
Role of α2-Adrenoceptor Subtypes in Suppression of L-Type Ca2+ Current in Mouse Cardiac Myocytes

 , ,
, ,
Abstract
:1. Introduction
2. Results
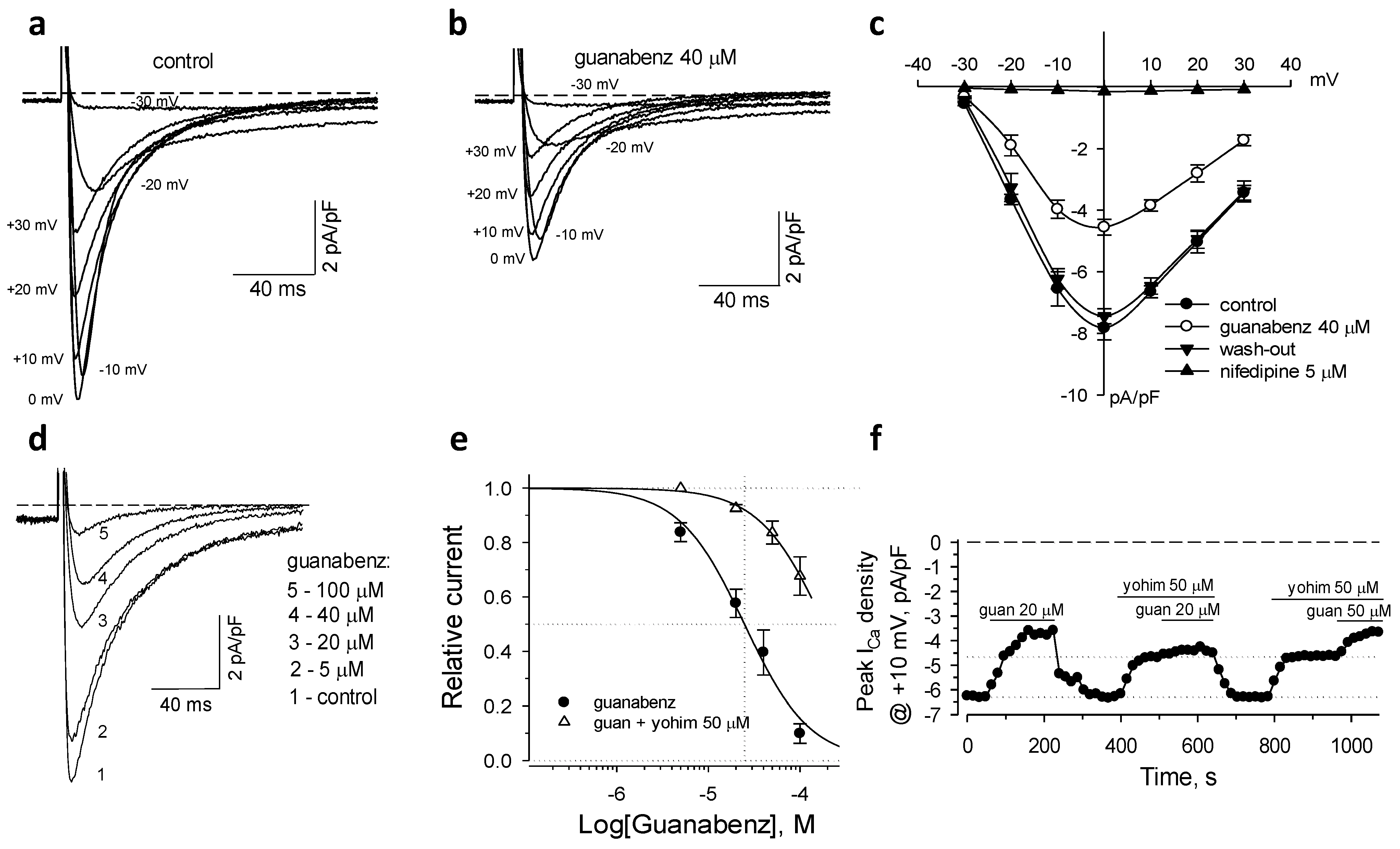
2.1. Activation of α2-ARs Inhibits L-Type Ca2+ Current
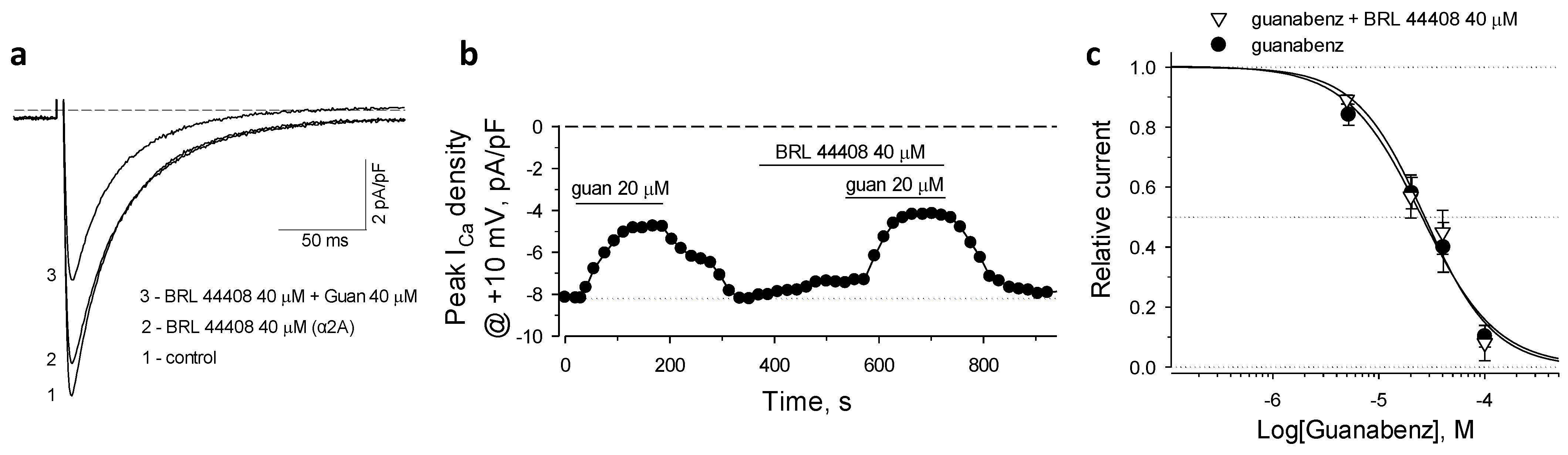
2.2. α2A-AR in Guanabenz-Induced Suppression of ICaL
2.3. α2B-AR in Guanabenz-Induced Suppression of ICaL
2.4. α2C-AR Mediates Guanabenz-Induced Suppression of ICaL
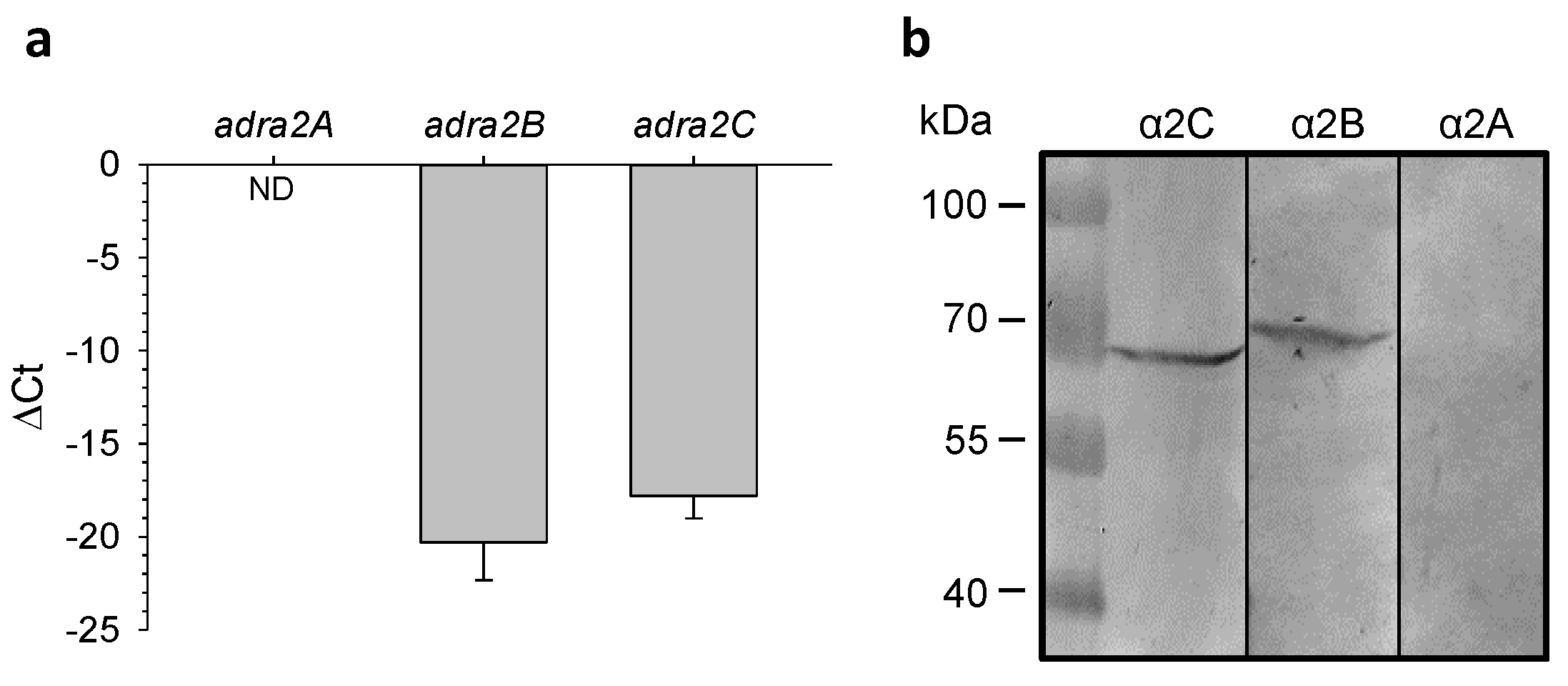
2.5. Expression of α2-AR Genes in Mouse Hearts
3. Discussion
4. Materials and Methods
4.1. Cell Isolation
4.2. Electrophysiology
4.3. RNA Isolation and RT-qPCR Assay
4.4. Western Blot
4.5. Drugs
4.6. Data Analysis and Presentation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alekseev, A.E.; Park, S.; Pimenov, O.Y.; Reyes, S.; Terzic, A. Sarcolemmal α2-Adrenoceptors in Feedback Control of Myocardial Response to Sympathetic Challenge. Pharmacol. Ther. 2019, 197, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Kokoz, Y.M.; Evdokimovskii, E.V.; Maltsev, A.V.; Nenov, M.N.; Nakipova, O.V.; Averin, A.S.; Pimenov, O.Y.; Teplov, I.Y.; Berezhnov, A.V.; Reyes, S.; et al. Sarcolemmal α2-Adrenoceptors Control Protective Cardiomyocyte-Delimited Sympathoadrenal Response. J. Mol. Cell. Cardiol. 2016, 100, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Averin, A.S.; Nakipova, O.V.; Kosarsky, L.S.; Pimenov, O.Y.; Galimova, M.H.; Nenov, M.N.; Berejnov, A.V.; Alekseev, A.E. Activation of Sarcolemmal α2 Adrenoceptors Supports Ca2+ Homeostasis and Prevents Ventricular Arrhythmia under Sympathetic Stress. Biophysics 2019, 64, 793–798. [Google Scholar] [CrossRef]
- Reyes, S.; Varagic, J.; VonCannon, J.; Cheng, C.P.; Ferrario, C.M. Novel Action of Cardiomyocyte α2-Adrenergic Receptors in Reversing Angiotensin II Mediated Cardiac Hypertrophy. Circulation 2018, 138 (Suppl. S1), A16308. [Google Scholar]
- Lomasney, J.W.; Lorenz, W.; Allen, L.F.; King, K.; Regan, J.W.; Yang-Feng, T.L.; Caron, M.G.; Lefkowitz, R.J. Expansion of the Alpha 2-Adrenergic Receptor Family: Cloning and Characterization of a Human Alpha 2-Adrenergic Receptor Subtype, the Gene for Which Is Located on Chromosome 2. Proc. Natl. Acad. Sci. USA 1990, 87, 5094–5098. [Google Scholar] [CrossRef] [Green Version]
- Bylund, D.B.; Blaxall, H.S.; Iversen, L.J.; Caron, M.G.; Lefkowitz, R.J.; Lomasney, J.W. Pharmacological Characteristics of Alpha 2-Adrenergic Receptors: Comparison of Pharmacologically Defined Subtypes with Subtypes Identified by Molecular Cloning. Mol. Pharmacol. 1992, 42, 1–5. [Google Scholar]
- MacDonald, E.; Kobilka, B.K.; Scheinin, M. Gene Targeting—Homing in on α2-Adrenoceptor-Subtype Function. Trends Pharmacol. Sci. 1997, 18, 211–219. [Google Scholar] [CrossRef]
- Ruuskanen, J.O.; Xhaard, H.; Marjamäki, A.; Salaneck, E.; Salminen, T.; Yan, Y.-L.; Postlethwait, J.H.; Johnson, M.S.; Larhammar, D.; Scheinin, M. Identification of Duplicated Fourth α2-Adrenergic Receptor Subtype by Cloning and Mapping of Five Receptor Genes in Zebrafish. Mol. Biol. Evol. 2004, 21, 14–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruuskanen, J.O.; Laurila, J.; Xhaard, H.; Rantanen, V.-V.; Vuoriluoto, K.; Wurster, S.; Marjamäki, A.; Vainio, M.; Johnson, M.S.; Scheinin, M. Conserved Structural, Pharmacological and Functional Properties among the Three Human and Five Zebrafish α2-Adrenoceptors: Human and Fish α2-Receptor Comparative Pharmacology. Br. J. Pharmacol. 2005, 144, 165–177. [Google Scholar] [CrossRef] [Green Version]
- Céspedes, H.A.; Zavala, K.; Vandewege, M.W.; Opazo, J.C. Evolution of the α2 -Adrenoreceptors in Vertebrates: ADRA2D Is Absent in Mammals and Crocodiles. Gen. Comp. Endocrinol. 2017, 250, 85–94. [Google Scholar] [CrossRef]
- Hein, L.; Altman, J.D.; Kobilka, B.K. Two Functionally Distinct α2-Adrenergic Receptors Regulate Sympathetic Neurotransmission. Nature 1999, 402, 181–184. [Google Scholar] [CrossRef]
- Philipp, M.; Brede, M.; Hein, L. Physiological Significance of α2-Adrenergic Receptor Subtype Diversity: One Receptor Is Not Enough. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R287–R295. [Google Scholar] [CrossRef] [Green Version]
- Uys, M.M.; Shahid, M.; Harvey, B.H. Therapeutic Potential of Selectively Targeting the α2C-Adrenoceptor in Cognition, Depression, and Schizophrenia—New Developments and Future Perspective. Front. Psychiatry 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.Z.; Davies, M.F.; Kingery, W.S.; Patterson, A.J.; Limbird, L.E.; Maze, M. Nitrous Oxide Produces Antinociceptive Response via Alpha2B and/or Alpha2C Adrenoceptor Subtypes in Mice. Anesthesiology 1999, 90, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, C.A.; Stone, L.S.; Wilcox, G.L. Pharmacological Profiles of Alpha 2 Adrenergic Receptor Agonists Identified Using Genetically Altered Mice and Isobolographic Analysis. Pharmacol. Ther. 2009, 123, 224–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlicker, E.; Feuerstein, T. Human Presynaptic Receptors. Pharmacol. Ther. 2017, 172, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.D.; Trendelenburg, A.U.; MacMillan, L.; Bernstein, D.; Limbird, L.; Starke, K.; Kobilka, B.K.; Hein, L. Abnormal Regulation of the Sympathetic Nervous System in Alpha2A-Adrenergic Receptor Knockout Mice. Mol. Pharmacol. 1999, 56, 154–161. [Google Scholar] [CrossRef] [Green Version]
- Trendelenburg, A.U.; Klebroff, W.; Hein, L.; Starke, K. A Study of Presynaptic Alpha2-Autoreceptors in Alpha2A/D-, Alpha2B- and Alpha2C-Adrenoceptor-Deficient Mice. Naunyn Schmiedebergs Arch. Pharmacol. 2001, 364, 117–130. [Google Scholar] [CrossRef]
- Brede, M.; Nagy, G.; Philipp, M.; Sørensen, J.B.; Lohse, M.J.; Hein, L. Differential Control of Adrenal and Sympathetic Catecholamine Release by α2 -Adrenoceptor Subtypes. Mol. Endocrin. 2003, 17, 1640–1646. [Google Scholar] [CrossRef] [Green Version]
- Quaglia, W.; Del Bello, F.; Giannella, M.; Piergentili, A.; Pigini, M. α2C-Adrenoceptor Modulators: A Patent Review. Expert. Opin. Ther. Pat. 2011, 21, 455–481. [Google Scholar] [CrossRef]
- Kaur, M.; Singh, P.M. Current Role of Dexmedetomidine in Clinical Anesthesia and Intensive Care. Anesth. Essays. Res. 2011, 5, 128–133. [Google Scholar] [CrossRef] [Green Version]
- Kaye, A.D.; Chernobylsky, D.J.; Thakur, P.; Siddaiah, H.; Kaye, R.J.; Eng, L.K.; Harbell, M.W.; Lajaunie, J.; Cornett, E.M. Dexmedetomidine in Enhanced Recovery After Surgery (ERAS) Protocols for Postoperative Pain. Curr. Pain Headache Rep. 2020, 24, 21. [Google Scholar] [CrossRef]
- Lorenz, W.; Lomasney, J.W.; Collins, S.; Regan, J.W.; Caron, M.G.; Lefkowitz, R.J. Expression of Three Alpha 2-Adrenergic Receptor Subtypes in Rat Tissues: Implications for Alpha 2 Receptor Classification. Mol. Pharmacol. 1990, 38, 599–603. [Google Scholar]
- Ibacache, M.; Sanchez, G.; Pedrozo, Z.; Galvez, F.; Humeres, C.; Echevarria, G.; Duaso, J.; Hassi, M.; Garcia, L.; Díaz-Araya, G.; et al. Dexmedetomidine Preconditioning Activates Pro-Survival Kinases and Attenuates Regional Ischemia/Reperfusion Injury in Rat Heart. Biochim. Biophys. Acta 2012, 1822, 537–545. [Google Scholar] [CrossRef] [Green Version]
- Maltsev, A.V.; Kokoz, Y.M.; Evdokimovskii, E.V.; Pimenov, O.Y.; Reyes, S.; Alekseev, A.E. Alpha-2 Adrenoceptors and Imidazoline Receptors in Cardiomyocytes Mediate Counterbalancing Effect of Agmatine on NO Synthesis and Intracellular Calcium Handling. J. Mol. Cell. Cardiol. 2014, 68, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhou, C.-L.; Xia, Z.-Y.; Wang, L. Effects of Dexmedetomidine on L-Type Calcium Current in Rat Ventricular Myocytes. Acta Cardiol. Sin. 2013, 29, 175–180. [Google Scholar]
- Yoshikawa, Y.; Hirata, N.; Kawaguchi, R.; Tokinaga, Y.; Yamakage, M. Dexmedetomidine Maintains Its Direct Cardioprotective Effect Against Ischemia/Reperfusion Injury in Hypertensive Hypertrophied Myocardium. Anesth. Analg. 2018, 126, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A. Chapter Two—Arrestins in the Cardiovascular System: An Update. In Progress in Molecular Biology and Translational Science; Teplow, D.B., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 159, pp. 27–57. [Google Scholar] [CrossRef]
- Liggett, S.B.; Ostrowski, J.; Chesnut, L.C.; Kurose, H.; Raymond, J.R.; Caron, M.G.; Lefkowitz, R.J. Sites in the Third Intracellular Loop of the Alpha 2A-Adrenergic Receptor Confer Short Term Agonist-Promoted Desensitization. Evidence for a Receptor Kinase-Mediated Mechanism. J. Biol. Chem. 1992, 267, 4740–4746. [Google Scholar] [CrossRef]
- Eason, M.G.; Liggett, S.B. Subtype-Selective Desensitization of Alpha 2-Adrenergic Receptors. Different Mechanisms Control Short and Long Term Agonist-Promoted Desensitization of Alpha 2C10, Alpha 2C4, and Alpha 2C2. J. Biol. Chem. 1992, 267, 25473–25479. [Google Scholar] [CrossRef]
- Devedjian, J.-C.; Esclapez, F.; Denis-Pouxviel, C.; Paris, H. Further Characterization of Human α2-Adrenoceptor Subtypes: [3H]RX821002 Binding and Definition of Additional Selective Drugs. Eur. J. Pharmacol. 1994, 252, 43–49. [Google Scholar] [CrossRef]
- Bylund, D.B.; Ray-Prenger, C.; Murphy, T.J. Alpha-2A and Alpha-2B Adrenergic Receptor Subtypes: Antagonist Binding in Tissues and Cell Lines Containing Only One Subtype. J. Pharmacol. Exp. Ther. 1988, 245, 600–607. [Google Scholar] [PubMed]
- Sallinen, J.; Höglund, I.; Engström, M.; Lehtimäki, J.; Virtanen, R.; Sirviö, J.; Wurster, S.; Savola, J.-M.; Haapalinna, A. Pharmacological Characterization and CNS Effects of a Novel Highly Selective α2C-Adrenoceptor Antagonist JP-1302. Br. J. Pharmacol. 2007, 150, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anti-α2B-Adrenergic Receptor (Extracellular) Antibody. Available online: https://www.alomone.com/p/anti-2b-adrenoceptor-extracellular/AAR-021 (accessed on 2 February 2021).
- Anti-Alpha 2C Adrenergic Receptor/ADRA2C Antibody (Ab46536). Archived Datasheet (PDF). Available online: https://www.abcam.com/alpha-2c-adrenergic-receptoradra2c-antibody-ab46536.html (accessed on 2 February 2021).
- Gurevich, E.V.; Gurevich, V.V. GRKs as Modulators of Neurotransmitter Receptors. Cells 2021, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Hurt, C.M.; Sorensen, M.W.; Angelotti, T. Common α2A and α2C Adrenergic Receptor Polymorphisms Do Not Affect Plasma Membrane Trafficking. Naunyn Schmiedebergs Arch. Pharmacol. 2014, 387, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, S.K. Seven-Transmembrane Receptors and Ubiquitination. Circ. Res. 2007, 100, 1142–1154. [Google Scholar] [CrossRef] [Green Version]
- Hara, M.; Zhou, Z.-Y.; Hemmings, H.C., Jr. α2-Adrenergic Receptor and Isoflurane Modulation of Presynaptic Ca2+ Influx and Exocytosis in Hippocampal Neurons. Anesthesiology 2016, 125, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zefirov, T.L.; Khisamieva, L.I.; Ziyatdinova, N.I.; Zefirov, A.L. Selective Blockade of α2-Adrenoceptor Subtypes Modulates Contractility of Rat Myocardium. Bull. Exp. Biol. Med. 2016, 162, 177–179. [Google Scholar] [CrossRef]
- Baier, M.J.; Klatt, S.; Hammer, K.P.; Maier, L.S.; Rokita, A.G. Ca2+/Calmodulin-Dependent Protein Kinase II Is Essential in Hyperacute Pressure Overload. J. Mol. Cell. Cardiol. 2020, 138, 212–221. [Google Scholar] [CrossRef]
- Ackers-Johnson, M.; Li, P.Y.; Holmes, A.P.; O’Brien, S.-M.; Pavlovic, D.; Foo, R.S. A Simplified, Langendorff-Free Method for Concomitant Isolation of Viable Cardiac Myocytes and Nonmyocytes From the Adult Mouse Heart. Circ. Res. 2016, 119, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Alekseev, A.E.; Markevich, N.I.; Korystova, A.F.; Terzic, A.; Kokoz, Y.M. Comparative Analysis of the Kinetic Characteristics of L-Type Calcium Channels in Cardiac Cells of Hibernators. Biophys J. 1996, 70, 786–797. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Primer Sequence | GenBank Index | Localization |
|---|---|---|---|
| Adra2a_F | TTTCCCCTGTGCCTAACTGC | NM_007417.5 | 3072–3091 |
| Adra2a_R | TGGCTTTATACACGGGGCTG | 2250–2269 | |
| Adra2a_P | FAM-ACAGCGATGGACCAAGGCAGAAGG-BHQ1 | 2222–2246 | |
| Adra2b_F | TTCAACCTCGCAGAGAGCAG | NM_009633.4 | 2728–2747 |
| Adra2b_R | CTCTAGCGCATTTCCCCCAT | 2834–2815 | |
| Adra2b_P | GCCTGCCGCCT-R6G-ACTTGCAGCAGGG-BHQ1 | 2758–2781 | |
| Adra2c_F | AGTTGCCAGAACCGCTCTTT | NM_007418.3 | 2546–2565 |
| Adra2c_R | GAGCGCCTGAAGTCCTGATT | 2648–2629 | |
| Adra2c_P | Cy3-TGCAACAGTTCGCTCAACCCGGT-BHQ2 | 2590–2612 | |
| GAPDH_F | GGGTCCCAGCTTAGGTTCAT | NM_001289726.1 | 32–51 |
| GAPDH_R | CCCAATACGGCCAAATCCGT | 131–112 | |
| GAPDH_P | Cy5-CAGGAGAGTGTTTCCTCGTCCCGT-BHQ2 | 62–85 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evdokimovskii, E.V.; Jeon, R.; Park, S.; Pimenov, O.Y.; Alekseev, A.E. Role of α2-Adrenoceptor Subtypes in Suppression of L-Type Ca2+ Current in Mouse Cardiac Myocytes. Int. J. Mol. Sci. 2021, 22, 4135. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084135
Evdokimovskii EV, Jeon R, Park S, Pimenov OY, Alekseev AE. Role of α2-Adrenoceptor Subtypes in Suppression of L-Type Ca2+ Current in Mouse Cardiac Myocytes. International Journal of Molecular Sciences. 2021; 22(8):4135. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084135
Chicago/Turabian StyleEvdokimovskii, Edward V., Ryounghoon Jeon, Sungjo Park, Oleg Y. Pimenov, and Alexey E. Alekseev. 2021. "Role of α2-Adrenoceptor Subtypes in Suppression of L-Type Ca2+ Current in Mouse Cardiac Myocytes" International Journal of Molecular Sciences 22, no. 8: 4135. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084135