
Core RNA Interference Genes Involved in miRNA and Ta-siRNA Biogenesis in Hops and Their Expression Analysis after Challenging with Verticillium nonalfalfae



Abstract
:1. Introduction
2. Results
2.1. Characterization and Structural Analysis of RNAi Genes
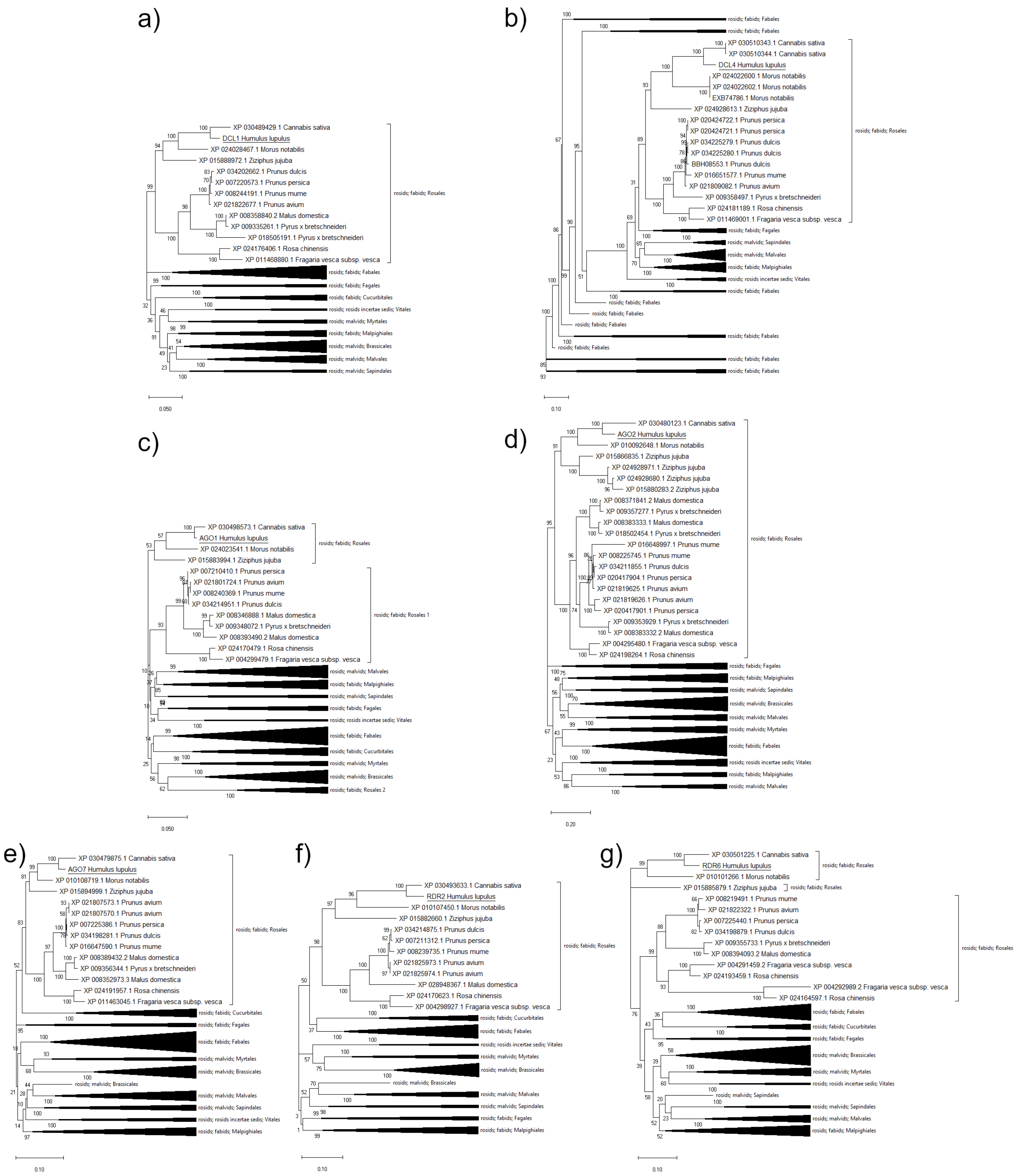
2.2. Phylogenetic Analysis
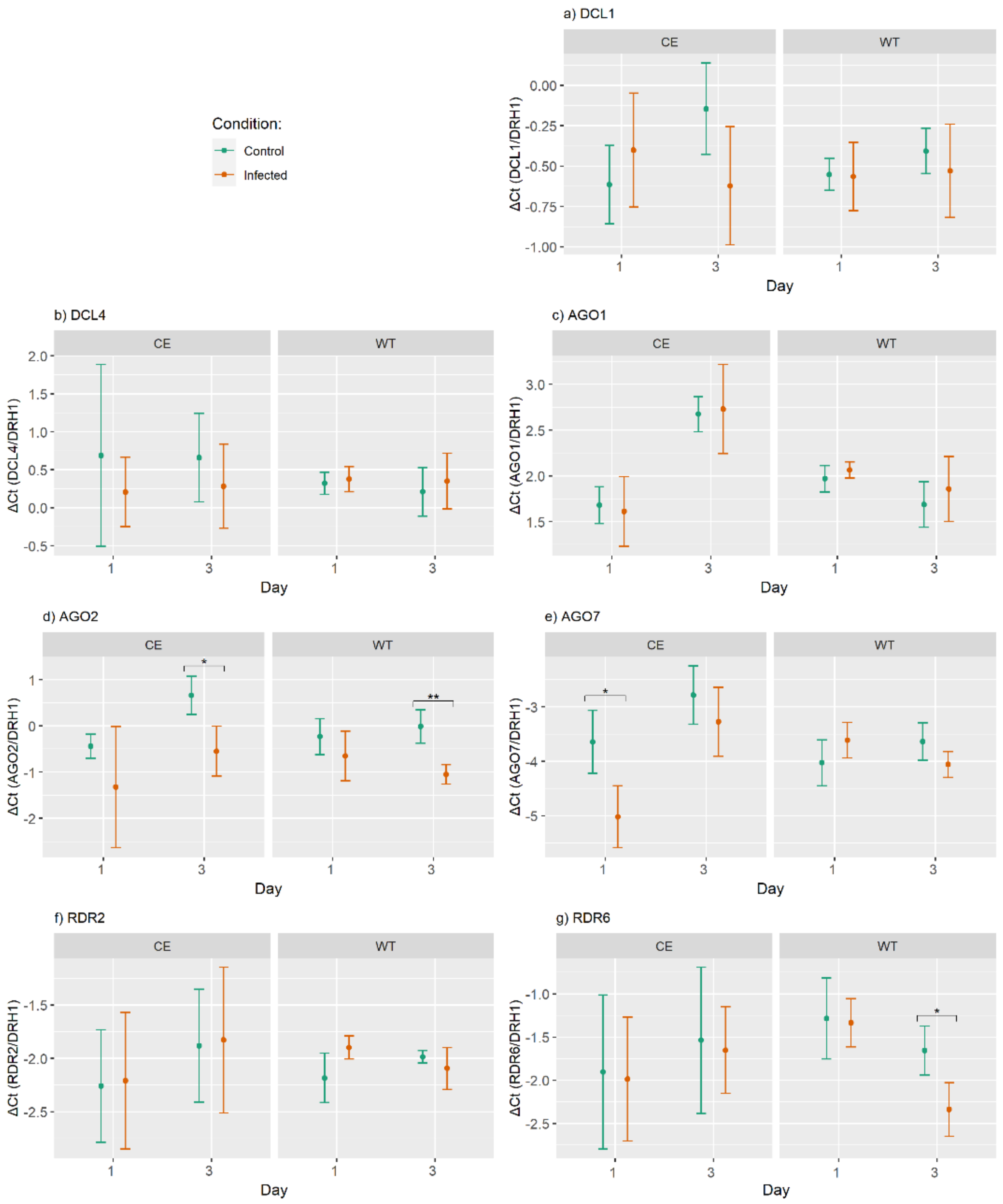
2.3. Differential Expression of RNAi Genes
3. Discussion
4. Materials and Methods
4.1. Identification of RNAi Genes
4.2. Phylogenetic Analysis
4.3. Hop Inoculation Experiment
4.4. Total RNA Isolation and qPCR Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shabalina, S.A.; Koonin, E.V. Origins and evolution of eukaryotic RNA interference. Trends Ecol. Evol. 2008, 23, 578–587. [Google Scholar] [CrossRef] [Green Version]
- Axtell, M.J. Classification and Comparison of Small RNAs from Plants. Ann. Rev. Plant Biol. 2013, 64, 137–159. [Google Scholar] [CrossRef] [Green Version]
- Khraiwesh, B.; Zhu, J.K.; Zhu, J.H. Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. BBA Gene Regul. Mech. 2012, 1819, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, A.W. Ribonuclease III mechanisms of double-stranded RNA cleavage. Wires RNA 2014, 5, 31–48. [Google Scholar] [CrossRef]
- Kurihara, Y.; Watanabe, Y. Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc. Natl. Acad. Sci. USA 2004, 101, 12753–12758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasciolli, V.; Mallory, A.C.; Bartel, D.P.; Vaucheret, H. Partially redundant functions of Arabidopsis DICER-like enzymes and a role for DCL4 in producing trans-acting siRNAs. Curr. Biol. 2005, 15, 1494–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, E.; Xie, Z.X.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuperus, J.T.; Carbonell, A.; Fahlgren, N.; Garcia-Ruiz, H.; Burke, R.T.; Takeda, A.; Sullivan, C.M.; Gilbert, S.D.; Montgomery, T.A.; Carrington, J.C. Unique functionality of 22-nt miRNAs in triggering RDR6-dependent siRNA biogenesis from target transcripts in Arabidopsis. Nat. Struct. Mol. Biol. 2010, 17, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhang, Q.; Yan, K.; Zhou, M.M. Structural insights into piRNA recognition by the human PIWI-like 1 PAZ domain. Proteins 2011, 79, 2004–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, X.F.; Qi, Y.J. RNAi in Plants: An Argonaute-Centered View. Plant Cell 2016, 28, 272–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumberger, N.; Baulcombe, D.C. Arabidopsis ARGONAUTE1 is an RNA Slicer that selectively recruits microRNAs and short interfering RNAs. Proc. Natl. Acad. Sci. USA 2005, 102, 11928–11933. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, T.A.; Howell, M.D.; Cuperus, J.T.; Li, D.W.; Hansen, J.E.; Alexander, A.L.; Chapman, E.J.; Fahlgren, N.; Allen, E.; Carrington, J.C. Specificity of ARGONAUTE7-miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell 2008, 133, 128–141. [Google Scholar] [CrossRef] [Green Version]
- Mi, S.J.; Cai, T.; Hu, Y.G.; Chen, Y.; Hodges, E.; Ni, F.R.; Wu, L.; Li, S.; Zhou, H.; Long, C.Z.; et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 2008, 133, 116–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenackers, B.; De Cooman, L.; De Vos, D. Chemical transformations of characteristic hop secondary metabolites in relation to beer properties and the brewing process: A review. Food Chem. 2015, 172, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Hrncic, M.K.; Spaninger, E.; Kosir, I.J.; Knez, Z.; Bren, U. Hop Compounds: Extraction Techniques, Chemical Analyses, Antioxidative, Antimicrobial, and Anticarcinogenic Effects. Nutrients 2019, 11, 257. [Google Scholar] [CrossRef] [Green Version]
- Savary, S.; Ficke, A.; Aubertot, J.N.; Hollier, C. Crop losses due to diseases and their implications for global food production losses and food security. Food Secur. 2012, 4, 519–537. [Google Scholar] [CrossRef]
- Radisek, S.; Jakse, J.; Simoncic, A.; Javornik, B. Characterization of Verticillium albo-atrum field isolates using pathogenicity data and AFLP analysis. Plant Dis. 2003, 87, 633–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadeta, K.; Thomma, B.P.H.J. The xylem as battleground for plant hosts and vascular wilt pathogens. Front. Plant Sci. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, G.Z. Origin and evolution of the plant immune system. New Phytol. 2019, 222, 70–83. [Google Scholar] [CrossRef] [Green Version]
- Segers, G.C.; Zhang, X.M.; Deng, F.Y.; Sun, Q.H.; Nuss, D.L. Evidence that RNA silencing functions as an antiviral defense mechanism in fungi. Proc. Natl. Acad. Sci. USA 2007, 104, 12902–12906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jesenicnik, T.; Stajner, N.; Radisek, S.; Jakse, J. RNA interference core components identified and characterised in Verticillium nonalfalfae, a vascular wilt pathogenic plant fungi of hops. Sci. Rep. 2019, 9, 8651. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.S.; Zhang, Z.; Liu, Y. RNA interference pathways in fungi: Mechanisms and functions. Ann. Rev. Microbiol. 2012, 66, 305–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, C.L.; Zhao, J.H.; Guo, H.S. Trans-Kingdom RNA Silencing in Plant-Fungal Pathogen Interactions. Mol. Plant 2018, 11, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhao, Y.L.; Zhao, J.H.; Wang, S.; Jin, Y.; Chen, Z.Q.; Fang, Y.Y.; Hua, C.L.; Ding, S.W.; Guo, H.S. Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nat. Plants 2016, 2. [Google Scholar] [CrossRef]
- Weiberg, A.; Wang, M.; Lin, F.M.; Zhao, H.W.; Zhang, Z.H.; Kaloshian, I.; Huang, H.D.; Jin, H.L. Fungal Small RNAs Suppress Plant Immunity by Hijacking Host RNA Interference Pathways. Science 2013, 342, 118–123. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.Y.; Xu, Y.P.; Zhao, L.; Li, S.S.; Cai, X.Z. Tight regulation of the interaction between Brassica napus and Sclerotinia sclerotiorum at the microRNA level. Plant Mol. Biol. 2016, 92, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Ellendorff, U.; Fradin, E.F.; de Jonge, R.; Thomma, B.P.H.J. RNA silencing is required for Arabidopsis defence against Verticillium wilt disease. J. Exp. Bot. 2009, 60, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Pokorn, T.; Radisek, S.; Javornik, B.; Stajner, N.; Jakse, J. Development of hop transcriptome to support research into host-viroid interactions. PLoS ONE 2017, 12, e0184528. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.Q.; van Velzen, R.; Bakker, F.T.; Sattarian, A.; Li, D.Z.; Yip, T.S. Molecular phylogenetics and character evolution of Cannabaceae. Taxon 2013, 62, 473–485. [Google Scholar] [CrossRef]
- Zhang, S.D.; Soltis, D.E.; Yang, Y.; Li, D.Z.; Yi, T.S. Multi-gene analysis provides a well-supported phylogeny of Rosales. Mol. Phylogenet. Evol. 2011, 60, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.P.; Feng, Y.; Zhu, Z.J. Dicer-like (DCL) proteins in plants. Funct. Integr. Genom. 2009, 9, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Arora, R.; Lama, T.; Nijhawan, A.; Khurana, J.P.; Tyagi, A.K.; Kapoor, S. Genome-wide identification, organization and phylogenetic analysis of Dicer-like, Argonaute and RNA-dependent RNA Polymerase gene families and their expression analysis during reproductive development and stress in rice. BMC Genom. 2008, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Liu, M.; Tang, M.; Dong, B.; Wu, D.; Zhang, Z.; Zhou, B. Repression of microRNA biogenesis by silencing of OsDCL1 activates the basal resistance to Magnaporthe oryzae in rice. Plant Sci. 2015, 237, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Blevins, T.; Rajeswaran, R.; Shivaprasad, P.V.; Beknazariants, D.; Si-Ammour, A.; Park, H.S.; Vazquez, F.; Robertson, D.; Meins, F.; Hohn, T.; et al. Four plant Dicers mediate viral small RNA biogenesis and DNA virus induced silencing. Nucleic Acids Res. 2006, 34, 6233–6246. [Google Scholar] [CrossRef] [Green Version]
- Moura, M.O.; Fausto, A.K.S.; Fanelli, A.; Guedes, F.A.D.; Silva, T.F.; Romanel, E.; Vaslin, M.F.S. Genome-wide identification of the Dicer-like family in cotton and analysis of the DCL expression modulation in response to biotic stress in two contrasting commercial cultivars. BMC Plant Biol. 2019, 19. [Google Scholar] [CrossRef] [Green Version]
- Shen, D.; Suhrkamp, I.; Wang, Y.; Liu, S.Y.; Menkhaus, J.; Verreet, J.A.; Fan, L.J.; Cai, D.G. Identification and characterization of microRNAs in oilseed rape (Brassica napus) responsive to infection with the pathogenic fungus Verticillium longisporum using Brassica AA (Brassica rapa) and CC (Brassica oleracea) as reference genomes. New Phytol. 2014, 204, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Meins, F.; Si-Ammour, A.; Blevins, T. RNA silencing systems and their relevance to plant development. Annu. Rev. Cell Dev. Biol. 2005, 21, 297–318. [Google Scholar] [CrossRef]
- Kidner, C.A.; Martienssen, R.A. The role of ARGONAUTE1 (AGO1) in meristem formation and identity. Dev. Biol. 2005, 280, 504–517. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.X.; Kasschau, K.D.; Carrington, J.C. Negative feedback regulation of Dicer-Like1 in Arabidopsis by microRNA-guided mRNA degradation. Curr. Biol. 2003, 13, 784–789. [Google Scholar] [CrossRef] [Green Version]
- Vaucheret, H.; Vazquez, F.; Crete, P.; Bartel, D.P. The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev. 2004, 18, 1187–1197. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.M.; Zhao, H.W.; Gao, S.; Wang, W.C.; Katiyar-Agarwal, S.; Huang, H.D.; Raikhel, N.; Jin, H.L. Arabidopsis Argonaute 2 Regulates Innate Immunity via miRNA393*-Mediated Silencing of a Golgi-Localized SNARE Gene, MEMB12. Mol. Cell 2011, 42, 356–366. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.B.; Jovel, J.; Udomporn, P.; Wang, Y.; Wu, Q.F.; Li, W.X.; Gasciolli, V.; Vaucheret, H.; Ding, S.W. The 21-Nucleotide, but Not 22-Nucleotide, Viral Secondary Small Interfering RNAs Direct Potent Antiviral Defense by Two Cooperative Argonautes in Arabidopsis thaliana. Plant Cell 2011, 23, 1625–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Xu, L.; Wang, H.; Yuan, Z.; Cao, X.F.; Yang, Z.N.; Zhang, D.B.; Xu, Y.Q.; Huang, H. The putative RNA-dependent RNA polymerase RDR6 acts synergistically with ASYMMETRIC LEAVES1 and 2 to repress BREVIPEDICELLUS and microRNA165/166 in Arabidopsis leaf development. Plant Cell 2005, 17, 2157–2171. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Singh, S.; Panigrahi, K.C.S.; Reski, R.; Sarkar, A.K. Balanced activity of microRNA166/165 and its target transcripts from the class III homeodomain-leucine zipper family regulates root growth in Arabidopsis thaliana. Plant Cell Rep. 2014, 33, 945–953. [Google Scholar] [CrossRef]
- Singh, A.; Roy, S.; Singh, S.; Das, S.S.; Gautam, V.; Yadav, S.; Kumar, A.; Singh, A.; Samantha, S.; Sarkar, A.K. Phytohormonal crosstalk modulates the expression of miR166/165s, target Class III HD-ZIPs, and KANADI genes during root growth in Arabidopsis thaliana. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Progar, V.; Jakse, J.; Stajner, N.; Radisek, S.; Javornik, B.; Berne, S. Comparative transcriptional analysis of hop responses to infection with Verticillium nonalfalfae. Plant Cell Rep. 2017, 36, 1599–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandelc, S.; Timperman, I.; Radisek, S.; Devreese, B.; Samyn, B.; Javornik, B. Comparative proteomic profiling in compatible and incompatible interactions between hop roots and Verticillium albo-atrum. Plant Physiol. Biochem. 2013, 68, 23–31. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Jakse, J.; Cerenak, A.; Radisek, S.; Satovic, Z.; Luthar, Z.; Javornik, B. Identification of quantitative trait loci for resistance to Verticillium wilt and yield parameters in hop (Humulus lupulus L.). Theor. Appl. Genet. 2013, 126, 1431–1443. [Google Scholar] [CrossRef]
- Flajsman, M.; Radisek, S.; Javornik, B. Pathogenicity Assay of Verticillium nonalfalfae on Hop Plants. Bio Protoc. 2017, 7. [Google Scholar] [CrossRef]
- Kunej, U.; Mikulic-Petkovsek, M.; Radisek, S.; Stajner, N. Changes in the Phenolic Compounds of Hop (Humulus lupulus L.) Induced by Infection with Verticillium nonalfalfae, the Causal Agent of Hop Verticillium Wilt. Plants 2020, 9, 841. [Google Scholar] [CrossRef] [PubMed]
- Stajner, N.; Cregeen, S.; Javornik, B. Evaluation of Reference Genes for RT-qPCR Expression Studies in Hop (Humulus lupulus L.) during Infection with Vascular Pathogen Verticillium albo-atrum. PLoS ONE 2013, 8, e068228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- R Foundation for Statistical Computing. R: A Language and Environment for Statistical Computing; Version 3.5.1; R Foundation for Statistical Computing: Vienna, Austria, 2017; Available online: https://www.R-project.org/ (accessed on 22 November 2019).
- rstatix: Pipe-Friendly Framework for Basic Statistical Tests. 2020. Available online: https://cran.r-project.org/web/packages/rstatix/index.html (accessed on 3 December 2020).




{kind=link}
{kind=link}
{kind=link}
| Protein | Number of Introns | Transcript Length (nt) | CDS Length (aa) | pI | Mw (Da) |
|---|---|---|---|---|---|
| DCL1 | 19 | 6497 | 1984 | 6.03 | 222,623.18 |
| DCL4 | 24 | 5281 | 1645 | 6.14 | 184,947.75 |
| AGO1 | 21 | 4519 | 1035 | 9.22 | 114,542.48 |
| AGO2 | 2 | 4333 | 1038 | 9.41 | 115,025.66 |
| AGO7 | 2 | 3485 | 1029 | 9.33 | 117,545.61 |
| RDR2 | 3 | 3743 | 941 | 7.03 | 106,793.04 |
| RDR6 | 2 | 4270 | 1204 | 6.66 | 137,281.43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kunej, U.; Jakše, J.; Radišek, S.; Štajner, N. Core RNA Interference Genes Involved in miRNA and Ta-siRNA Biogenesis in Hops and Their Expression Analysis after Challenging with Verticillium nonalfalfae. Int. J. Mol. Sci. 2021, 22, 4224. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084224
Kunej U, Jakše J, Radišek S, Štajner N. Core RNA Interference Genes Involved in miRNA and Ta-siRNA Biogenesis in Hops and Their Expression Analysis after Challenging with Verticillium nonalfalfae. International Journal of Molecular Sciences. 2021; 22(8):4224. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084224
Chicago/Turabian StyleKunej, Urban, Jernej Jakše, Sebastjan Radišek, and Nataša Štajner. 2021. "Core RNA Interference Genes Involved in miRNA and Ta-siRNA Biogenesis in Hops and Their Expression Analysis after Challenging with Verticillium nonalfalfae" International Journal of Molecular Sciences 22, no. 8: 4224. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22084224