim, T.W.; Acevedo-Canabal, A.; Bohn, L.M. Toward directing opioid receptor signaling to refine opioid therapeutics. Biol. Psychiatry 2020, 87, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnaturi, R.; Chiechio, S.; Salerno, L.; Rescifina, A.; Pittala, V.; Cantarella, G.; Tomarchio, E.; Parenti, C.; Pasquinucci, L. Progress in the development of more effective and safer analgesics for pain management. Eur. J. Med. Chem. 2019, 183, 111701. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Jones, A.; Olson, K.R.; Peng, K.; Wehrman, T.; Park, A.; Mallari, R.; Nebalasca, D.; Young, S.W.; Xiao, S.H. A homogeneous enzyme fragment complementation-based beta-arrestin translocation assay for high-throughput screening of G-protein-coupled receptors. J. Biomol. Screen. 2008, 13, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Kroeze, W.K.; Sassano, M.F.; Huang, X.P.; Lansu, K.; McCorvy, J.D.; Giguere, P.M.; Sciaky, N.; Roth, B.L. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 2015, 22, 362–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, L.; Parent, S.; Caron, M.; Legault, M.; Joly, E.; Angers, S.; Bouvier, M.; Brown, M.; Houle, B.; Menard, L. The BRET2/arrestin assay in stable recombinant cells: A platform to screen for compounds that interact with G protein-coupled receptors (GPCRS). J. Recept. Signal. Transduct. Res. 2002, 22, 533–541. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Audet, M.; Garneau, P.; Pelletier, J.; Bouvier, M. High-throughput screening of G protein-coupled receptor antagonists using a bioluminescence resonance energy transfer 1-based beta-arrestin2 recruitment assay. J. Biomol. Screen. 2005, 10, 463–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barak, L.S.; Ferguson, S.S.; Zhang, J.; Caron, M.G. A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J. Biol. Chem. 1997, 272, 27497–27500. [Google Scholar] [CrossRef] [Green Version]
- Dixon, A.S.; Schwinn, M.K.; Hall, M.P.; Zimmerman, K.; Otto, P.; Lubben, T.H.; Butler, B.L.; Binkowski, B.F.; Machleidt, T.; Kirkland, T.A.; et al. NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem. Biol. 2016, 11, 400–408. [Google Scholar] [CrossRef]
- Kenakin, T.; Watson, C.; Muniz-Medina, V.; Christopoulos, A.; Novick, S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 2012, 3, 193–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Westhuizen, E.T.; Breton, B.; Christopoulos, A.; Bouvier, M. Quantification of ligand bias for clinically relevant beta2-adrenergic receptor ligands: Implications for drug taxonomy. Mol. Pharmacol. 2014, 85, 492–509. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Precursor | Opioid Peptide | Copies of Peptide | Structure | Other Peptides |
|---|---|---|---|---|
| Proenkephalin (PENK) | [Leu]5enkephalin | 1 | Tyr-Gly-Gly-Phe-Leu | Synenkephalin |
| [Met]5enkephalin | 4 | Tyr-Gly-Gly-Phe-Met | ||
| [Met]5enkephalin-Arg6-Gly7-Leu8(Octapeptide) | 1 | Tyr-Gly-Gly-Phe-Met-Arg-Gly-Leu | ||
| [Met]5enkephalin-Arg6-Phe7 (Heptapeptide) | 1 | Tyr-Gly-Gly-Phe-Met-Arg-Ph | ||
| Prodynorphin (PDYN) | Dynorphin A1-17 | 1 | Tyr-Gly-Gly-Phe-Leu-Arg-Arg-Ile-Arg-Pro-Lys-Leu-Lys-Trp-Asp-Asn-Gln | α-neoendorphin, β-neoendorphin, Big dynorphin, Leumorphin |
| Dynorphin B1-13 | 1 | Tyr-Gly-Gly-Phe-Leu-Arg-Arg-Gln-Phe-Lys-Val-Val-Thr | ||
| Pro-opiomelanocortin (POMC) | βh-Endorphin1-31 | 1 | Tyr-Gly-Gly-Phe-Met-Thr-Ser-Glu-Lys-Ser-Gln-Thr-Pro-Leu-Val-Thr-Leu-Phe-Lys-Asn-Ala-Ile-Ile-Lys-Asn-Ala-Tyr-Lys-Lys-Gly-Glu | γ-MSH, ACTH, α-MSH, CLIP, β-LPH, γ-LPH, β-MSH |
| Unknown | Endomorphin-1 | Tyr-Pro-Trp-Phe-NH2 | ||
| Endomorphin-2 | Tyr-Pro-Phe-Phe-NH2 |
| Ligand | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ki Value (nM) | DAMGO | Morphine | Fentanyl | Methadone | M6G | β-Endorphin | Dynorphin A | Endomorphin 1 | Endomorphin 2 | [Met]5Enkephalin-Arg6-Phe7 | Refs. |
| Mouse | |||||||||||
| mMOR-1 | 1.8 ± 0.5 | 5.3 ± 2.0 | 2.3 ± 1.0 | 1.4 ± 0.1 | 5.2 ± 1.8 | 11 ± 2.9 | 11 ± 0.5 | 2.1 ± 0.8 | 4.2 ± 1.8 | 4.1 ± 1.0 | [73,100] |
| mMOR-1A | 1.0 ± 0.3 | 3.1 ± 0.5 | 1.5 ± 0.6 | 0.7 ± 0.1 | 5.0 ± 1.5 | 4.3 ± 1.0 | 8.2 ± 2.8 | 3.5 ± 1.3 | [73,77] | ||
| mMOR-1C | 0.93 ± 0.2 | 2.4 ± 0.6 | 1.2 ± 0.4 | 0.5 ± 0.1 | 4.1 ± 1.2 | 5.8 ± 0.5 | 5.6 ± 0.8 | 1.4 ± 0.4 | 1.6 ± 0.2 | 2.1 ± 0.7 | [73,100] |
| mMOR-1D | 0.71 ± 0.1 | 1.5 ± 0.2 | 3.3 ± 1.5 | 1.4 ± 0.1 | 4.8 ± 0.8 | 1.7 ± 0.4 | 2.2 ± 0.8 | 1.8 ± 0.3 | 2.0 ± 0.3 | 3.7 ± 1.2 | [73,100] |
| mMOR-1E | 1.2 ± 0.5 | 2.3 ± 0.4 | 1.2 ± 0.5 | 0.7 ± 0.3 | 5.6 ± 0.7 | 5.0 ± 1.2 | 8.9 ± 1.1 | 2.4 ± 0.1 | 4.4 ± 0.8 | 4.4 ± 0.9 | [73,100] |
| mMOR-1B1 | 1.4 ± 0.2 | 5.3 ± 1.0 | 10 ± 1.6 | 6.8 ± 3.2 | 15 ± 7.1 | 11 ± 5.6 | 12 ± 1.5 | [75] | |||
| mMOR-1B2 | 1.3 ± 0.1 | 3.9 ± 0.4 | 8.4 ± 1.3 | 4.9 ± 1.7 | 34 ± 18 | 5.0 ± 1.8 | 8.4 ± 1.1 | [75] | |||
| mMOR-1B3 | 1.8 ± 0.9 | 1.5 ± 0.5 | 3.9 ± 1.3 | 3.1 ± 1.4 | 8.7 ± 1.8 | 3.2 ± 0.6 | 3.2 ± 0.8 | [75] | |||
| mMOR-1B5 | 1.0 ± 0.3 | 1.4 ± 0.6 | 5.2 ± 0.1 | 5.7 ± 1.2 | 8.9 ± 2.3 | 4.3 ± 0.8 | 11 ± 1.8 | [75] | |||
| mMOR-1F | 1.1 ± 0.2 | 2.9 ± 0.5 | 1.7 ± 0.5 | 1.3 ± 0.2 | 9.6 ± 0.8 | 6.0 ± 1.6 | 12 ± 1.0 | 2.9 ± 0.5 | 4.1 ± 1.3 | 3.9 ± 0.8 | [73,78] |
| mMOR-1O | 3.3 ± 1.2 | 2.7 ± 0.6 | 17 ± 1.0 | 16 ± 5.3 | 58 ± 26 | [77] | |||||
| mMOR-1P | 0.8 ± 0.3 | 1.2 ± 0.8 | 11 ± 3.4 | 5.9 ± 2.4 | 103 ± 23 | [77] | |||||
| Rat | |||||||||||
| rMOR-1 | 3.3 ± 0.6 | 5.6 ± 0.8 | 17 ± 2.2 | 3.7 ± 0.4 | 12 ± 3.0 | 4.1 ± 0.7 | 8.0 ± 2.0 | [74] | |||
| rMOR-1A | 6.0 ± 0.9 | 8.0 ± 0.4 | 26 ± 2.1 | 11 ± 0.6 | 23 ± 1.6 | 6.5 ± 0.3 | 12 ± 0.6 | [74] | |||
| rMOR-1C1 | 4.5 ± 0.9 | 7.4 ± 0.3 | 25 ± 2.4 | 8.8 ± 0.5 | 13 ± 2.3 | 3.9 ± 0.1 | 10 ± 0.6 | [74] | |||
| rMOR-1D | 4.7 ± 1.2 | 7.4 ± 0.5 | 21 ± 1.8 | 8.5 ± 0.6 | 11 ± 1.7 | 3.9 ± 0.4 | 7.5 ± 0.4 | [74] | |||
| Human | |||||||||||
| hMOR-1 | 1.2 ± 0.2 | 2.2 ± 0.9 | 10 ± 0.3 | 15 ± 11.0 | 87 ± 14 | 4.2 ± 1.4 | 15 ± 7.1 | [76] | |||
| hMOR-1B1 | 1.2 ± 0.4 | 2.4 ± 1.1 | 5.0 ± 0.2 | 7.8 ± 1.5 | 19 ± 6.6 | 3.8 ± 0.8 | 5.4 ± 0.6 | [76] | |||
| hMOR-1B2 | 5.2 ± 1.4 | 11 ± 3.5 | 42 ± 7.9 | 25 ± 5.1 | 49 ± 22 | 12 ± 0.1 | 20 ± 1.3 | [76] | |||
| hMOR-1B3 | 1.8 ± 0.5 | 3.2 ± 0.6 | 16 ± 1.2 | 8.2 ± 2.2 | 14 ± 2.3 | 4.9 ± 1.5 | 6.3 ± 1.5 | [76] | |||
| hMOR-1B4 | 2.3 ± 0.6 | 5.5 ± 1.7 | 23 ± 7.4 | 16 ± 0.4 | 71 ± 30 | 9.9 ± 2.3 | 23 ± 2.0 | [76] | |||
| hMOR-1B5 | 2.1 ± 0.4 | 3.9 ± 0.9 | 12 ± 2.6 | 10 ± 3.4 | 53 ± 23 | 4.6 ± 0.3 | 9.6 ± 3.0 | [76] | |||
| hMOR-1O | 2.2 ± 0.6 | 2.0 ± 0.7 | 16 ± 2.6 | 25 ± 8.5 | [79] | ||||||
| hMOR-1X | 2.1 ± 0.2 | 2.7 ± 1.0 | 17 ± 5.3 | 187 ± 27 | [79] | ||||||
| hMOR-1Y | 2.5 ± 0.8 | 4.3 ± 1.7 | 8.3 ± 2.2 | 8.4 ± 1.8 | 25 ± 13 | 5.1 ± 1.1 | 9.4 ± 3.0 | [76] | |||
| Ligand | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DAMGO | Morphine | β-Endorphin | Dynorphin A | Endomorphin 1 | Endomorphin 2 | [Met]5Enkephalin-Arg6-Phe7 | Ref. | ||||||||
| EC50 (nM) | %Max | EC50 (nM) | %Max | EC50 (nM) | %Max | EC50 (nM) | %Max | EC50 (nM) | %Max | EC50 (nM) | %Max | EC50 (nM) | %Max | ||
| Mouse | |||||||||||||||
| mMOR-1 | 68 ± 4 | 100 | 23 ± 2 | 102 ± 5 | 64 ± 7 | 97 ± 2 | 34 ± 9 | 109 ± 7 | 26 ± 4 | 98 ± 8 | 72 ± 11 | 124 ± 8 | 53 ± 3 | 118 ± 15 | [73] |
| mMOR-1A | 70 ± 3 | 100 | 19 ± 4 | 91 ± 2 | 111 ± 27 | 83 ± 3 | 150 ± 36 | 73 ± 6 | 42 ± 13 | 69 ± 2 | 97 ± 28 | 76 ± 3 | 133 ± 9 | 75 ± 4 | [73] |
| mMOR-1C | 62 ± 4 | 100 | 23 ± 5 | 75 ± 4 | 123 ± 19 | 44 ± 3 | 140 ± 19 | 76 ± 10 | 83 ± 20 | 68 ± 15 | 122 ± 46 | 62 ± 15 | 60 ± 17 | 51 ± 2 | [73] |
| mMOR-1D | 62 ± 6 | 100 | 82 ± 34 | 99 ± 3 | 73 ± 18 | 105 ± 6 | 100 ± 41 | 102 ± 6 | 47 ± 21 | 94 ± 8 | 137 ± 24 | 92 ± 5 | 170 ± 16 | 94 ± 3 | [73] |
| mMOR-1E | 48 ± 4 | 100 | 41 ± 13 | 116 ± 4 | 113 ± 25 | 130 ± 3 | 113 ± 9 | 129 ± 9 | 80 ± 4 | 85 ± 9 | 52 ± 26 | 86 ± 8 | 131 ± 19 | 94 ± 10 | [73] |
| mMOR-1B1 | 39 ± 8 | 100 | 100 ± 38 | 104 ± 38 | 113 ± 47 | 69 ± 21 | 137 ± 69 | 83 ± 23 | 57 ± 23 | 68 ± 19 | 197 ± 95 | 90 ± 0 | [75] | ||
| mMOR-1B2 | 85 ± 18 | 100 | 76 ± 13 | 82 ± 8 | 163 ± 22 | 84 ± 5 | 210 ± 25 | 81 ± 6 | 126 ± 29 | 82 ± 8 | 187 ± 23 | 92 ± 4 | [75] | ||
| mMOR-1B3 | 100 ± 14 | 100 | 51 ± 6 | 91 ± 3 | 75 ± 19 | 93 ± 2 | 147 ± 56 | 90 ± 6 | 99 ± 1 | 97 ± 2 | 110 ± 6 | 80 ± 3 | [75] | ||
| mMOR-1B5 | 89 ± 13 | 100 | 53 ± 4 | 87 ± 7 | 83 ± 27 | 80 ± 4 | 197 ± 32 | 75 ± 3 | 89 ± 13 | 86 ± 7 | 155 ± 8 | 82 ± 4 | [75] | ||
| mMOR-1F | 50 ± 6 | 100 | 44 ± 17 | 76 ± 13 | 26 ± 6 | 74 ± 7 | 40 ± 8 | 73 ± 3 | 44 ± 18 | 113 ± 5 | 68 ± 18 | 107 ± 4 | 29 ± 9 | 94 ± 16 | [73] |
| mMOR-1O | 60 ± 19 | 100 | 85 ± 31 | 66 ± 23 | 6 ± 1 | 141 ± 8 | [77] | ||||||||
| mMOR-1P | 133 ± 23 | 100 | 58 ± 9 | 115 ± 23 | 24 ± 5 | 55 ± 3 | [77] | ||||||||
| Rat | |||||||||||||||
| rMOR-1 | 12 ± 3 | 100 | 4 ± 2 | 105.58 | 14 ± 4 | 137.34 | [74] | ||||||||
| rMOR-1A | 13 ± 5 | 100 | 13 ± 5 | 100.57 | 15 ± 3 | 116.48 | [74] | ||||||||
| rMOR-1C1 | 74 ± 22 | 100 | 48 ± 4 | 154.94 | 54 ± 8 | 161.80 | [74] | ||||||||
| rMOR-1D | 125 ± 26 | 100 | 91 ± 14 | 146.02 | 100 ± 26 | 128.32 | [74] | ||||||||
| Human | |||||||||||||||
| hMOR-1 | 120 ± 17 | 100 | 21 ± 4 | 97.57 | 4 ± 1 | 68.75 | 296 ± 16 | 36.46 | [76] | ||||||
| hMOR-1A | 161 ± 21 | 100 | 30 ± 2 | 121.31 | 8 ± 2 | 71.31 | 36 ± 1 | 63.93 | [76] | ||||||
| hMOR-1B1 | 255 ± 46 | 100 | 41 ± 5 | 64.41 | 25 ± 6 | 57.97 | 63 ± 17 | 50.51 | [76] | ||||||
| hMOR-1B2 | 1028 ± 68 | 100 | 77 ± 9 | 80.00 | 73 ± 10 | 97.84 | 292 ± 66 | 97.84 | [76] | ||||||
| hMOR-1B3 | 549 ± 86 | 100 | 86 ± 19 | 65.44 | 33 ± 11 | 61.78 | 98 ± 27 | 39.38 | [76] | ||||||
| hMOR-1B4 | 341 ± 65 | 100 | 38 ± 5 | 71.68 | 19 ± 2 | 65.32 | 58 ± 14 | 40.75 | [76] | ||||||
| hMOR-1B5 | 936 ± 233 | 100 | 90 ± 18 | 61.46 | 55 ± 2 | 92.01 | 158 ± 15 | 81.60 | [76] | ||||||
| hMOR-1Y | 571 ± 255 | 100 | 100 ± 20 | 88.05 | 43 ± 3 | 73.18 | 100 ± 21 | 77.26 | [76] | ||||||
| Ligand | ||||||||
|---|---|---|---|---|---|---|---|---|
| DAMGO | Morphine | β-Endorphin | Dynorphin A | Endomorphin-1 | Endomorphin-2 | [Met]5Enkephalin-Arg6-Phe7 | Refs. | |
| EC50/Ki | EC50/Ki | EC50/Ki | EC50/Ki | EC50/Ki | EC50/Ki | EC50/Ki | ||
| Mouse | ||||||||
| mMOR-1 | 38 | 4 | 6 | 3 | 12 | 17 | 13 | [73,100] |
| mMOR-1A | 70 | 6 | 26 | 18 | 38 | [73,77] | ||
| mMOR-1C | 67 | 10 | 21 | 25 | 59 | 76 | 29 | [73,100] |
| mMOR-1D | 87 | 55 | 43 | 45 | 26 | 69 | 46 | [73,100] |
| mMOR-1E | 40 | 18 | 23 | 13 | 33 | 12 | 30 | [73,100] |
| mMOR-1B1 | 28 | 19 | 17 | 9 | 5 | 16 | [75] | |
| mMOR-1B2 | 65 | 19 | 33 | 6 | 25 | 22 | [75] | |
| mMOR-1B3 | 56 | 34 | 24 | 17 | 31 | 34 | [75] | |
| mMOR-1B5 | 89 | 38 | 15 | 22 | 21 | 15 | [75] | |
| mMOR-1F | 45 | 15 | 4 | 3 | 15 | 17 | 7 | [73,78] |
| mMOR-1O | 18 | 31 | 0.4 | [77] | ||||
| mMOR-1P | 166 | 48 | 4 | [77] | ||||
| Rat | ||||||||
| rMOR-1 | 4 | 1 | 3 | [74] | ||||
| rMOR-1A | 2 | 1 | 2 | [74] | ||||
| rMOR-1C1 | 16 | 5 | 14 | [74] | ||||
| rMOR-1D | 27 | 11 | 26 | [74] | ||||
| Human | ||||||||
| hMOR-1 | 100 | 10 | 0.3 | 3 | [76,79] | |||
| hMOR-1B1 | 213 | 17 | 3 | 3 | [76] | |||
| hMOR-1B2 | 198 | 7 | 3 | 6 | [76] | |||
| hMOR-1B3 | 305 | 27 | 4 | 7 | [76] | |||
| hMOR-1B4 | 148 | 7 | 1 | 0.8 | [76] | |||
| hMOR-1B5 | 4 | 23 | 6 | 3 | [76] | |||
| hMOR-1Y | 228 | 23 | 5 | 4 | [76] | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abrimian, A.; Kraft, T.; Pan, Y.-X. Endogenous Opioid Peptides and Alternatively Spliced Mu Opioid Receptor Seven Transmembrane Carboxyl-Terminal Variants. Int. J. Mol. Sci. 2021, 22, 3779. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073779
Abrimian A, Kraft T, Pan Y-X. Endogenous Opioid Peptides and Alternatively Spliced Mu Opioid Receptor Seven Transmembrane Carboxyl-Terminal Variants. International Journal of Molecular Sciences. 2021; 22(7):3779. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073779
Chicago/Turabian StyleAbrimian, Anna, Tamar Kraft, and Ying-Xian Pan. 2021. "Endogenous Opioid Peptides and Alternatively Spliced Mu Opioid Receptor Seven Transmembrane Carboxyl-Terminal Variants" International Journal of Molecular Sciences 22, no. 7: 3779. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22073779